
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of a unique 3d/4f heterometallic square prismatic box-like coordination cage†
Li
Li
a,
Yingjie
Zhang
b,
Maxim
Avdeev
b,
Leonard F.
Lindoy
c,
David G.
Harman
d,
Rongkun
Zheng
 e,
Zhenxiang
Cheng
f,
Janice R.
Aldrich-Wright
a and
Feng
Li
*a
e,
Zhenxiang
Cheng
f,
Janice R.
Aldrich-Wright
a and
Feng
Li
*a
aSchool of Science and Health, University of Western Sydney, Penrith, NSW 2751, Australia. E-mail: feng.li@westernsydney.edu.au
bAustralian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia
cSchool of Chemistry, The University of Sydney, NSW 2006, Australia
dMolecular Medicine Research Group, School of Medicine, Building 30, University of Western Sydney, Goldsmith Avenue, Campbelltown, NSW 2560, Australia
eSchool of Physics, The University of Sydney, NSW 2006, Australia
fInstitute for Superconducting & Electronic Materials (ISEM), University of Wollongong, North Wollongong, NSW 2500, Australia
First published on 23rd May 2016
Abstract
We present the synthesis and characterization of a unique, slightly distorted square prismatic, box-like coordination cage of type [Cu6Dy8L8(MeOH)8(H2O)6](NO3)12·χsolvent obtained via the supramolecular assembly between a non-centrosymmetric Dy(III) metalloligand and Cu(II) nitrate. Magnetic susceptibility measurements indicate that the complex behaves as a single-molecule magnet.
Both homo- and heterometallo-coordination cages have continued to receive considerable attention over recent years,1,2 motivated by a range of potential applications as new materials for gas adsorption,3 drug delivery,4 catalysis,5 magnetic materials,6–10 and host–guest phenomena11,12 as well as their use as ‘molecular flasks’.13 Although a number of coordination cage types have been developed, examples of heterometallic systems incorporating paramagnetic metal centres are rare10,14,15 and the design and successful construction of these systems still represent a significant challenge.
In previous studies, our group14 and others3,10,15–21 have reported the structures of a range of homonuclear M14 and heteronuclear M′6/M′′8 metallocages where M, M′ and M′′ are mono-valent, divalent and/or trivalent d-block metal ions typically displaying four to six coordination geometries in their respective cage products. In these species, which typically display high symmetry, eight 6-coordinate metal centres occupy the corners of each cage, while a further six metals are positioned on or above each of the six faces to yield either distorted face-centred cubic or rhombo-dodecahedral arrangements of the overall fourteen metal centres. While the metal ions employed so far to prepare such cages have included Co(II)/(III), Cr(III), Ni(II), Fe(II)/(III), Cu(I)/(II), Ru(II), Pd(II) and Pt(II), to the best of our knowledge no such homonuclear (M14) or heteronuclear (M′6/M′′8) cages incorporating lanthanide(III) ions have been reported. Apart from the potential of new materials of this type for generating potentially useful properties such as unusual fluorescence22 or magnetic23,24 behaviour, the use of a Ln(III) ion with its propensity for adopting a coordination number greater than six appeared likely to result in a topological change away from cubic in the product cage. Anticipation of such an outcome provided a further motivation for undertaking the present study.
As an extension of our previous studies of a heteronuclear cage incorporating only 3d transition metals,14 we now describe the design and preparation of the new discrete 3d/4f coordination cage (1; [Cu6Dy8L8(MeOH)8(H2O)6](NO3)12·χsolvent) derived from a robust non-centrosymmetric Dy(III)-containing metalloligand. Excluding Ln-oxo-3d bridged systems, 1 appears to be the first example of such a 3d–4f coordination cage (see below). We also report its characterization and demonstrate that this cage system shows single molecular magnet behaviour.
