
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic rare-earth/platinum complexes ligated by phosphinoamides†
Franziska
Völcker
and
Peter W.
Roesky
*
Institut für Anorganische Chemie, Karlsruher Institut für Technologie (KIT), Engesserstr. 15, Geb. 30.45, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 17th March 2016
Abstract
The heterometallic early-late 5d/4f binuclear phosphinoamido Ln/Pt(0) complexes [(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Ln(μ-Cl)Li(THF)3] (Ln = Y (1a), Lu (1b)) were obtained by reaction of [Li(THF)4][(Ph2PNPh)4Ln] (Ln = Y, Lu) with the Pt(0) complex [Pt(tBu3P)2] in the presence of LiCl. In the absence of LiCl the corresponding Ln/Pt(0) complexes [(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Ln{η2-(Ph2PNPh)}][Li(THF)4] (Ln = Y (2a), Lu (2b)) were isolated. Both kind of complexes decompose in solution. The Pt(0) complex [Pt(Ph2PNHPh)4] (3) was identified as one of these decomposition products.
Introduction
Homo- and heterobimetallic complexes have been studied for catalytic application and small molecule activation over recent years.1,2 Some of these compounds show cooperative and synergistic effects that can arise from the simultaneous or consecutive action of different metal centers in different media.2 Inspired by nature, the concept of cooperative bimetallic catalysis has been employed for the synthesis of numerous artificial catalysts. The emerging field of homo- and heterobimetallic and polymetallic catalyst systems has been summarized in several review articles recently.2–6 Within this area heterobimetallic early/late complexes7,8 having interactions between a hard Lewis acidic early metal atom and a soft Lewis basic late metal atom are of interest because they feature significantly different reaction sites. This makes them interesting for potential applications in catalysis.9 For the synthesis of heterobimetallic early/late complexes carbonyl-, hydride-, halide-, selenide-, and thiolate ligands were used as bridging ligands.1 Recently, Thomas et al. reported heterobimetallic transition metal complexes with metal-to-metal bonds bridged by phosphinoamido ligands.9 The known combinations of metals realized by this concept are bimetallic complexes, e.g. Co/Ti,10,11 Co/Zr,9,12–16 Co/Hf,17 Pt/Zr,18 V/Fe,19 Nb/Co,20 Ta/Co,20 and Co/U.21In contrast to transition metal complexes, heterobimetallic early/late complexes7,8 containing rare-earth metals are far less common. Thus, only few complexes of the rare-earth elements with non-supported metal-to-metal bonds to a transition metal were reported.17 Examples include complexes with Lu–Ru,22 Ln–Re (Ln = La, Sm, Yb, Lu),20,23–25 Nd–Fe,26 and Yb–Fe bonds.27
Heterobimetallic compounds, which have a rare-earth metal atom and a rhodium, palladium, or platinum atom in close proximity (distance of less than 3.5 Å) are also not very common. Kempe et al. reported some Nd/Rh and Nd/Pd complexes,28,29 in which the metals are brought closely together by bis(aminopyridinato) ligands. Hou et al. recently reported heterobimetallic rare-earth metal/platinum complexes. In these half-sandwich rare-earth metal alkyl complexes Cp ligands with a phosphine side arm were used.30
As seen by these few examples one of the big challenges that still remain is the synthesis of heterobimetallic early/late complexes containing rare-earth metals. Lately, we reported the synthesis of the heterometallic early-late 4d/4f bi- and trinuclear phosphinoamido Ln/Pd(0) complexes [(Ph2PNHPh)Pd{μ-(Ph2PNPh)}3Ln(μ-Cl)Li(THF)3] (Ln = Y (Ia), Lu (Ib)) and [Li(THF)4][{(Ph2PNHPh)Pd}2{μ-(Ph2PNPh)}4Ln] (Ln = Y (IIa), Lu (IIb)) (Scheme 1).31,32 The latter compounds are the first early/late trimetallic phosphinoamido complexes. Compounds Ia,b and IIa,b were obtained by reaction of [Li(THF)4][(Ph2PNPh)4Ln] (Ln = Y, Lu)33–36 with the palladium allyl complex [Pd2(C3H5)2Cl2]. A reduction of the palladium atoms was observed upon the formation of the bi- and trimetallic compounds Ia,b and IIa,b. Although the metal atoms are forced into close proximity by the phosphinoamido ligands, quantum chemical calculations of [(Ph2PNHPh)Pd{μ-(Ph2PNPh)}3Lu(μ-Cl)Li(THF)3] showed only weak metal-to-metal interactions.31,32
 | ||
| Scheme 1 Bi- and trinuclear phosphinoamido Ln/Pd(0) complexes.31 | ||
Motivated by these initial results, we were interested in extending our studies on heterobimetallic early/late complexes containing rare-earth metals. Herein, we now report heterobimetallic rare-earth metal/platinum complexes bridged by phosphinoamido ligands.
