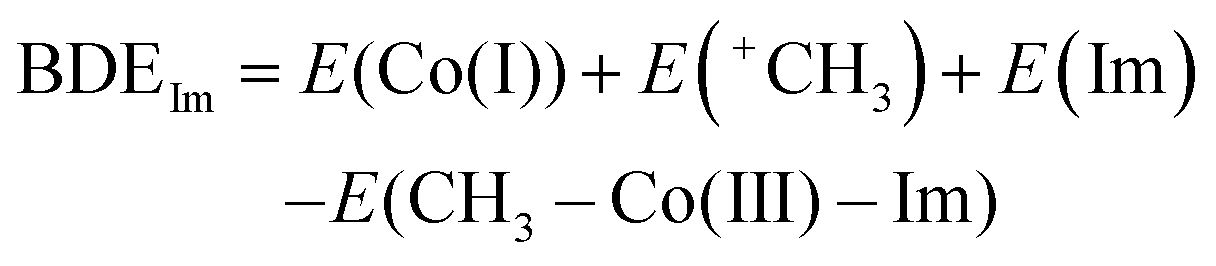DOI:
10.1039/C5DT04109K
(Paper)
Dalton Trans., 2016,
45, 3277-3284
Intraprotein transmethylation via a CH3–Co(III) species in myoglobin reconstituted with a cobalt corrinoid complex†
Received
19th October 2015
, Accepted 30th November 2015
First published on 1st December 2015
Abstract
Myoglobin reconstituted with a cobalt tetradehydrocorrin derivative, rMb(Co(TDHC)), was investigated as a hybrid model to replicate the reaction catalyzed by methionine synthase. In the heme pocket, CoI(TDHC) is found to react with methyl iodide to form the methylated cobalt complex, CH3–CoIII(TDHC), although it is known that a similar nucleophilic reaction of a cobalt(I) tetradehydrocorrin complex does not proceed effectively in organic solvents. Furthermore, we observed a residue- and regio-selective transmethylation from the CH3–CoIII(TDHC) species to the Nε2 atom of the His64 imidazole ring in myoglobin at 25 °C over a period of 48 h. These findings indicate that the protein matrix promotes the model reaction of methionine synthase via the methylated cobalt complex. A theoretical calculation provides support for a plausible reaction mechanism wherein the axial histidine ligation stabilizes the methylated cobalt complex and subsequent histidine-flipping induces the transmethylation via heterolytic cleavage of the Co–CH3 bond in the hybrid model.
Introduction
Cobalamin-dependent methionine synthase catalyzes an essential methylation of homocysteine to form methionine in most mammals and bacteria.1–10 The enzyme is a large modular protein (ca. 146 kDa) with four distinct functional domains. One domain contains methylcobalamin as a cofactor coordinated to a histidine residue.11 In the catalytic cycle, the enzyme promotes two kinds of methyl group transfers: (i) methylation of cob(I)alamin by N-methyl tetrahydrofolate as a methyl group donor, and (ii) methionine synthesis from homocysteine with the methylcobalamin intermediate. Compared to the non-catalyzed reactions, both reactions are ca. 105-fold accelerated by the catalytic activity of the enzyme.12,13 Elucidation of the mechanisms of these enzymatic reactions is challenging because of the complicated enzyme structures and the dynamic conformational changes occurring during activation of the cofactor and substrates.2,8,14–24 In this context, cobalt complexes have been investigated as enzyme models with the objective of understanding how the organometallic Co–C bond is formed and homolytically cleaved in several cobalamin-dependent enzymes.25–38 However the examples of model reactions of heterolytic Co–C bond cleavage, which is seen in methionine synthase, are quite limited30,31 because alkylated cobalt(III) complexes including methylcobalamin are generally less reactive with nucleophiles.25–29,32
Cobalt tetra- and di-dehydrocorrins have been synthesized as representative cobalamin models with tetrapyrrole macrocyclic ligands.36–38 It is known that the former has a relatively positive Co(II)/(I)-redox potential and the Co(I) species does not have sufficient nucleophilicity to promote attack by alkyl halides in organic solvents.36,37 The latter is found to have a relatively negative Co(II)/(I)-redox potential, and successive reactions of the obtained R–Co species with a nucleophile have not been reported.38 In contrast to these models, it is known that the transmethylation from methylated cobalt phthalocyanine to nucleophiles occurs. This is rationalized by the expectation that Co(I) species would be a good leaving group.32–35 However, there are few appropriate enzyme models for investigation of the role of the protein matrix, which is expected to stabilize the reaction intermediates and initiate the SN2-like transmethylation.1–9,25–38