The triprotoned form of the metalloligand [DyH3L(NO3)]2+ was prepared as reported previously.25 It exists as two enantiomers in both the solid state and solution, due to the screw coordination arrangement of the (achiral) distorted H3L tripodal ligand around Dy(III) ion.25 The discrete heterometallic Cu(II)6/Dy(III)8 box-like coordination cage, [Cu6Dy8(MeOH)8(H2O)6L8](NO3)12·χsolvent was synthesized via a self-assembly procedure employing the pre-assembled metalloligand,25 [DyH3L(NO3)]2+ (where H3L = tris[2-((imidazol-4-yl)methylidene)amino)ethyl]amine) and Cu(II) nitrate in methanol under basic conditions. Diffusion of diethyl ether vapour into the methanol solution afforded purple plate-shaped crystals of 1 in 69% yield.
An X-ray diffraction study showed that cage 1 crystallizes in the monoclinic space group C2/c (Table S1†). Dy(III) ions define the eight corners of the box-like structure while Cu(II) ions occupy the centres of its six faces (Fig. 1). Each complex is chiral, with either ΛΛΛΛΛΛΛΛ or ΔΔΔΔΔΔΔΔ configurations at the Dy(III) centres. Moreover, the chirality of [DyL(MeOH)] centres is communicated through the Cu(II) “propellers”, such that all Dy(III) centres have the same handedness within an individual cage, with the crystal structure containing an equal ratio of each of the cage enantiomers.
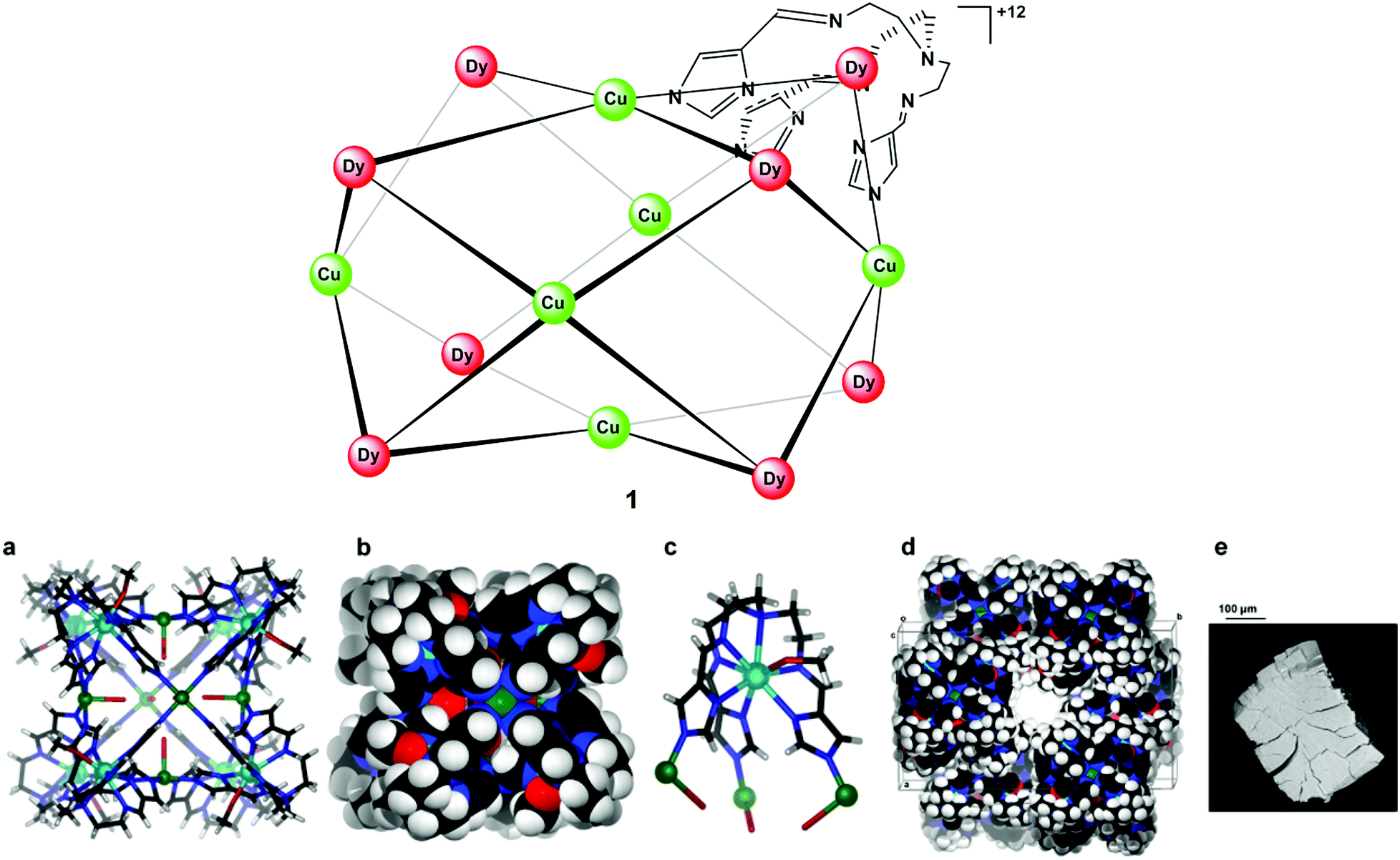 | ||
| Fig. 1 A crystal structure of cage 1. Colour codes: C-black, Cu-green, Dy-cyan, H-white, N-blue and O-red. (a) Single-crystal structure of the heterometallic cage 1 viewed from slightly off the C4 symmetry axes; (b) space-filling model of the structure; (c) the coordination environment of the respective Dy(III) sites; (d) schematic representation of part of the crystal packing of 1 viewed down the c-axis and showing the top of one of the one-dimensional channels present in the crystal structure; (e) SEM image showing the shape of a crystal of 1. | ||