Results and discussion
The palladium complexes Ia,b were obtained most efficiently by the addition of LiCl to the reaction mixture giving the desired complexes. In a similar protocol, the reaction of [Li(THF)4][(Ph2PNPh)4Ln] (Ln = Y, Lu) with the Pt(0) precursor [Pt(P(tBu)3)2] in the presence of LiCl resulted after crystallization in the Ln/Pt(0) complexes [(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Ln(μ-Cl)Li(THF)3] (Ln = Y (1a), Lu (1b)) in moderate yields (Scheme 2). Formally, the Pt atom inserts into the weak Ln–P bonds forming the desired heterobimetallic complexes. We presume that some decomposition occurs and Ph2PN(H)Ph, which is coordinated to the Pt atom, is formed as a side-product. In contrast to the formation of the Pd(0) complexes Ia,b, which were obtained only in a reductive approach from a Pd(II) source, compounds 1a,b are directly accessible from a Pt(0) precursor. By using Pt(II) salts as starting material no traceable products were obtained.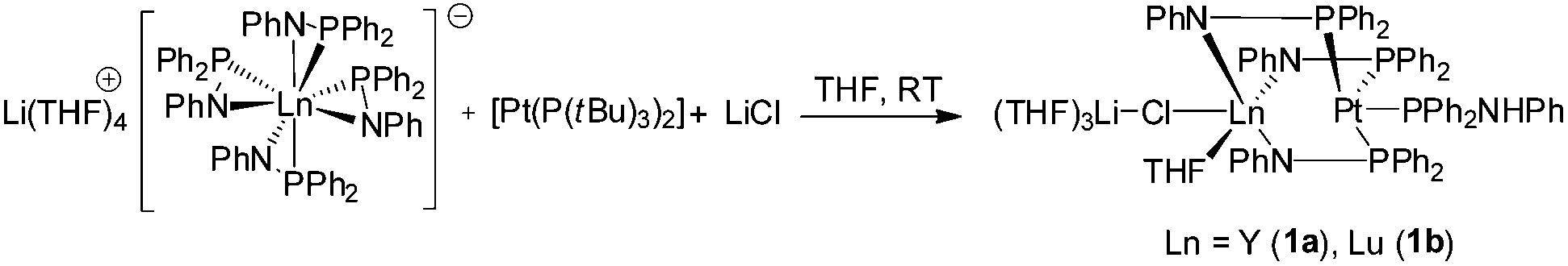 | ||
| Scheme 2 Synthesis of 1a,b. | ||
Although the reactions leading to 1a and 1b seem, at first glance, quiet similar, their accessibility is significantly different. Whereas 1a was obtained straight forward in a reproducible way, the preparation of 1b is more difficult.
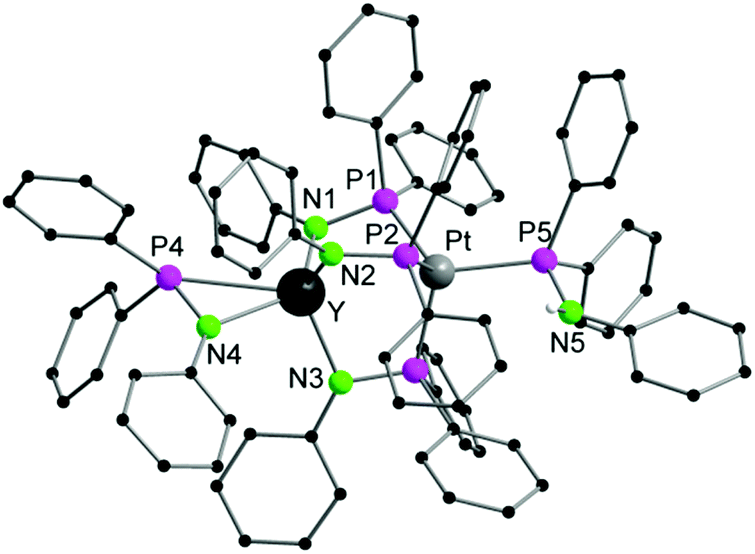
Single crystals of 1a,b suitable for X-ray diffraction were obtained by crystallization from THF/toluene/pentane (Fig. 1 and S1†). Compounds 1a,b crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule of the complexes in the asymmetric unit. Furthermore, one molecule of THF and toluene were localized each in the asymmetric unit. The toluene molecule in 1a showed a strong disorder and was thus suppressed by using Olex solvent mask.37 In both compounds, the Ln and the Pt atoms are forced in close proximity by three bridging μ-(Ph2PNPh) ligands. As expected the soft P atom binds to the Pt atom, whereas the hard nitrogen atoms coordinate to the rare-earth metal atom. In both compounds, the Li atom is coordinated to the Ln atom via a μ-Cl bridge. The rare-earth atoms are thus five-fold coordinated by three Ph2PNPh ligands, one molecule of THF and the chlorine atom. A distorted trigonal bipyramidal coordination polyhedron with the THF oxygen atom and N3 in the axis is formed. The Ln–N bond distances (1a: 2.291(3) Å–2.347(3); 1b: 2.242(6)–2.297(6) Å) are in the range of Ia,b (av. 2.317 Å (Ia), 2.262 Å (Ib)).31 The Ln–Cl bond lengths are 2.6525(14) Å (1a), 2.601(2) Å (1b)). The Pt atom is four-fold coordinated by the phosphorous atoms of three μ-(Ph2PNPh) ligands and one Ph2PNHPh ligand. A distorted tetrahedral coordination polyhedron is formed by the four P atoms around the Pt atom. The Pt–P bond distances are av. 2.3354 Å (1a), 2.328 Å (1b). Although the Pt atom has a slightly larger van-der-Waals radius in comparison to Pd, the Ln–Pt distances in 1a,b (3.0063(8) Å (1a), 2.9523(9) Å (1b)) are in the range of those in the Ln–Pd complexes Ia,b (2.9898(6) Å (Ia), 2.9031(11) Å (Ib)). This is clearly showing that the three μ-(Ph2PNPh) ligands are forcing the metal atoms into close proximity.