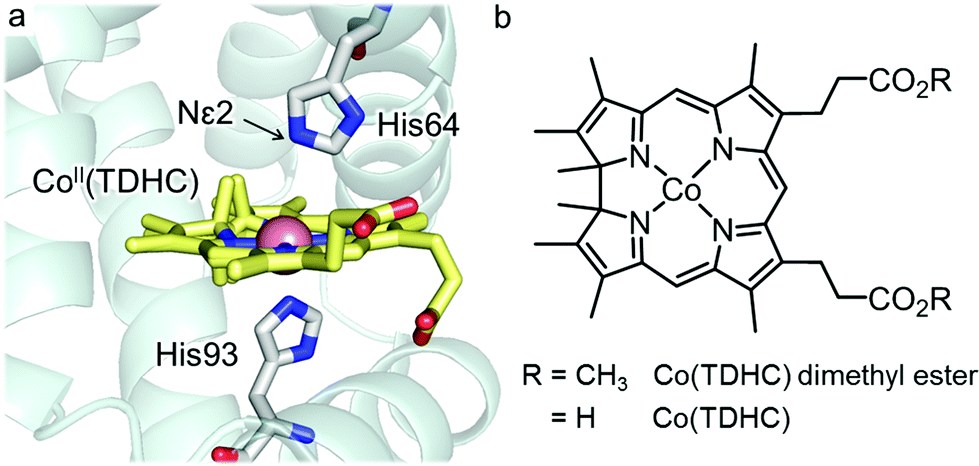
In our previous work, myoglobin reconstituted with cobalt tetradehydrocorrin Co(TDHC) as shown in Fig. 1 was prepared and a four-coordinate Co(I) species was detected in the protein matrix.39 Here, in an effort to replicate the reaction mechanism of methionine synthase, we demonstrate the spontaneous key reactions promoted by the enzyme and by the reconstituted protein, rMb(Co(TDHC)): (i) the formation of the CH3–Co(III) species from the supernucleophilic Co(I) species and (ii) subsequent intraprotein transmethylation in the myoglobin heme pocket.39
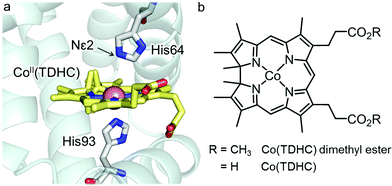 |
| | Fig. 1 (a) Crystal structure of rMb(CoII(TDHC)) (PDB code: 3WFT). (b) Chemical structures of Co(TDHC) dimethyl ester and Co(TDHC). | |
Results and discussion
The reaction of Co(I) species with methyl iodide in the reconstituted protein
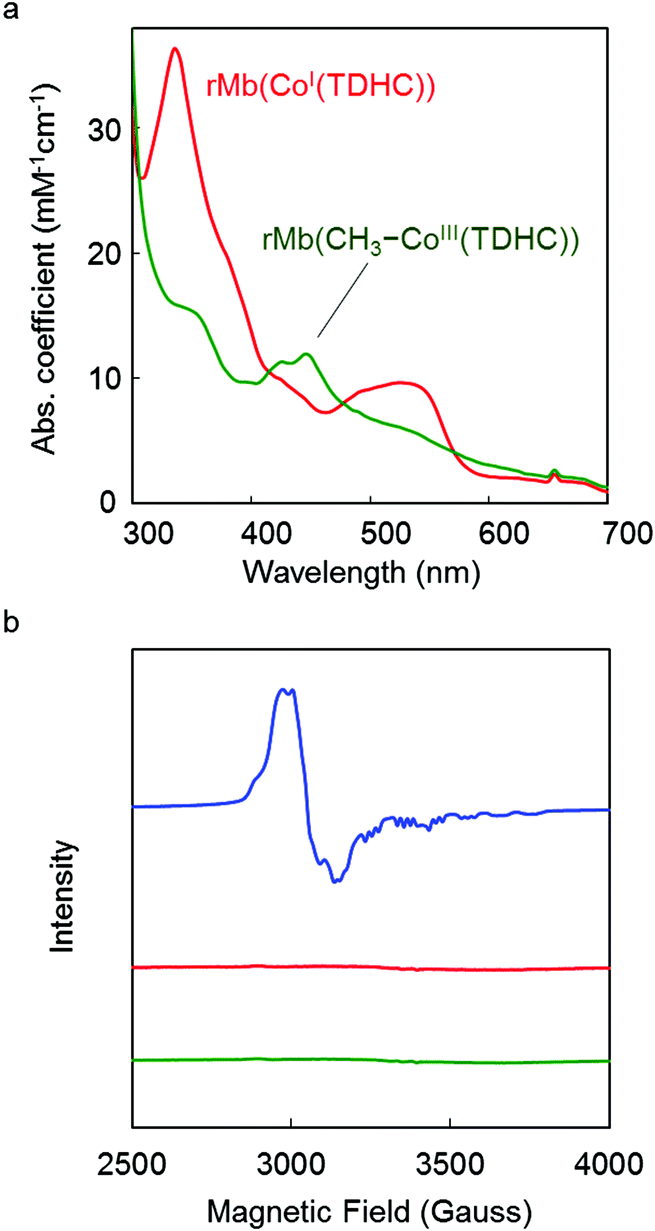
Addition of methyl iodide to a solution of rMb(CoI(TDHC)) leads to a new absorption maxima at 425 nm and 445 nm with disappearance of the 530 nm absorption within 2 h (Fig. 2a) despite the continued absence of EPR signals (Fig. 2b). The resulting species is photoactive and gradually converts to the Co(II) species upon irradiation with a Xe-lamp under aerobic conditions. The process is found to be accelerated upon the addition of 4-hydroxy-TEMPO (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl, free radical) as a radical quencher (Fig. S1 in the ESI†). These findings generally support the fact that the Co(I) species reacts with methyl iodide to form the Co–C bond under anaerobic conditions.38,40–42 In this reaction, a nucleophilic pathway is more plausible than a radical pathway which requires an electron transfer from the Co(I) species to methyl iodide, because rMb(Co(TDHC)) has the relevantly positive Co(I)/Co(II)-redox potential (E = −0.13 V vs. NHE).39,43,44
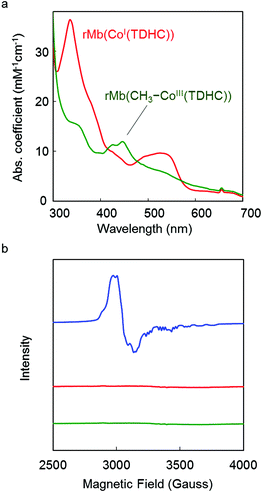 |
| | Fig. 2 (a) UV-vis spectra of reconstituted myoglobins in 0.1 M potassium phosphate buffer (pH 7.0) at 25 °C. Red and green spectra are of the rMb(CoI(TDHC)) and rMb(CH3–CoIII(TDHC)) species, respectively. (b) EPR spectra of reconstituted myoglobins (0.5 mM) in 0.1 M potassium phosphate buffer solution (pH 7.0) at 10 K. The blue spectrum is obtained by rMb(CoII(TDHC)).39 Red and green spectra represent rMb(CoI(TDHC)) and rMb(CH3–CoIII(TDHC)) species, respectively. | |
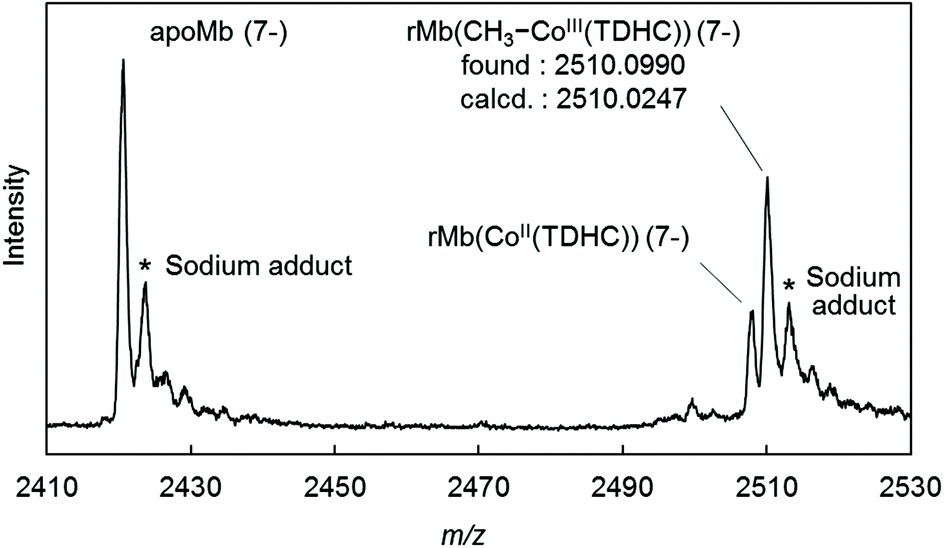
The CD spectrum of the new species also suggests formation of the Co–C bond in the protein. In the visible region, the positive Cotton effect is observed (Fig. S2 in the ESI†). This effect is also observed in native myoglobin and reconstituted myoglobins.45,46 Moreover, the CD spectrum in the far-UV region is completely consistent with the spectra of rMb(CoII(TDHC)) and rMb(CoI(TDHC)). This rules out dissociation of the methylated cofactor from the heme pocket (Fig. S3 in the ESI†). The ESI-TOF mass spectrum of the species in the negative ion mode confirms the generation of the methylated cobalt corrinoid complex in the protein, rMb(CH3–CoIII(TDHC)) (Fig. 3 and Fig. S4 in the ESI†).47Fig. 3 indicates the characteristic peak at m/z = 2510.10, which is consistent with the mass number of the 7− charge state for rMb(CH3–CoIII(TDHC)) (calculated m/z = 2510.02). These findings indicate that the nucleophilic Co(I) species reacts with methyl iodide to yield the CH3–CoIII(TDHC) complex in the heme pocket of myoglobin.