As anticipated, the structure is not a cube but shows a deviation from cubic symmetry that reflects the presence of the non-centrosymmetric 8-coordinate Dy(III) corners that direct the structure towards a slightly distorted square prismatic box with the unequal sides having mean lengths of 8.99 ± 0.06 and 9.65 ± 0.02 Å, respectively, with the ‘corner’ angles between sides being close to right angles (89.62–90.47°); alternate corners of the box show a slight non-equivalence (Fig. S1†). Angles between the coordination vectors defined by the N–Dy–N angles at each corner of the square prism, where N is the copper-bound imidazolate donor atom, are 89.02°, 67.60°, 80.18° (Dy1); 87.10°, 67.35°, 79.06° (Dy2); 88.17°, 67.32°, 77.60° (Dy3); and 87.91°, 69.61°, 79.30° (Dy4), respectively, for the four crystallographically distinct Dy(III) centres.
Scanning electron microscopy (SEM) photograph indicated that the crystals of nanocage 1 undergo rapid decay due to loss of solvent (Fig. 1e). In addition, scanning electron microscopy-energy-dispersive spectroscopy (SEM-EDS) analysis of 1 confirmed that, as expected, the ratio of Dy(III) and Cu(II) inside the cage is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Fig. S2†).
:3 (Fig. S2†).
Electrospray ionization high resolution mass spectrometry (ESI-HRMS) provided evidence that heterometallic 1 persists in solution. When 1 was dissolved in Milli-Q water, the major ions in the ESI-HRMS occurred at m/z values corresponding to {[Cu6Dy8L8](NO3)8}4+; {[Cu6Dy8L8](NO3)7}5+; {[Cu6Dy8L8](NO3)6}6+ (Fig. S3†), consistent with the loss of four, five and six nitrate anions from the heterometallic cluster, respectively. A few minor charge species were also easily identified, corresponding to the loss of two {[Cu6Dy8L8](NO3)10}2+, three {[Cu6Dy8L8](NO3)9}3+, seven {[Cu6Dy8L8](NO3)5}7+ and eight nitrates {[Cu6Dy8L8](NO3)6}6+. In addition, a singly charged species of type {[Cu6Dy8L8](NO3)11}1+, resulting from the loss of one nitrate, was observed at low intensity when the source cone voltage was increased (Fig. S4†). The expected isotope patterns for the various charged species (Fig. S4–S11†) were also observed, with the isotopic distributions for all the above eight species being in excellent agreement with their simulated patterns.