with one molecule of the complexes in the asymmetric unit. Furthermore, one molecule of THF and toluene were localized each in the asymmetric unit. The toluene molecule in 1a showed a strong disorder and was thus suppressed by using Olex solvent mask.37 In both compounds, the Ln and the Pt atoms are forced in close proximity by three bridging μ-(Ph2PNPh) ligands. As expected the soft P atom binds to the Pt atom, whereas the hard nitrogen atoms coordinate to the rare-earth metal atom. In both compounds, the Li atom is coordinated to the Ln atom via a μ-Cl bridge. The rare-earth atoms are thus five-fold coordinated by three Ph2PNPh ligands, one molecule of THF and the chlorine atom. A distorted trigonal bipyramidal coordination polyhedron with the THF oxygen atom and N3 in the axis is formed. The Ln–N bond distances (1a: 2.291(3) Å–2.347(3); 1b: 2.242(6)–2.297(6) Å) are in the range of Ia,b (av. 2.317 Å (Ia), 2.262 Å (Ib)).31 The Ln–Cl bond lengths are 2.6525(14) Å (1a), 2.601(2) Å (1b)). The Pt atom is four-fold coordinated by the phosphorous atoms of three μ-(Ph2PNPh) ligands and one Ph2PNHPh ligand. A distorted tetrahedral coordination polyhedron is formed by the four P atoms around the Pt atom. The Pt–P bond distances are av. 2.3354 Å (1a), 2.328 Å (1b). Although the Pt atom has a slightly larger van-der-Waals radius in comparison to Pd, the Ln–Pt distances in 1a,b (3.0063(8) Å (1a), 2.9523(9) Å (1b)) are in the range of those in the Ln–Pd complexes Ia,b (2.9898(6) Å (Ia), 2.9031(11) Å (Ib)). This is clearly showing that the three μ-(Ph2PNPh) ligands are forcing the metal atoms into close proximity.
 | ||
| Fig. 1 Solid-state structure of 1a. Carbon bound hydrogen atoms are omitted for clarity. Compound 1b is isostructural. Selected bond lengths [Å], angles [°]: 1a: Pt–Y 3.0063(8), Pt–P1 2.3206(13), Pt–P2 2.3363(12), Pt–P3 2.3368(11), Pt–P4 2.3477(12), Y–Cl 2.6525(14), Y–O1 2.471(3), Y–N1 2.347(3), Y–N2 2.291(3), Y–N3 2.325(3), Cl–Li 2.353(2), P1–N1 1.678(3), P2–N2 1.665(3), P3–N3 1.676(3), P4–N4 1.677(4); P1–Pt–P2 116.56(4), P1–Pt–P3 106.95(5), P1–Pt–P4 104.66(4), P2–Pt–P3 106.03(4), P2–Pt–P4 113.45(5), P3–Pt–P4 108.87(4), N1–Y–N2 130.76(12), N1–Y–N3 108.52(11), N2–Y–N3 97.97(12), N1–Y–Cl 106.17(9), N2–Y–Cl 116.47(9), N3–Y–Cl 86.40(9), O1–Y–Cl 81.61(8), N1–Y–O1 81.56(11), N2–Y–O1 81.44(11), N3–Y–O1 166.19(11). 1b (see Fig. S1†) Pt–Lu 2.9523(9), Pt–P1 2.321(2), Pt–P2 2.337(2), Pt–P3 2.307(2), Pt–P4 2.324(2), Lu–Cl 2.601(2), Lu–O1 2.442(6), Lu–N1 2.269(6), Lu–N2 2.242(6), Lu–N3 2.297(6), Lu–P1 3.085(2), Lu–P2 3.107(2), Lu–P3 3.109(2), Cl–Li 2.31(2), P1–N1 1.658(7), P2–N2 1.650(6), P3–N3 1.664(6), P4–N4 1.683(7); P1–Pt–P2 106.10(8), P1–Pt3–P1 106.85(8), P1–Pt–P4 107.98(9), P2–Pt–P3 116.32(8), P3–Pt–P4 104.49(8), N1–Lu–N2 97.7(2), N1–Lu–N3 110.1(2), N2–Lu–N3 131.5(2), N1–Lu–Cl 85.5(2), N2–Lu–Cl 116.2(2), N3–Lu–Cl 105.2(2), N1–Lu–Cl 85.5(2), N2–Lu–Cl 116.2(2), N3–Lu–O1 81.4(2). | ||
Since traces of LiCl within the starting material [Li(THF)4][(Ph2PNPh)4Y] (Ln = Y, Lu) resulted in the reaction with [Pt(P(tBu)3)2] in 1a, we improved our synthesis of [Li(THF)4][(Ph2PNPh)4Ln]. [Li(THF)4][(Ph2PNPh)4Ln]33–36 is obtained by the reaction of LnCl3 with LiPPh2NPh in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio. Usually, the product can be directly obtained by crystallization from THF/n-pentane. But obviously the bulk material is sometimes contaminated with traces of LiCl. By extraction of the crude product with toluene before crystallization, the contamination of the product with LiCl is avoided. The desired product is thus obtained in higher purity.
:4 molar ratio. Usually, the product can be directly obtained by crystallization from THF/n-pentane. But obviously the bulk material is sometimes contaminated with traces of LiCl. By extraction of the crude product with toluene before crystallization, the contamination of the product with LiCl is avoided. The desired product is thus obtained in higher purity.
Reaction of very pure [Li(THF)4][(Ph2PNPh)4Ln] (Ln = Y, Lu) with [Pt(P(tBu)3)2] gave the bimetallic compounds [(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Ln{η2-(Ph2PNPh)}][Li(THF)4] (Ln = Y (2a), Lu (2b)) as yellow crystals (Scheme 3). The formation of 2a,b is similar to 1a,b. Again, the Pt(0) atom formally inserts into three of the Ln–P bonds. The fourth PPh2NPh ligand remains in a η2-coordination mode on the Ln atom. The Pt atom forms a similar coordination polyhedron as observed in 1a,b.