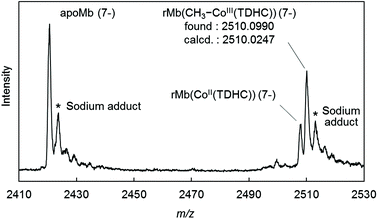 |
| | Fig. 3 ESI mass spectrum (negative mode) of rMb(CH3–CoIII(TDHC)), prepared upon addition of methyl iodide to rMb(CoI(TDHC)) at 4 °C in 0.1 M ammonium acetate buffer (pH 6.7). The voltage difference between the gas capillary exit and the 1st skimmer is 20 V. The desolvation temperature is 180 °C. The asterisks identify sodium adducts of the apoprotein and rMb(CH3–CoIII(TDHC)). | |
In contrast to rMb(CoI(TDHC)), no spectral change of CoI(TDHC) dimethyl ester (Fig. 1b) was observed in CH2Cl2 upon the addition of methyl iodide, regardless of the presence of imidazole (1 mM, 40 eq.) as an axial ligand (Fig. S5 in the ESI†). The time-course plots of spectral changes at 525 nm clearly indicate that the Co(I) complex in the protein (see Fig. S6 in the ESI†) is more reactive with methyl iodide than the Co(I) complex in CH2Cl2.
Bond-dissociation energy of methylated complexes
As discussed above, the protein environment is essential for the formation of the CH3–Co(III) complex, which indicates that the Co–C bond is stabilized in the heme pocket. To explain the stabilization of methylated Co(III) complexes by axial coordination of imidazole,48 we computed the heterolytic bond-dissociation energy (BDE)49,50 with density functional theory (DFT) calculations (Table 1 and Fig. S7 the ESI†). As listed in Table 1, the BDE of the methylated Co(III) complexes can be increased by the coordination of imidazole. The BDEs of the Co–N(Im) bonds for the methylated Co(TDHC′) and Co(corrin) complexes49 were computed to be 14.2 and 15.9 kcal mol−1, respectively, which indicates that the cobalt complexes are significantly stabilized by the coordination of imidazole.51 Thus, it appears that the stability of the CH3–Co(III) species is derived from His93 ligation in the myoglobin matrix.52
Table 1 DFT-computed heterolytic bond-dissociation energy (BDE) and stabilization energy provided by imidazole ligation in the gas phase (kcal mol−1)
| Cobalt complex |
BDE |
Stabilization energyc |
|
The value of the heterolytic BDE for the imidazole-coordinated CH3–Co complex consists of the dissociation of the CH3–Co and Co–N(Im) bonds which provides the three fragments: methyl cation, imidazole and a corresponding tetra-coordinated Co(I) complex.
The value of the heterolytic BDE for the Co–CH3 bond.
The stabilization energy is evaluated by the BDE value of the Co–N(Im) bond in the imidazole-coordinated CH3–Co(III) complex.
|
| CH3–Co(TDHC′)–Ima |
151.0 |
14.2 |
| CH3–Co(TDHC′)b |
136.8 |
| CH3–Co(corrin)–Ima |
169.6 |
15.9 |
| CH3–Co(corrin)b |
153.7 |
Intraprotein transmethylation from the CH3–CoIII(TDHC) complex
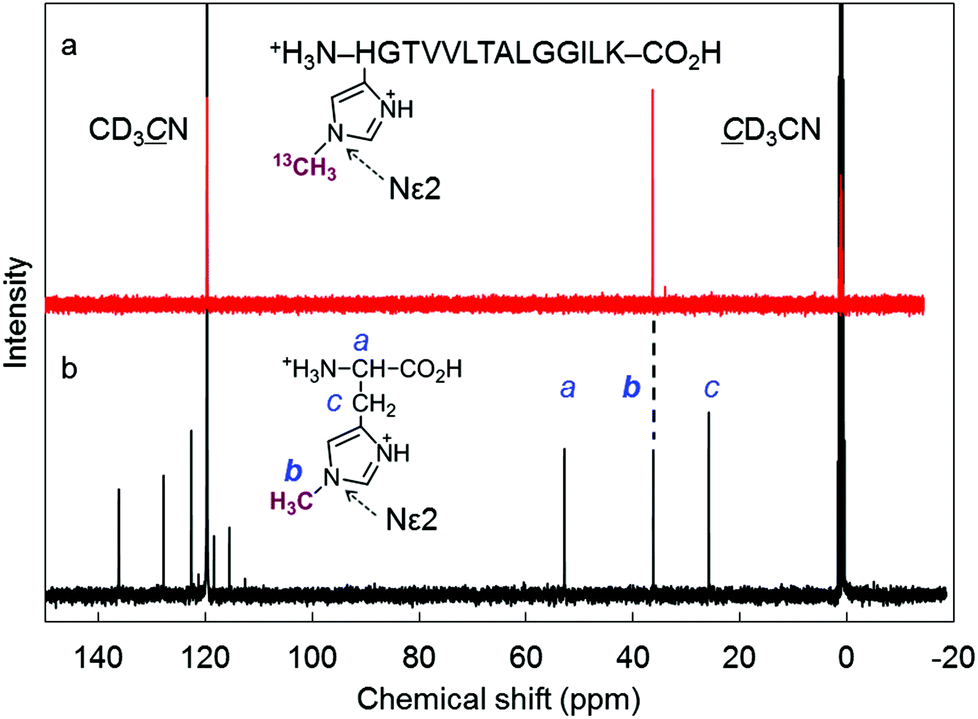
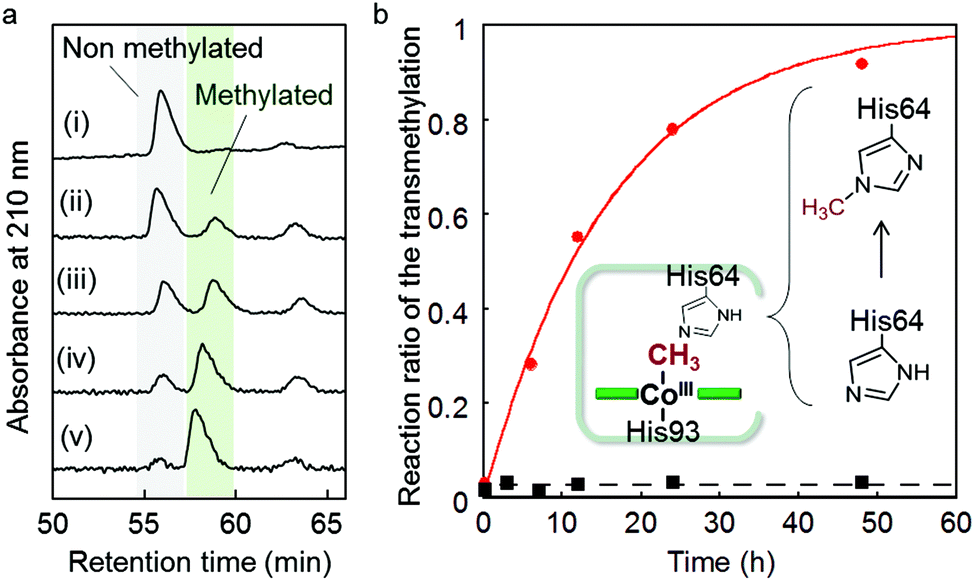
We found that the deconvoluted peak intensity of the apoprotein at 16950.5, detected by ESI-TOF MS, gradually decreases after addition of methyl iodide to the protein solution of rMb(CoI(TDHC)), and then a new peak at 16964.2 appears. Surprisingly, the in-source decay mode of MALDI-TOF MS shows an increase of the sole His64 mass number by +14 over 24 h (Fig. S8 in the ESI†). The fragment including the His64 residue, ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) GTVVLTALGGILK, was obtained by trypsin digestion and characterized by ESI-TOF MS and NMR spectroscopy (see Fig. 4 and Fig. S9 in the ESI†). In the 1H and 13C NMR spectra, the characteristic peaks at 4.05 and 36.31 ppm are respectively assigned to the hydrogen and carbon of the methyl group that was transferred to the His64 residue. These values are completely consistent with those of the authentic sample, Nε2-methylated histidine as shown in Fig. 4 and Fig. S9 in the ESI.