The absorption spectrum of 1 in the solid state over 2000–200 nm is shown in Fig. S12.† The spectrum in the UV-vis region (Fig. S12a†) shows an intense broad absorption band at 560 nm and a lower intensity band at 330 nm, the former arising from the d–d transitions characteristic of the Cu(II) ion and the latter assigned to the π–π* transitions associated with the bridged imidazolate ligand. The spectrum in the vis-NIR region (Fig. S12b and c†) exhibits nine distinct peaks attributable to the f–f transitions arising from the Dy(III) ion with the six expected absorption bands at 1637 (6H15/2), 1295 (6H15/2), 1107 (6H9/2), 910 (6H7/2), 810 (6H5/2) and 760 (6H3/2) nm.26 The additional bands at 1420, 1727 and 1920 nm can also be assigned to the f–f transition between 6H11/2 and the ground state, 6H15/2. The extensive splitting of the 6H11/2 state is due to the presence of multiple Dy(III) centres in cage 1. The upper states of Dy(III) ions for cage 1 together with the values from Chandrasekhar et al.26 are summarized in Table S2.†
FT-IR spectrum and Raman spectrum of 1 were recorded at room temperature (Fig. S13 and S14†). The FT-IR spectrum shows absorptions in the region 1600–1500 cm−1 that are typical for imidazolate-imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching modes. The FT-IR and Raman spectrum of 1 are very similar and also in accord with the presence of CN groups.
N) stretching modes. The FT-IR and Raman spectrum of 1 are very similar and also in accord with the presence of CN groups.
Zero-field-cooled DC magnetic susceptibility was measured over the temperature range 2.5–300 K under a field of 1000 Oe. Analysis of the data with the Curie–Weiss law using the range 200–300 K indicated almost solely paramagnetic behaviour to yield θ = 0.29(13) K (Fig. 2). The χT plot above 100 K is nearly constant at a value of 110 emu K mol−1 (Fig. 2), from which the effective magnetic moment can be derived: 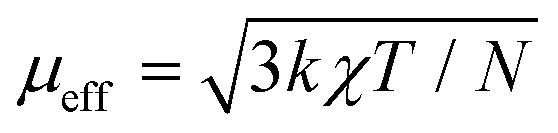 , where k, N and μB are the Boltzmann constant, Avogadro constant and Bohr magneton, respectively. The measured μeff = 29.6μB is in good agreement with the expected value 30.4μB for eight Dy(III) (S = 5/2, L = 5, J = 15/2,