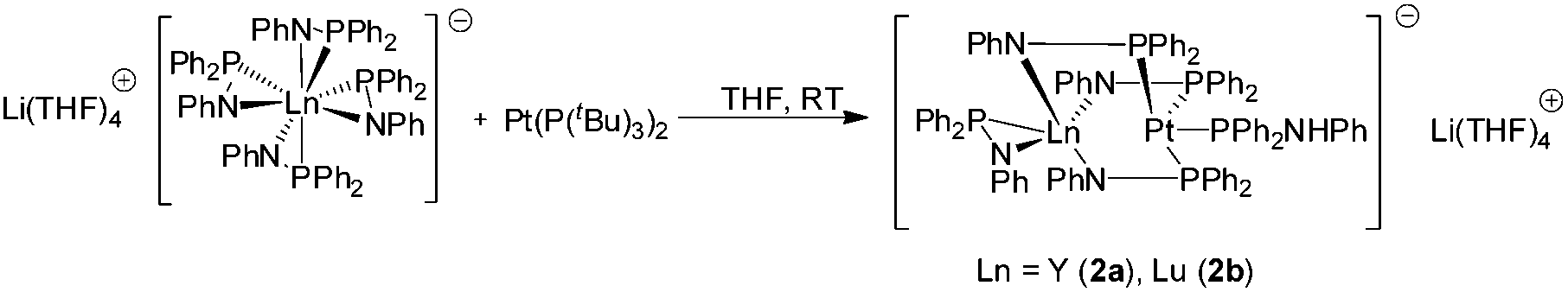 | ||
| Scheme 3 Synthesis of 2a,b. | ||
As seen for the formation of compounds 1a,b, there is a difference in reactivity. Upon workup, 2a was obtained as pure material in single crystalline form and complete characterization was possible. In contrast, the reaction leading to 2b is not quantitative. Even after prolonged reaction times the workup of 2b resulted in a mixture of the desired product and [Li(THF)4][(Ph2PNPh)4Lu]. The solid-state structures of both 2a and 2b were established by single crystal X-ray diffraction but for 2b no further analytical data could be collected.
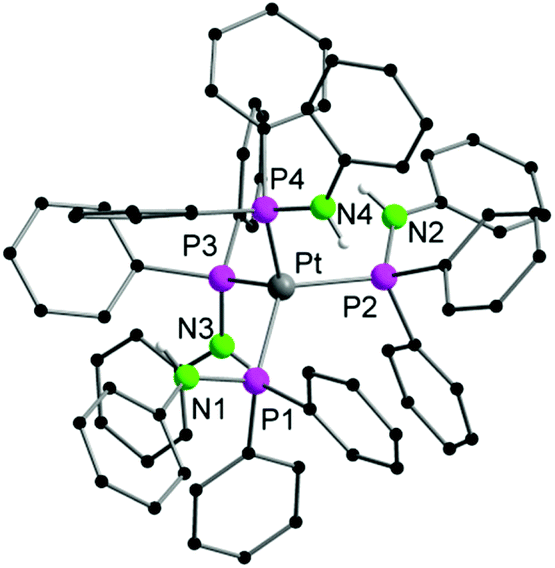
Compound 2a crystallizes in the monoclinic space group Cc with one molecule of the complexes in the asymmetric unit. Although the X-ray data collected from 2b was very poor, its composition was deduced from the difference Fourier map (Fig. S2†). Bond angles and distances of 2b thus are not discussed. Compounds 2a,b consist of a [(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Ln{η2-(Ph2PNPh)}]− anion and a [Li(THF)4]+ cation (Fig. 2). As seen in 1a,b the Pt atom in 2a,b is four fold-coordinated by three μ-(Ph2PNPh) and one Ph2PNHPh ligand resulting in a distorted tetrahedral coordination polyhedron, which is formed by the four P atoms around the Pt atom. Since the central part of the [(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Ln{η2-(Ph2PNPh)}]− anion is similar to 1a,b the Y–Pt distance in 2a (3.032(2) Å) is as expected. The main difference between 2a,b and 1a,b is the coordination sphere of the Ln atom. Instead of a molecule of THF and a chlorine atom, which are bound to the Ln atom in 1a,b, a η2-coordinated PPh2NPh ligand is bound to the lanthanide atom in 2a,b. Additionally, the N atoms of three bridging μ-(Ph2PNPh) ligands coordinate to the Ln atom resulting in a five-fold coordinated metal atom. The Ln–N bonds in 2a range from 2.285(12) Å to 2.301(2) Å.
 | ||
| Fig. 2 Solid-state structure of the anion of 2a. Carbon bound hydrogen atoms are omitted for clarity. Compound 2b is similar. Selected bond lengths [Å], angles [°]: 2a: Pt–Y 3.032(2), Pt–P1 2.367(4), Pt–P2 2.301(4), Pt–P3 2.330(4), Pt–P5 2.371(4), Y–P4 2.931(4), Y–N1 2.285(12), Y–N2 2.309(13), Y–N3 2.300(15), Y–N4 2.315(15), P1–N1 1.659(13), P2–N2 1.649(13), P3–N3 1.663(14), P4–N4 1.643(12); P1–Pt–P5 114.73(2), P2–Pt–P1 107.47(14), P2–Pt–P3 111.18(15), P2–Pt–P5 105.54(15), P3–Pt–P1 109.77(14), P3–Pt–P5 108.12(15), N4–Y–P4 34.0(3), N1–Y–N2 114.9(5), N1–Y–N3 103.0(5), N1–Y–N4 126.2(4), N3–Y–N2 120.0(5), N3–Y–N4 99.1(5). | ||
As observed for Ia,b, also the Pt complexes 1a,b and 2a,b decompose in solution. This is one explanation for the low yields and the formation of the protonated ligand PPh2NHPh. The 31P{1H} NMR spectra show fast decomposition of all compounds in various solvents. In d8-THF a large number of signals were observed. In C6D6 major signals with the corresponding 195Pt satellites were observed in the 31P{1H} NMR spectra of 1a,b and 2a (Fig. S3–S5†). However, the ratio varies from sample to sample and further decomposition signals were sometimes also observed as well. As a result of the fast decomposition, there remain significant uncertainties of the correct assignment of the signals. A 1H, 195Pt HMBC NMR spectrum was also not conclusive.