†53 The NMR study provides a strong indication that the methylation reaction selectively occurs at the Nε2-position of the His64 imidazole ring. In a control experiment, it was found that addition of a large amount of methyl iodide (ca. 640 equiv.) to native myoglobin and to myoglobin reconstituted with cobalt protoporphyrin IX leads to non-selectively monomethylated protein on the surface in both cases with 50% conversion over 24 h and failure to produce the methylated His64 residue. These results indicate that the Nε2 atom of the His64 imidazole nucleophilically attacks the activated methyl group bound to the cobalt atom prior to residue- and regio-selective transfer of the methyl group within the heme pocket of myoglobin.
GTVVLTALGGILK, was obtained by trypsin digestion and characterized by ESI-TOF MS and NMR spectroscopy (see Fig. 4 and Fig. S9 in the ESI†). In the 1H and 13C NMR spectra, the characteristic peaks at 4.05 and 36.31 ppm are respectively assigned to the hydrogen and carbon of the methyl group that was transferred to the His64 residue. These values are completely consistent with those of the authentic sample, Nε2-methylated histidine as shown in Fig. 4 and Fig. S9 in the ESI.†53 The NMR study provides a strong indication that the methylation reaction selectively occurs at the Nε2-position of the His64 imidazole ring. In a control experiment, it was found that addition of a large amount of methyl iodide (ca. 640 equiv.) to native myoglobin and to myoglobin reconstituted with cobalt protoporphyrin IX leads to non-selectively monomethylated protein on the surface in both cases with 50% conversion over 24 h and failure to produce the methylated His64 residue. These results indicate that the Nε2 atom of the His64 imidazole nucleophilically attacks the activated methyl group bound to the cobalt atom prior to residue- and regio-selective transfer of the methyl group within the heme pocket of myoglobin.
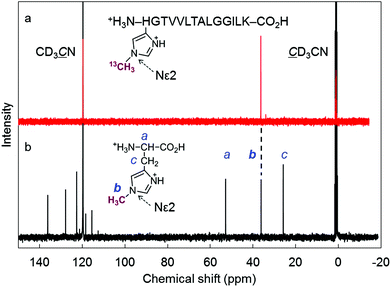 |
| | Fig. 4 (a) 13C NMR (150 MHz, D2O/CD3CN (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)) spectrum of the H(13CH3)GTVVLTALGGILK fragment after trypsin digestion of rMb(CH3–CoIII(TDHC)). The fragment was protonated because the eluent (H2O/CH3CN) containing 0.1% TFA was used for the HPLC purification. The chemical shift of the 13C-enriched methyl group was determined to be 36.31 ppm. (b) 13C NMR (100 MHz, D2O/CD3CN (9:1) containing 1% TFA) spectrum of Nε2-methylated histidine as an authentic sample.53 (The chemical shift of the methyl group bounded to the Nε2 position was determined to be 36.20 ppm.) Each 13C signal was assigned using the HMQC technique. :1)) spectrum of the H(13CH3)GTVVLTALGGILK fragment after trypsin digestion of rMb(CH3–CoIII(TDHC)). The fragment was protonated because the eluent (H2O/CH3CN) containing 0.1% TFA was used for the HPLC purification. The chemical shift of the 13C-enriched methyl group was determined to be 36.31 ppm. (b) 13C NMR (100 MHz, D2O/CD3CN (9:1) containing 1% TFA) spectrum of Nε2-methylated histidine as an authentic sample.53 (The chemical shift of the methyl group bounded to the Nε2 position was determined to be 36.20 ppm.) Each 13C signal was assigned using the HMQC technique. | |
Kinetic study of the intraprotein transmethylation
The first-order rate constant of the transmethylation is determined to be 0.062 h−1 for rMb(Co(TDHC)) by conversion of the digested fragments evaluated by HPLC, while the transmethylation does not occur in native myoglobin under the same conditions (Fig. 5 and Fig. S10 in the ESI†). In contrast, methylcobalamin does not provide any spectral changes owing to the transmethylation in 0.1 M phosphate buffer (pH 7.0) in the presence of histidine over 12 h (Fig. S11 in the ESI†). These findings may be explained by the proximity effect and the low heterolytic BDE of the CH3–Co(TDHC′)–Im (151.0 kcal mol−1) in contrast to the cobalamin model (169.6 kcal mol−1) (Table 1).