, where k, N and μB are the Boltzmann constant, Avogadro constant and Bohr magneton, respectively. The measured μeff = 29.6μB is in good agreement with the expected value 30.4μB for eight Dy(III) (S = 5/2, L = 5, J = 15/2, 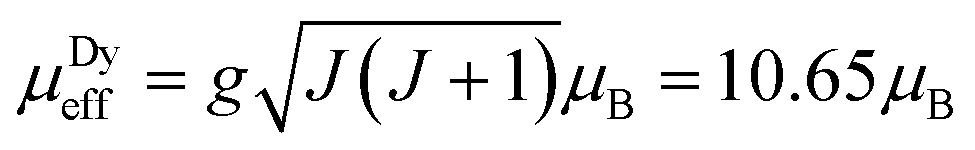 ) and six spin-only Cu(II) non-interacting ions (S = 1/2,
) and six spin-only Cu(II) non-interacting ions (S = 1/2, 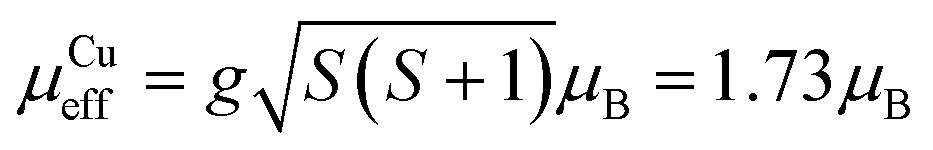 ). As shown in Fig. 2, χT decreases with a lowering of the temperature below 100 K, with the magnetic moment of the ions starting to be affected by zero-field splitting (ZFS).24χT reaches minimum at 11 K and then rapidly increases with further lowering of the temperature and reaches 121 emu K mol−1 at 3 K, indicating the effect of ferromagnetic coupling between the Dy(III) and Cu(II) metal centers.24 The ZFS and ferromagnetic coupling predict single-molecule-magnet (SMM) behavior for this cage. Magnetization curves were measured at a series of temperatures and plotted as a function of the field-temperature ratio H/T (Fig. 2). The magnetization rises rapidly with the ramping field and then continues to increase gradually. The maximum magnetic moment per molecule reaches 51.9μB at T = 2.5 K and H = 9 T, but is not saturated. The 51.9μB is significantly smaller than the nominal value of 86μB for eight Dy(III) (μDyz = gJμB = 10μB) and six spin-only Cu(II) (μCuz = gSμB = 1μB), but close to the values found in highly magnetically anisotropic systems in which Dy(III) contribution saturated around 5–5.2μB per atom.27–29
). As shown in Fig. 2, χT decreases with a lowering of the temperature below 100 K, with the magnetic moment of the ions starting to be affected by zero-field splitting (ZFS).24χT reaches minimum at 11 K and then rapidly increases with further lowering of the temperature and reaches 121 emu K mol−1 at 3 K, indicating