One of the decomposition products could be identified. From a saturated solution of 1a in d8-THF the Pt(0) complex [Pt(PPh2NHPh)4] (3) crystallized once in a NMR tube. Although 3 was not fully characterized and the X-ray data collected was poor, its composition was deduced from the difference Fourier map (Fig. 3) giving thus some insight into the decomposition pathway. Moreover, 3 was also identified by ESI-MS spectroscopy of a solution of 1a. In 3, the Pt(0) atom is four-fold coordinated by the phosphorous atoms of four PPh2NHPh ligands in a tetrahedral fashion. The only slightly related phosphinoamido structure of platinum reported in the literature is found in the Pt(I) species [Pt(NPhPPh2)(HNPhPPh2)]2, having a Pt–Pt bond.38
 | ||
| Fig. 3 Solid-state structure of the anion of 3. Carbon bound hydrogen atoms are omitted for clarity. | ||
Experimental32
All manipulations of air-sensitive materials were performed with the rigorous exclusion of oxygen and moisture in Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−3 torr) line, or in an argon-filled MBraun glove box. Elemental analyses were carried out with an Elementar vario Micro Cube. Hydrocarbon solvents were predried using an MBraun solvent purification system (SPS-800) and then they were degassed, dried and stored in vacuo over LiAlH4. Tetrahydrofuran was distilled under nitrogen from potassium before storage over LiAlH4. Deuterated solvents were obtained from Euro-Isotop (99.5 atom% D) and were degassed, dried and stored in vacuo over Na/K alloy in resealable flasks. 1H and 31P{1H} NMR spectra were recorded on a Bruker Avance II 300 MHz or Avance 400 MHz. 1H chemical shifts were referenced to the residual deuterated solvents and are reported relative to tetramethylsilane, 31P{1H} was referenced to external 85% phosphoric acid. IR spectra were obtained on a Bruker Tensor 37 FTIR spectrometer equipped with a room temperature DLaTGS detector and a diamond ATR (attenuated total reflection) unit. Raman spectra were recorded on a Bruker MultiRAM spectrometer.
195Pt-NMR spectra were recorded at 300 K on a Bruker Avance II 600 spectrometer using a double-resonance 1H-BBI probe head. Platinum frequencies were determined by an ultrabroadband version of a gradient selected 1H,195Pt-HMBC.39 Due to the very large chemical shift range of platinum complexes (15000 ppm, 1.94 MHz @ 14.1 T), it is not possible to cover this range in one conventional experiment. Conventional experiments are acquired with hard pulses that can excite bandwidth of about 50 kHz. Broadband spectra are achieved via application of broadband saturation pulses on platinum, which have been designed by optimal control derived optimizations.40–45
[Pt(P(tBu)3)2] (ABCR) was used as purchased from commercial sources without further purification. [Li(THF)4][(Ph2PNPh)4Ln] (Ln = Y, Lu),31,34 LnCl3 (Ln = Y, Lu),46 and LiPh2PNPh38,47 were prepared according to literature procedure.
[(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Y(μ-Cl)Li(thf)3] (1a)
15 ml THF was condensed at −78 °C onto a mixture of 186 mg (0.125 mmol) [Li(THF)4][(Ph2PNPh)4Y], 75 mg (0.125 mmol) bis(tri-tert-butylphosphine)platinum(0) and 5.3 mg (0.125 mmol) anhydrous lithium chloride. The orange solution was stirred for 3 h at ambient temperature. After the reaction period the solution was concentrated and 10 ml toluene was condensed onto the mixture. After filtration, the THF/toluene mixture was layered with toluene and with n-pentane. After one day 50 mg (0.035 mmol, 28%) of yellow crystals were formed. In a few cases the formation of colorless crystals of [Li(THF)4][(Ph2PNPh)4Y] was observed, which could be separated from the yellow crystals in the glove box.IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3380 (w), 3044 (w), 2866 (w), 1598 (m), 1494 (m), 1472 (m), 1431 (m), 1389 (m), 1280 (m), 1225 (m), 1181 (w), 1155 (w), 1092 (m), 1066 (m), 1028 (m), 996 (m), 894 (m), 741 (s), 691 (s), 617 (m), 593 (s), 508 (s), 463 (s), 429 (s). – Raman: (cm−1) = 3060 (m), 3023 (w), 2886 (w), 1585 (vs), 1570 (m), 1479 (w), 1445 (w), 1433 (w), 1393 (w), 1324 (w), 1282 (w), 1246 (w), 1183 (w), 1155 (w), 1088 (s), 1030 (s), 1001 (vs), 927 (w), 849 (w), 794 (w), 769 (w), 741 (w), 695 (w), 641 (w), 619 (w), 597 (w), 530 (m), 509 (w), 486 (w), 463 (w), 442 (w), 430 (w), 408 (w), 272 (w), 227 (w), 187 (m), 170 (m). – C88H93ClLiN4O4P4PtY (1720.98): calc. C 61.41, H 5.45, N 3.26; exp. C 61.38, H 5.45, N 2.95.