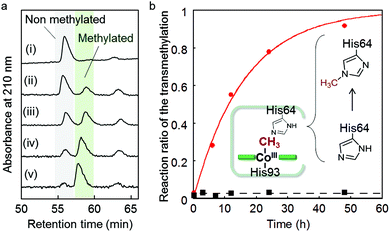 |
| | Fig. 5 (a) HPLC traces for the digested peptide fragment (HGTVVLTALGGILK) of rMb(Co(TDHC)) after reaction with methyl iodide in 0.1 M potassium phosphate buffer (pH 7.0) at 25 °C for 0, 6, 12, 24, 48 h (i–v). (b) Time courses of the conversion of His64 to methylated His64. Red and black plots represent the data for rMb(Co(TDHC)) and native myoglobin, respectively. Conditions: [protein] = 25 μM, [sodium dithionite] = 0.6 mM, [CH3I] = 16 mM. | |
Theoretical calculation of the transmethylation in myoglobin
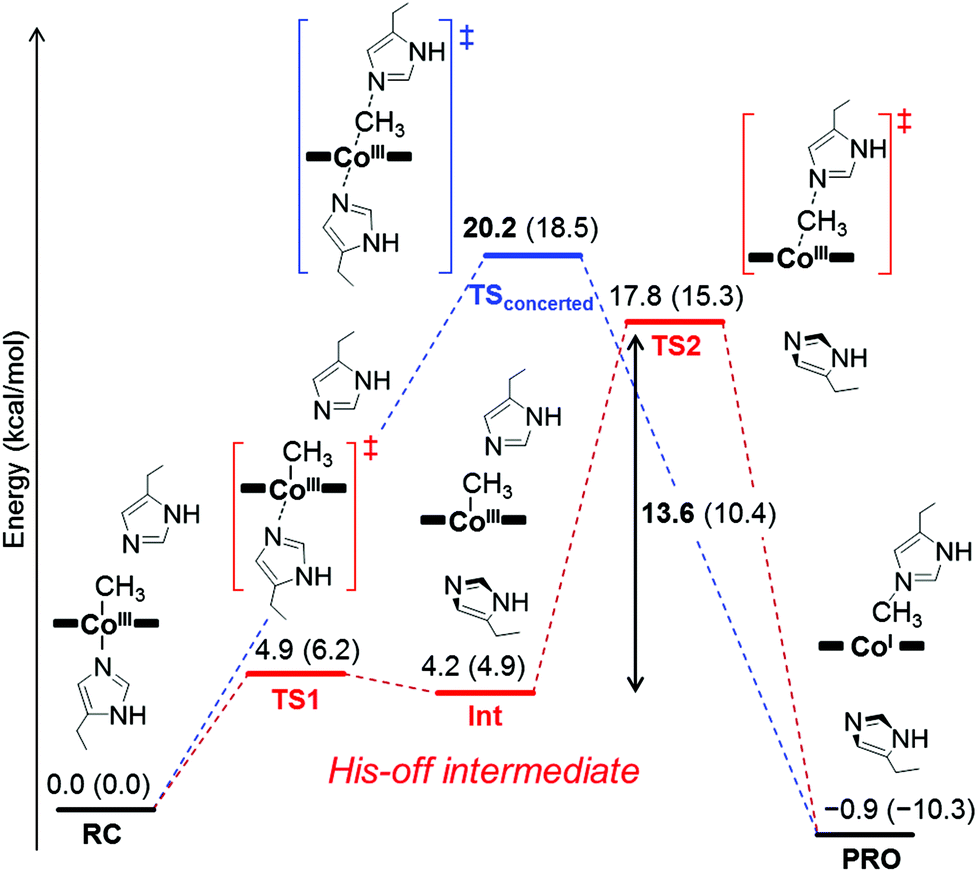
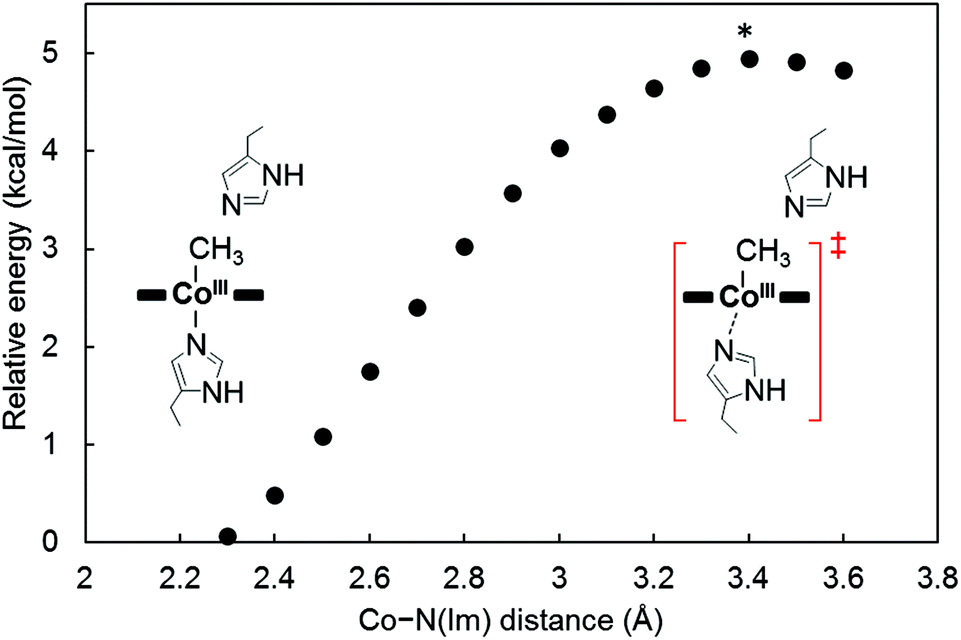
To investigate the mechanism of transmethylation in the protein matrix, nucleophilic stepwise and concerted pathways were considered in DFT computations as shown in Fig. 6.23,54 A base-off CH3–Co(III) complex is produced in the stepwise mechanism. In the calculations, we used a simplified active site model, in which the two carbon atoms corresponding to the α-carbons of the His64 and His93 residues and the two β-carbons of TDHC pyrroles conjugating each of the propionate side chains are fixed at the position of the crystal structure of rMb(CoII(TDHC)) (Fig. S12 in the ESI†).49,55 As shown in Fig. 7 and 8, the axial imidazole is readily displaced from the CH3–Co(III) complex with an activation barrier of 4.9 kcal mol−1 to produce a penta-coordinated CH3–Co(III) intermediate (RC → TS1 → Int). As discussed above, the Co–C bond is weakened in Int, and therefore, the activation barrier of 13.6 kcal mol−1 in the stepwise pathway is lower than that of the concerted pathway (20.2 kcal mol−1). These theoretical results demonstrate that the stepwise pathway via the base-off CH3–Co(III) complex activated by His93 flipping in rMb(Co(TDHC)) is more plausible than the concerted reaction. We found that the barriers for the methyl transfer are reduced and the final product is significantly stabilized by the dielectric effect of the protein environment. This is because the cobalt complexes are highly polarized in the course of the methyl transfer. In the crystal structure, the Nδ1 atom of the His64 is hydrogen-bonded to a water molecule (Fig. S6 in the ESI†),39 and therefore, the barriers can be further reduced by the partial or full deprotonation of the methylated histidine.23
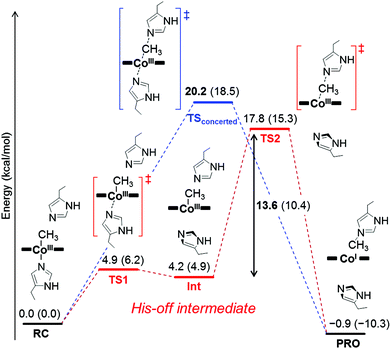 |
| | Fig. 6 Energy diagrams for the stepwise reaction (red line) and the concerted reaction (blue line) for the transmethylation by Co(TDHC′). Energy values in the gas phase relative to CH3–CoIII(TDHC′)–Im are indicated. The relative energies in parentheses include the effect of a dielectric constant of 4.0. | |
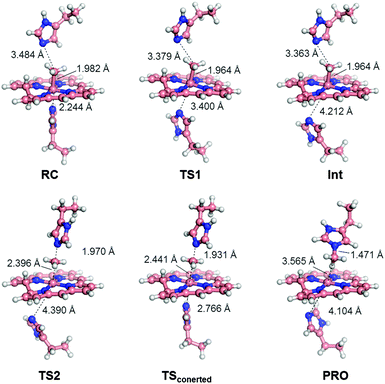 |
| | Fig. 7 Optimized structures of key species in each transmethylation pathway: reaction complex (RC), transition state (TS1, TS2, TSconcerted), intermediate (Int) and product (PRO) in Fig. 6. The stepwise and concerted pathways consist of RC → TS1 → Int → TS2 → PRO and RC → TSconcerted → PRO, respectively. | |
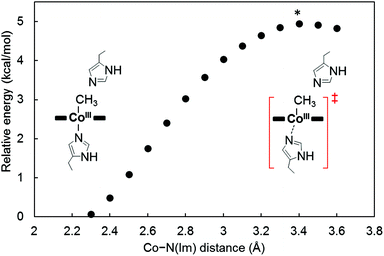 |