the effect of ferromagnetic coupling between the Dy(III) and Cu(II) metal centers.24 The ZFS and ferromagnetic coupling predict single-molecule-magnet (SMM) behavior for this cage. Magnetization curves were measured at a series of temperatures and plotted as a function of the field-temperature ratio H/T (Fig. 2). The magnetization rises rapidly with the ramping field and then continues to increase gradually. The maximum magnetic moment per molecule reaches 51.9μB at T = 2.5 K and H = 9 T, but is not saturated. The 51.9μB is significantly smaller than the nominal value of 86μB for eight Dy(III) (μDyz = gJμB = 10μB) and six spin-only Cu(II) (μCuz = gSμB = 1μB), but close to the values found in highly magnetically anisotropic systems in which Dy(III) contribution saturated around 5–5.2μB per atom.27–29
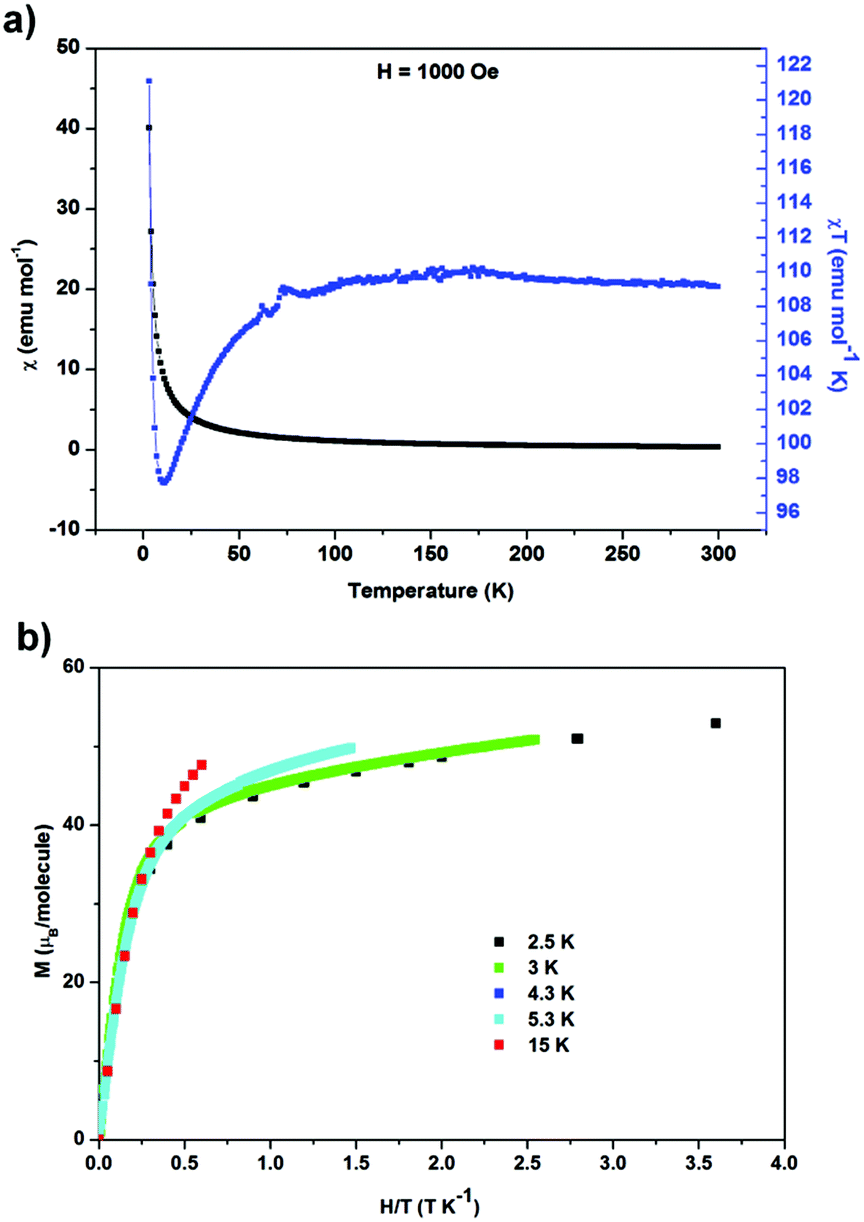 | ||
| Fig. 2 (a) Magnetic susceptibility χ and χT product as functions of temperature; (b) isothermal magnetization as a function of field-temperature ratio at various temperatures. | ||
AC magnetic susceptibility was measured in an ac magnetic field of 3.0 Oe, oscillating at frequencies ranging from 100 to 5000 Hz at temperatures of 2.5–10.0 K to confirm the possible SMM behaviour (Fig. 3). Indeed, the out-of-phase susceptibility, χ′′, demonstrated characteristic frequency dependence. The faster experimental timescale enables the observation of the slow relaxation at higher temperatures. The blocking temperature is below 2.5 K as it was not observed in our temperature range (Fig. 3), which implies the corresponding energy barrier is below 42 cm−1.
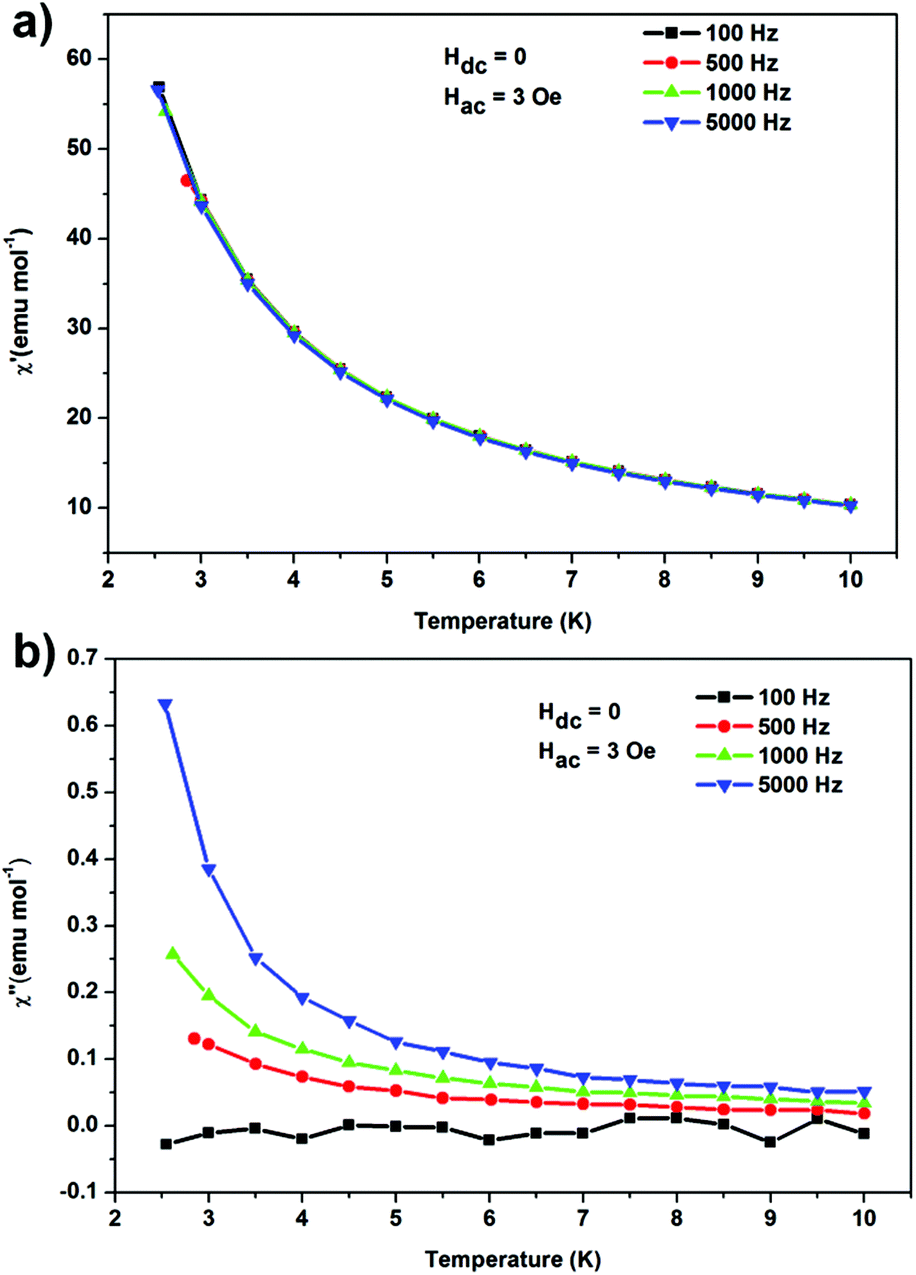 | ||
| Fig. 3 Temperature dependence of the in- (a) and out-of-phase (b) components of the AC susceptibility for frequencies of 100–5000 kHz. | ||