(cm−1) = 3380 (w), 3044 (w), 2866 (w), 1598 (m), 1494 (m), 1472 (m), 1431 (m), 1389 (m), 1280 (m), 1225 (m), 1181 (w), 1155 (w), 1092 (m), 1066 (m), 1028 (m), 996 (m), 894 (m), 741 (s), 691 (s), 617 (m), 593 (s), 508 (s), 463 (s), 429 (s). – Raman: (cm−1) = 3060 (m), 3023 (w), 2886 (w), 1585 (vs), 1570 (m), 1479 (w), 1445 (w), 1433 (w), 1393 (w), 1324 (w), 1282 (w), 1246 (w), 1183 (w), 1155 (w), 1088 (s), 1030 (s), 1001 (vs), 927 (w), 849 (w), 794 (w), 769 (w), 741 (w), 695 (w), 641 (w), 619 (w), 597 (w), 530 (m), 509 (w), 486 (w), 463 (w), 442 (w), 430 (w), 408 (w), 272 (w), 227 (w), 187 (m), 170 (m). – C88H93ClLiN4O4P4PtY (1720.98): calc. C 61.41, H 5.45, N 3.26; exp. C 61.38, H 5.45, N 2.95.
[(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Lu(μ-Cl)Li(thf)3] (1b)
15 ml THF was condensed at −78 °C onto a mixture of 200 mg (0.127 mmol) [Li(THF)4][(Ph2PNPh)4Lu], 76 mg (0.127 mmol) bis(tri-tert-butylphosphine)platinum(0) and 2.7 mg (0.063 mmol) anhydrous lithium chloride. The orange solution was stirred over night at ambient temperature. After the reaction period the solution was concentrated and 10 ml toluene was condensed onto the mixture. After filtration, the THF/toluene mixture was layered with toluene and with n-pentane. After one day 36 mg (0.020 mmol, 16%) of yellow crystals were formed. Additionally the formation of colorless crystals of [Li(THF)4][(PPh2NPh)4Lu] was observed, which could be separated from the yellow crystals in the glove box. When the product was recrystallized several times, crystals of 2b were also formed.IR (ATR): (cm−1) = 3381 (w), 3047 (w), 2972 (w), 2865 (w), 1598 (m), 1494 (m), 1473 (m), 1432 (m), 1390 (m), 1280 (s), 1226 (m), 1181 (m), 1155 (w), 1092 (m), 1066 (m), 1028 (m), 996 (m), 894 (s), 741 (s), 691 (vs), 630 (m), 617 (m), 593 (s), 507 (vs), 475 (s), 464 (vs), 430 (s). – Raman: (cm−1) = 3057 (m), 2980 (w), 2881 (w), 1585 (vs), 1570 (m), 1481 (w), 1445 (w), 1277 (w), 1239 (w), 1181 (w), 1156 (w), 1091 (s), 1030 (s), 1001 (vs), 918 (w), 793 (w), 741 (w), 695 (w), 642 (w), 619 (w), 597 (w), 530 (m), 480 (w), 439 (w), 407 (w), 269 (w), 227 (w), 170 (m). – EA: C88H93ClLiN4O4P4PtLu (1807.04): calc: C 58.49, H 5.19, N 3.10; exp: C 58.37, H 5.09, N 2.95.
Improved synthesis of [Li(thf)4][Ln(Ph2PNPh)4] (Ln = Y, Lu) to avoid traces of LiCl34
[(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Y{η2-(Ph2PNPh)}][Li(thf)4] (2a)
15 ml of THF was condensed at −78 °C onto a mixture of 186 mg (0.125 mmol) [Li(thf)4][(Ph2PNPh)4Y] and 75 mg (0.125 mmol) bis(tri-tert-butylphosphine)-platinum(0). The mixture was stirred for 4 h at ambient temperature. After the reaction period the solution was concentrated and 10 ml of toluene was condensed onto the mixture. After filtration, the THF/toluene mixture was layered with toluene and with n-pentane. After one day 23 mg (0.012 mmol, 9%) yellow crystals were formed.IR (ATR): (cm−1) = 3381 (w), 3045 (w), 2924 (w), 2854 (w), 1598 (m), 1494 (m), 1472 (m), 1432 (m), 1389 (m), 1280 (m), 1226 (m), 1180 (w), 1155 (w), 1092 (m), 1066 (m), 1027 (m), 996 (m), 894 (m), 822 (w), 740 (s), 691 (s) 617 (m), 593 (s), 507 (s), 464 (s), 429 (s). – Raman: (cm−1) = 3056 (s), 1585 (vs), 1476 (w), 1433 (w), 1396 (w), 1284 (w), 1234 (w), 1186 (w), 1158 (w), 1093 (m), 1031 (m), 1000 (vs), 898 (w), 800 (w), 769 (w), 698 (w), 619 (w), 525 (w), 487 (w), 412 (w), 255 (w), 225 (w), 194 (w), 173 (w). – EA: C106H109LiN5O4P5PtY (1962.83): calc: C 64.86, H 5.60, N 3.57; exp: C 64.11, H 5.77, N 3.21.