| | Fig. 8 Energy barrier for the deligation of imidazole from the CH3–CoIII(TDHC′) estimated by the DFT calculations. The maximum point marked with asterisk is identified as a transition state in the stepwise pathway. | |
Conclusions
We demonstrated the reactivity of the methionine synthase model conjugated between cobalt tetradehydrocorrin and apomyoglobin. The heme pocket of myoglobin is found to enhance the nucleophilicity of the Co(I) species and provide a methylated Co(III) species stabilized by the ligation. Furthermore, the residue- and regio-selective transmethylation of the Nε2 atom of the His64 residue was observed at 25 °C. In support of the experimental results, our theoretical calculations indicate that His-on/off switching regulates the reactivity of the CH3–Co(III) species for both the formation and the transmethylation. The moderate coordination of His93 to the cobalt atom in TDHC induces the transmethylation via the His-off species,52 because there is sufficient space to allow flipping of the imidazole ring in the proximal site39 and because dynamic structural changes of the enzyme would likely induce deligation in the cobalamin-binding domain.8 Further evaluation of the present system will contribute to the elucidation of the reaction mechanisms promoted by the structurally complex cobalamin-dependent enzymes.
Experimental
Materials
All reagents of the highest guaranteed grade available were obtained from commercial sources and were used as received unless otherwise indicated. Distilled water was demineralized using a Barnstead NANOpure DIamond™ or Millipore Integral3 apparatus. Syntheses of tetradehydrocorrin dimethyl ester cobalt complex, Co(TDHC) dimethyl ester, and tetradehydrocorrin cobalt complex, Co(TDHC) were reported in our previous paper.39 Native horse heart myoglobin (Sigma Aldrich) was purified with a cation exchange CM-52 cellulose column. The apoprotein was prepared according to Teale's method.56,57 The reconstituted protein, rMb(CoII(TDHC)), was obtained by the conventional method.39 A cobalt standard solution for ICP-OES was purchased from Wako Pure Chemical Industries.
Reduction of rMb(CoII(TDHC)) and following methylation
The following procedures were performed in a glove box (O2 < 0.1 ppm). A solution of sodium dithionite (10 mg mL−1, 10 μL, 0.57 μmol) in 0.1 M potassium phosphate buffer (pH 7.0) was added to 1 mL of rMb(CoII(TDHC)) solution (25 μM) in the same buffer at 25 °C. A solution of rMb(CoI(TDHC)) was immediately obtained.39 To the solution, methyl iodide (1 μL, 16 μmol) was added to yield an organometallic species, rMb(CH3–CoIII(TDHC)), within 2 h. The reaction was monitored by performing UV-vis spectral measurements at 25 °C.
EPR measurements
The measurements of EPR spectra were carried out at the X-band (9.35 GHz) microwave frequency with 100 kHz field modulation and 10 G of modulation amplitude. During EPR measurements, the sample temperature was maintained at 10 K by an Oxford Instruments ESR900 cryostat equipped with a turbo pump to lower the vapor pressure of the liquid He. Each protein solution (0.5 mM) in 0.1 M potassium phosphate buffer (pH 7.0) was placed in a 5 mm tube. The sample was quickly frozen in a cold pentane bath chilled with liquid N2.