In conclusion, we describe the efficient synthesis of a unique example of a new heterometallic mixed-valent cationic cage of type [Cu6Dy8(MeOH)8(H2O)6L8]12+ incorporating eight metalloligands of type [DyL(MeOH)], with the square prismatic structure having been unambiguously characterized by X-ray crystallography, ESI mass spectrometry, FT-IR, and UV–vis–NIR spectroscopy. The results illustrate how non-centrosymmetric corner units can result in modification of the shape of the resulting cage structure from the usual cubic arrangement generated when centosymmetric corner units are employed, which has been reported by Clever30 using a non-centrosymmetric organic ligand. Further, the magnetic characterization shows the slow magnetic relaxation, characteristic of single-molecule magnets. As typical of a polymetallic lanthanide-based cage compound with large number of magnetic centers,31 the SMM behaviour of the title material is also rather weak. However, distortion of the cage from the cubic shape using non-centrosymmetric ligands as demonstrated in this work opens the new way to further increase overall anisotropy of magnetic cages and thus utilize large total spin of the high-nuclear lanthanide SMM systems. The complex behaviours of χT reveal the ZFS and ferromagnetic coupling between the Dy(III) and Cu(II) metal centres at low temperature.
In spite of several M′6/M′′8-based metallocages now reported,10,15–211 represents the first example of such a heteronuclear cage structure incorporating Ln(III) as one of the metal ions composing the heteronuclear pair. Further, in contrast to the previous systems of the above type based on symmetrical metalloligand building blocks, the present system involves the use of a non-symmetric metalloligand in coordination cage systems for the first time and we demonstrate that it leads to a square prismatic, box-like structure rather than the usual symmetrical cubic cage arrangement that is normal for such systems. The present result clearly opens the way for the use of coordination sphere ‘tuning’ of the metal centre in a metalloligand as a means for modulating (and directing) the topology of the resulting cage in a systematic manner. We are currently undertaking further investigations in this direction.