[(Ph2PNHPh)Pt{μ-(Ph2PNPh)}3Lu{η2-(Ph2PNPh)}][Li(thf)4] (2b)
15 ml of THF was condensed at −78 °C onto a mixture of 140 mg (0.089 mmol) [Li(thf)4][(Ph2PNPh)4Lu] and 53 mg (0.089 mmol) bis(tri-tert-butylphosphine)platinum(0). The mixture was stirred for 2 days at ambient temperature. After the reaction period the solution was concentrated and 10 ml of toluene was condensed onto the mixture. After filtration the THF/toluene mixture was layered with toluene and with n-pentane. After two times of recrystallization yellow (2b) and colourless crystals of [Li(THF)4][(PPh2NPh)4Lu] could be obtained, which could not be separated from each other.X-Ray-crystallographic studies of 1a, 1b, 2a, 2b and 3
A suitable crystal was covered in mineral oil (Aldrich) and mounted on a glass fiber. The crystal was transferred directly to the cold stream of a STOE IPDS 2 or STOE StadiVari diffractometer.All structures were solved using SHELXS-2013.48 The remaining non-hydrogen atoms were located from difference Fourier map calculations. The refinements were carried out by using full-matrix least-squares techniques on F, minimizing the function (Fo − Fc)2, where the weight is defined as 4Fo2/2(Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes using the program SHELXL-2013.48 Carbon-bound hydrogen atom positions were calculated. The locations of the largest peaks in the final difference Fourier map calculation as well as the magnitude of the residual electron densities in each case were of no chemical significance. Positional parameters, hydrogen atom parameters, thermal parameters, bond lengths and angles have been deposited as ESI.†
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary-publication no. CCDC 1450174–1450176.
Crystal data for 1a: C88H93ClLiN4O4P4PtY·C4H8O, M = 1793.03, a = 13.302(3) Å, b = 15.181(3) Å, c = 22.663(5) Å, α = 99.59(3)°, β = 98.88(3)°, γ = 99.85(3)°, V = 4366.3(16) Å3, T = 150(2) K, space group P, Z = 2, μ(Mo Kα) = 2.418 mm−1, 31469 reflections measured, 15963 independent reflections (Rint = 0.0478). The final R1 values were 0.0368 (I > 2σ(I)). The final wR(F2) values were 0.0918 (I > 2σ(I)). The final R1 values were 0.0451 (all data). The final wR(F2) values were 0.0947 (all data). The goodness of fit on F2 was 0.988.
Crystal data for 1b: C88H93ClLiLuN4O4P4Pt·C4H8O·C7H8, M = 1971.23, a = 13.388(3) Å, b = 15.201(3) Å, c = 22.573(5) Å, α = 99.85(3)°, β = 98.77(3)°, γ = 97.53(3)°, V = 4414.6(16) Å3, T = 210(2) K, space group P, Z = 2, μ(Mo Kα) = 2.853 mm−1, 34017 reflections measured, 17155 independent reflections (Rint = 0.0947). The final R1 values were 0.0469 (I > 2σ(I)). The final wR(F2) values were 0.0629 (I > 2σ(I)). The final R1 values were 0.1300 (all data). The final wR(F2) values were 0.0763 (all data). The goodness of fit on F2 was 0.649.
Crystal data for 2a: C90H76N5P5PtY·C16H32LiO4, M = 1961.76, a = 23.628(5) Å, b = 14.886(3) Å, c = 28.196(6) Å, β = 107.47(3)°, V = 9459(4) Å3, T = 209 K, space group Cc, Z = 4, μ(Mo Kα) = 2.22 mm−1, 36018 reflections measured, 18329 independent reflections (Rint = 0.1126). The final R1 values were 0.0639 (I > 2σ(I)). The final wR(F2) values were 0.1085 (I > 2σ(I)). The final R1 values were 0.1064 (all data). The final wR(F2) values were 0.1212 (all data). The goodness of fit on F2 was 0.864.
Crystal data for 3: C72H64N4P4Pt·6(C4H8O), M = 1736.86, a = 18.281(2) Å, b = 18.250(2) Å, c = 18.261(2) Å, α = 104.797(7)°, β = 104.736(9)°, γ = 119.060(9)°, V = 4603.9(9) Å3, T = 100 K, space group P, Z = 2. Due to the fast decomposition no full data set could be acquired.
Conclusions
As already noted in our communication on Ln/Pd complexes31 the chemistry of heterobimetallic Ln/Pt complexes supported by phosphinoamido ligands is significantly different from the well-established family of heterobimetallic transition metal complexes with phosphinoamido ligands. The most obvious differences are:1. The Ln/Pd and Ln/Pt phosphinoamido complexes, which were synthesized so far, are significantly less stable than phosphinoamido transition metal complexes, e.g. Co/Zr. This hampers an investigation of their reactivity.
2. As shown by our recently reported quantum chemical calculations for Ib,31 the short metal-to-metal distances are a result of ligand effects, rather than of metal–metal bonds.
The lack of significant metal-to-metal interaction is a result of the strong ionic bonding contribution generally observed in rare-earth chemistry. The lack of stability of the heterobimetallic rare-earth/platinum metal complexes supported by phosphinoamido compounds may also be a result of the difference in bonding compared to pure heterobimetallic transition metals.
Acknowledgements
This work was supported by the DFG-funded transregional collaborative research center SFB/TRR 88 “3MET” Project B3. Dr Benjamin Görling is acknowledged for performing Pt–NMR measurements.Notes and references
- D. W. Stephan, Coord. Chem. Rev., 1989, 95, 41–107 CrossRef CAS.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- M. Weiss and R. Peters, in Cooperative Catalysis, ed. R. Peters, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 227–262, DOI:10.1002/9783527681020.ch8.
- I. Bratko and M. Gomez, Dalton Trans., 2013, 42, 10664–10681 RSC.
- J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS.
- L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS.
- N. Wheatley and P. Kalck, Chem. Rev., 1999, 99, 3379–3420 CrossRef CAS PubMed.
- C. M. Thomas, Comments Inorg. Chem., 2011, 32, 14–38 CrossRef CAS.
- B. Wu, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Chem. Sci., 2015, 6, 2044–2049 RSC.
- B. Wu, K. M. Gramigna, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2015, 54, 10909–10917 CrossRef CAS PubMed.
- V. N. Setty, W. Zhou, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2011, 50, 4647–4655 CrossRef CAS PubMed.