Preparation of rMb(CH3–CoIII(TDHC)) for the ESI mass spectrometry experiment
The sample solution was prepared in degassed water prior to use in a nitrogen-purged glove box (O2 < 0.1 ppm). To a solution of rMb(CoII(TDHC)) (0.1 mM, 0.5 mL, 50 nmol) in 0.1 M ammonium acetate buffer (pH 6.7), sodium dithionite (57 mM, 1.6 mL, 91 nmol) and saturated CH3I (99 mM, 25 μL, 2.5 μmol) in the same buffer were added and the solution was allowed to stand for 1 h at 4 °C.
Identification of a methylated amino acid residue in the protein
Site-specific methylation in the protein was confirmed by the in-source decay mode on a Bruker autoflex III smartbeam mass spectrometer (MALDI-TOF MS). As a matrix solution, 1,5-diaminonaphthalene was saturated in a mixture of CH3CN and 0.1% TFA aqueous solution (1/1 v/v). After the methyl transfer reaction, the protein solution was purified by HiTrap Desalting (5 mL, GE Healthcare). To 1 μL of the concentrated protein solution (0.1 mM) was added 5.0 μL of the matrix solution, and then 1 μL of the mixed solution was placed on a sample plate. The solvent was evaporated under ambient conditions. All mass spectra were acquired in positive ion mode.
For the NMR study, the methylated protein was prepared by 13C-enriched methyl iodide. The concentrated protein solution (0.25 mM, 1 mL) was passed through a HiTrap Desalting column (GE Healthcare) equilibrated with 50 mM Tris-HCl buffer (pH 8.0) containing 6 M guanidine HCl. After denaturation of the protein by incubation of the collected solution at 95 °C for 20 min, the buffer was exchanged to 50 mM Tris-HCl buffer (pH 7.6) containing 1 mM CaCl2 using the same gel filtration column. To the solution, Sequencing Grade Modified Trypsin (Promega) (0.5 mg mL−1, 20 μL) was added and the mixture was incubated at 37 °C for 24 h. The peptide fragment containing methylated His64, H(13CH3)GTVVLTALGGILK, was isolated by HPLC with a YMC-Pack Pro C18 column (water/acetonitrile co-solvent containing 0.1% TFA). Purification was performed at a flow rate of 8 mL min−1 with a linear gradient of two eluents for 60 min. The isolated peptide was characterized by ESI-TOF MS: found m/z = 1392.854, calculated m/z = 1392.857 (z = 1+). 1H and 13C NMR spectra of the isolated peptide fragment in 10% acetonitrile-d3 in D2O solution were obtained on 600 MHz NMR instrument at 25 °C.
Computational chemistry
We used the Becke–Perdew (BP86)58,59 method implemented in the Gaussian 09 program. For all atoms, the 6-31G(d) basis set was used. This level of theory BP86/6-31G(d) serves as an appropriate platform for addressing the structural, electronic, and spectroscopic properties of cobalamin cofactors.60–66 All calculations were carried out in the gas phase unless otherwise noted. To explore the realistic reaction pathways, we used a truncated model of Co(TDHC) where all peripheral side chains of TDHC were replaced with hydrogens (Fig. S7b in the ESI†). The positions of four atoms marked with asterisk * in Fig. S12 in the ESI† are constrained at the coordinate of the crystal structure (PDB ID: 3WFT) to retain the structure of the active site. All of the transition states were characterized by an imaginary frequency mode that corresponds to the motion along the reaction coordinate under consideration. The dielectric effect of protein environment on the transmethylation reaction was estimated at the BP86/SV(P) level of theory by using the conductor-like screening model67 (COSMO) implemented in the TURBOMOLE program. The dielectric constant was chosen to be 4, which is a standard value that has been used in previous studies.23
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research provided by JSPS and MEXT (24655051, 15H00944, 15H05804, 22105013, 26104523), and the JSPS Japanese-German Graduate Externship. Y. M. appreciates support from the JSPS Research Fellowship for Young Scientists (14J00790).
Notes and references
-
B. Kräutler, in Metal Ions in Life Science, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Royal Society of Chemistry, Cambridge, 2009, ch. 1, vol. 6, pp. 1–51 Search PubMed.
-
R. G. Matthews, in Metal Ions in Life Science, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Royal Society of Chemistry, Cambridge, 2009, ch. 2, vol. 6, pp. 53–113 Search PubMed.
-
B. Kräutler and B. Puffer in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2012, ch. 117, vol. 25, pp. 131–263 Search PubMed.
-
B. Kräutler and S. Ostermann in The Porphyrin handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, ch. 68, vol. 11, pp. 229–276 Search PubMed.
- K. L. Brown, Chem. Rev., 2005, 105, 2075–2150 CrossRef CAS PubMed.
- R. Banerjee, C. Gherasim and D. Padovani, Curr. Opin. Chem. Biol., 2009, 13, 484–491 CrossRef CAS PubMed.
- K. Gruber, B. Puffer and B. Kräutler, Chem. Soc. Rev., 2011, 40, 4346–4363 RSC.
- R. G. Matthews, Acc. Chem. Res., 2001, 34, 681–689 CrossRef CAS PubMed.
- Y. Kung, N. Ando, T. I. Doukov, L. C. Blasiak, G. Bender, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Nature, 2012, 484, 265–269 CrossRef CAS PubMed.
- F. Zelder, Chem. Commun., 2015, 51, 14004–14017 RSC.
- C. L. Drennan, S. Huang, J. T. Drummond, R. G. Matthews and M. L. Ludwig, Science, 1994, 266, 1669–1674 CAS.
-
R. G. Matthews, in Chemistry and biochemistry of B12, ed. R. Banerjee, John Wiley & Sons. Inc., New York, 1999, ch. 27, pp. 681–706 Search PubMed.
- H. P. C. Hogenkamp, G. T. Bratt and A. T. Kotchevar, Biochemistry, 1987, 26, 4723–4727 CrossRef CAS PubMed.
- C. W. Goulding, D. Postigo and R. G. Matthews, Biochemistry, 1997, 36, 8082–8091 CrossRef CAS PubMed.
- J. S. Dorweiler, R. G. Finke and R. G. Matthews, Biochemistry, 2003, 42, 14653–14662 CrossRef CAS PubMed.
- V. Bandarian, M. L. Ludwig and R. G. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8156–8163 CrossRef CAS PubMed.
- N. Kumar and P. M. Kozlowski, J. Phys. Chem. B, 2013, 117, 16044–16057 CrossRef CAS PubMed.