The research described herein was supported by the Western Sydney University. The authors acknowledge AMCF facilities at the Western Sydney University and MX1 beamline at the Australian Synchrotron. L. L. also acknowledges Australian Postgraduate Award and the Western Sydney University Top-up Award.
Notes and references
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- M. B. Duriska, S. M. Neville, J. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 8919–8922 CrossRef PubMed.
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef PubMed.
- H. Li, Y.-F. Han, Y.-J. Lin, Z.-W. Guo and G.-X. Jin, J. Am. Chem. Soc., 2014, 136, 2982–2985 CrossRef PubMed.
- S. Kang, H. Zheng, T. Liu, K. Hamachi, S. Kanegawa, K. Sugimoto, Y. Shiota, S. Hayami, M. Mito, T. Nakamura, M. Nakano, M. L. Baker, H. Nojiri, K. Yoshizawa, C. Duan and O. Sato, Nat. Commun., 2015, 6, 5955 CrossRef PubMed.
- F. Li, N. F. Sciortino, J. K. Clegg, S. M. Neville and C. J. Kepert, Aust. J. Chem., 2014, 67, 1625–1628 CrossRef.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Letard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549–2552 CrossRef PubMed.
- D. Li, S. Parkin, G. Wang, G. T. Yee, R. Clérac, W. Wernsdorfer and S. M. Holmes, J. Am. Chem. Soc., 2006, 128, 4214–4215 CrossRef PubMed.
- S. Wang, J.-L. Zuo, H.-C. Zhou, H. J. Choi, Y. Ke, J. R. Long and X.-Z. You, Angew. Chem., Int. Ed., 2004, 43, 5940–5943 CrossRef PubMed.
- R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H.-C. Yao and F. Li, Inorg. Chem., 2014, 53, 688–690 CrossRef PubMed.
- S. Sanz, H. M. O'Connor, E. M. Pineda, K. S. Pedersen, G. S. Nichol, O. Mønsted, H. Weihe, S. Piligkos, E. J. L. McInnes, P. J. Lusby and E. K. Brechin, Angew. Chem., Int. Ed., 2015, 54, 6761–6764 CrossRef PubMed.
- K. Li, L.-Y. Zhang, C. Yan, S.-C. Wei, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456–4459 CrossRef PubMed.
- X.-P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062–16065 CrossRef CAS PubMed.
- M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Létard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549–2552 CrossRef CAS PubMed.
- H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343–7345 CrossRef CAS PubMed.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed.
- M. Ibrahim, V. Mereacre, N. Leblanc, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2015, 54, 15574–15578 CrossRef CAS PubMed.
- J.-L. Liu, J.-Y. Wu, Y.-C. Chen, V. Mereacre, A. K. Powell, L. Ungur, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2014, 53, 12966–12970 CrossRef CAS PubMed.
- C. Kachi-Terajima, K. Yanagi, T. Kaziki, T. Kitazawa and M. Hasegawa, Dalton Trans., 2011, 40, 2249–2256 RSC.
- A. V. Chandrasekhar, A. Radhapathy, B. J. Reddy, Y. P. Reddy, L. Ramamoorthy and R. V. S. S. N. Ravikumar, Opt. Mater., 2003, 22, 215–220 CrossRef CAS.
- J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed.
- J. Luzon, K. Bernot, I. J. Hewitt, C. E. Anson, A. K. Powell and R. Sessoli, Phys. Rev. Lett., 2008, 100, 247205 CrossRef PubMed.
- J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed.
- M. Han, R. Michel and G. H. Clever, Chem. – Eur. J., 2014, 20, 10640–10644 CrossRef CAS PubMed.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, SEM-EDS, ESI-Mass, FT-IR and Raman spectra, and the crystallographic data for this communication. CCDC 1440867. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01651k |
| This journal is © The Royal Society of Chemistry 2016 |