- B. P. Greenwood, G. T. Rowe, C.-H. Chen, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2009, 132, 44–45 CrossRef PubMed.
- C. M. Thomas, J. W. Napoline, G. T. Rowe and B. M. Foxman, Chem. Commun., 2010, 46, 5790–5792 RSC.
- B. P. Greenwood, S. I. Forman, G. T. Rowe, C.-H. Chen, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2009, 48, 6251–6260 CrossRef CAS PubMed.
- J. P. Krogman, B. M. Foxman and C. M. Thomas, Organometallics, 2015, 34, 3159–3166 CrossRef CAS.
- B. Oelkers, M. V. Butovskii and R. Kempe, Chem. – Eur. J., 2012, 18, 13566–13579 CrossRef CAS PubMed.
- B. G. Cooper, C. M. Fafard, B. M. Foxman and C. M. Thomas, Organometallics, 2010, 29, 5179–5186 CrossRef CAS.
- S. Kuppuswamy, T. M. Powers, J. P. Krogman, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Chem. Sci., 2013, 4, 3557–3565 RSC.
- C. Döring, A.-M. Dietel, M. V. Butovskii, V. Bezugly, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2010, 16, 10679–10683 CrossRef PubMed.
- J. W. Napoline, S. J. Kraft, E. M. Matson, P. E. Fanwick, S. C. Bart and C. M. Thomas, Inorg. Chem., 2013, 52, 12170–12177 CrossRef CAS PubMed.
- I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, N. I. Kirillova, A. K. Shestakova, I. N. Parshina, A. I. Gusev and G. K. I. Magomedov, J. Am. Chem. Soc., 1993, 115, 3156–3166 CrossRef CAS.
- M. V. Butovskii, O. L. Tok, V. Bezugly, F. R. Wagner and R. Kempe, Angew. Chem., Int. Ed., 2011, 50, 7695–7698 CrossRef CAS PubMed.
- M. V. Butovskii, C. Döring, V. Bezugly, F. R. Wagner, Y. Grin and R. Kempe, Nat. Chem., 2010, 2, 741–744 CrossRef CAS PubMed.
- M. V. Butovskii, O. L. Tok, F. R. Wagner and R. Kempe, Angew. Chem., Int. Ed., 2008, 47, 6469–6472 CrossRef CAS PubMed.
- P. L. Arnold, J. McMaster and S. T. Liddle, Chem. Commun., 2009, 818–820, 10.1039/b819072k.
- M. P. Blake, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 15358–15361 CrossRef CAS PubMed.
- A. Spannenberg, M. Oberthür, H. Noss, A. Tillack, P. Arndt and R. Kempe, Angew. Chem., Int. Ed., 1998, 37, 2079–2082 CrossRef CAS.
- R. Kempe, H. Noss and H. Fuhrmann, Chem. – Eur. J., 2001, 7, 1630–1636 CrossRef CAS.
- Y. Nakajima and Z. Hou, Organometallics, 2009, 28, 6861–6870 CrossRef CAS.
- F. Völcker, F. M. Mück, K. D. Vogiatzis, K. Fink and P. W. Roesky, Chem. Commun., 2015, 51, 11761–11764 RSC.
- F. Völcker, Disseration, Karlsruhe Institute of Technology Karlsruhe, 2015.
- F. Völcker, Y. Lan, A. K. Powell and P. W. Roesky, Dalton Trans., 2013, 42, 11471–11475 RSC.
- T. G. Wetzel, S. Dehnen and P. W. Roesky, Angew. Chem., Int. Ed., 1999, 38, 1086–1088 CrossRef CAS.
- P. W. Roesky, Heteroat. Chem., 2002, 13, 514–520 CrossRef CAS.
- T. Li, J. Jenter and P. W. Roesky, Z. Anorg. Allg. Chem., 2010, 636, 2148–2155 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. Fenske, B. Maczek and K. Maczek, Z. Anorg. Allg. Chem., 1997, 623, 1113–1120 CrossRef CAS.
- M. Enders, B. Görling, A. B. Braun, J. E. Seltenreich, L. F. Reichenbach, K. Rissanen, M. Nieger, B. Luy, U. Schepers and S. Bräse, Organometallics, 2014, 33, 4027–4034 CrossRef CAS.
- S. Ehni and B. Luy, J. Magn. Reson., 2013, 232, 7–17 CrossRef CAS PubMed.
- K. Kobzar, S. Ehni, T. E. Skinner, S. J. Glaser and B. Luy, J. Magn. Reson., 2012, 225, 142–160 CrossRef CAS PubMed.
- N. I. Gershenzon, T. E. Skinner, B. Brutscher, N. Khaneja, M. Nimbalkar, B. Luy and S. J. Glaser, J. Magn. Reson., 2008, 192, 235–243 CrossRef CAS PubMed.
- K. Kobzar, T. E. Skinner, N. Khaneja, S. J. Glaser and B. Luy, J. Magn. Reson., 2004, 170, 236–243 CrossRef CAS PubMed.
- T. E. Skinner, T. O. Reiss, B. Luy, N. Khaneja and S. J. Glaser, J. Magn. Reson., 2004, 167, 68–74 CrossRef CAS PubMed.
- T. E. Skinner, T. O. Reiss, B. Luy, N. Khaneja and S. J. Glaser, J. Magn. Reson., 2003, 163, 8–15 CrossRef CAS PubMed.
- M. D. Taylor and C. P. Carter, J. Inorg. Nucl. Chem., 1962, 24, 387–391 CrossRef CAS.
- M. T. Ashby and Z. Li, Inorg. Chem., 1992, 31, 1321–1322 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental and crystallographic studies. CCDC 1450174–1450176. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00417b |
| This journal is © The Royal Society of Chemistry 2016 |