- M. Alfonso-Prieto, X. Biarne, M. Kumar and C. Rovira, J. Phys. Chem. B, 2010, 114, 12965–12971 CrossRef CAS PubMed.
- N. Kumar, M. Alfonso-Prieto, C. Rovira, P. Lodowski, M. Jaworska and P. M. Kozlowski, J. Chem. Theory Comput., 2011, 7, 1541–1551 CrossRef CAS PubMed.
- K. Kornobis, K. Ruud and P. M. Kozlowski, J. Phys. Chem. A, 2013, 117, 863–876 CrossRef CAS PubMed.
- M. Koutmos, S. Datta, K. A. Pattridge, J. L. Smith and R. G. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18527–18532 CrossRef CAS PubMed.
- S. Datta, M. Koutmos, K. A. Pattridge, M. L. Ludwig and R. G. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4115–4120 CrossRef CAS PubMed.
- S.-L. Chen, M. R. A. Blomberg and P. E. M. Siegbahn, J. Phys. Chem. B, 2011, 115, 4066–4077 CrossRef CAS PubMed.
- P. M. Kozlowski, T. Kamachi, M. Kumar and K. Yoshizawa, J. Biol. Inorg. Chem., 2012, 17, 611–619 CrossRef CAS PubMed.
- L. D. Zydowsky, T. M. Zydowsky, E. S. Haas, J. W. Brown, J. N. Reeve and H. G. Floss, J. Am. Chem. Soc., 1987, 109, 7922–7923 CrossRef.
- C. Wedemeyer-Exl, T. Darbre and R. Keese, Helv. Chim. Acta, 1999, 82, 1173–1184 CrossRef CAS.
- L. Pan, H. Shimakoshi and Y. Hisaeda, Chem. Lett., 2009, 38, 26–27 CrossRef CAS.
- L. Pan, H. Shimakoshi, T. Masuko and Y. Hisaeda, Dalton Trans., 2009, 9898–9905 RSC.
- L. Pan, K. Tahara, T. Masuko and Y. Hisaeda, Inorg. Chim. Acta, 2011, 368, 194–199 CrossRef CAS.
- K. L. Brown, Dalton Trans., 2006, 1123–1133 RSC.
- Y. Hisaeda, T. Masuko, E. Hanashima and T. Hayashi, Sci. Technol. Adv. Mater., 2006, 7, 655–661 CrossRef CAS.
- W. Galezowski, P. N. Ibrahim and E. S. Lewis, J. Am. Chem. Soc., 1993, 115, 8660–8668 CrossRef CAS.
- W. Galezowski and E. S. Lewis, J. Phys. Org. Chem., 1994, 7, 90–95 CrossRef CAS.
- W. Galezowski, Inorg. Chem., 2005, 44, 5483–5494 CrossRef CAS PubMed.
- W. Galezowski, Inorg. Chem., 2005, 44, 1530–1546 CrossRef CAS PubMed.
- Y. Murakami and K. Aoyama, Bull. Chem. Soc. Jpn., 1976, 49, 683–688 CrossRef CAS.
- C.-J. Liu, A. Thompson and D. Dolphin, J. Inorg. Biochem., 2001, 83, 133–138 CrossRef CAS PubMed.
- Y. Murakami, Y. Aoyama and K. Tokunaga, J. Am. Chem. Soc., 1980, 102, 6736–6744 CrossRef CAS.
- T. Hayashi, Y. Morita, E. Mizohata, K. Oohora, J. Ohbayashi, T. Inoue and Y. Hisaeda, Chem. Commun., 2014, 50, 12560–12563 RSC.
- M. J. Kendrick and W. Al-Akhdar, Inorg. Chem., 1987, 26, 3971–3972 CrossRef CAS.
- D. Dolphin, A. W. Johnson and R. Rodrigo, Ann. N. Y. Acad. Sci., 1964, 112, 590–600 CrossRef CAS PubMed.
- H. Chen, H. Yan, L. Luo, X. Cui and W. Tang, J. Inorg. Biochem., 1997, 66, 219–225 CrossRef CAS.
- D. Lexa and J. M. Saveant, J. Am. Chem. Soc., 1976, 98, 2652–2658 CrossRef CAS PubMed.
- D. Occhialini, J. S. Kristensen, K. Daasbjerg and H. Lund, Acta Chem. Scand., 1992, 46, 474–481 CrossRef CAS.
- C. Li, K. Nishiyama and I. Taniguchi, Electrochim. Acta, 2000, 45, 2883–2888 CrossRef CAS.
- M. Nagai, Y. Nagai, K. Imai and S. Neya, Chirality, 2014, 442, 438–442 CrossRef PubMed.
- Furthermore, two additional peaks were observed at 625.221 and 2420.608, corresponding to CH3–CoIII(TDHC) and apoprotein, respectively, which are mainly derived from dissociation of the methylated cobalt complex from the protein matrix by CID (collision-induced dissociation) under the harsh ionization conditions. (Fig. S4 in the ESI†).
- B. Kräutler, Helv. Chim. Acta, 1987, 70, 1268–1278 CrossRef.
- As truncated models of Co(TDHC) and cobalamin, we used TDHC′ and corrin where all of the peripheral side chains were replaced with hydrogen atoms for DFT calculations (Fig. S7 in the ESI†).
- The heterolytic cleavage of the Co–C bond inevitably brings about the Co–N bond dissociation since Co(I) favours four-coordinate square planar geometry. Thus, the BDEIm and BDEIm-free for the Im-coordinated Co(III) complex and the Im-free Co(III) complex (penta-coordination), respectively, are defined by the following equations:
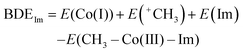 BDEIm-free = E(Co(I)) + E(+CH3) – E(CH3Co(III)) where E(X) is the energy of the optimized structure of X. In contrast, the homolytic BDE of CH3–CoIII(TDHC′) is higher than that of CH3–CoIII(TDHC′)–Im because the homolytic cleavage of the Co–C bond does not bring about the Co–N bond dissociation and stabilization of the Co(II) species induced by the axial ligation is greater than that of the CH3–Co(III) species (Table S1 in the ESI†).
BDEIm-free = E(Co(I)) + E(+CH3) – E(CH3Co(III)) where E(X) is the energy of the optimized structure of X. In contrast, the homolytic BDE of CH3–CoIII(TDHC′) is higher than that of CH3–CoIII(TDHC′)–Im because the homolytic cleavage of the Co–C bond does not bring about the Co–N bond dissociation and stabilization of the Co(II) species induced by the axial ligation is greater than that of the CH3–Co(III) species (Table S1 in the ESI†).
- In aqueous solution, the stabilization energy of the CH3–Co(III) complex by the imidazole ligation would not be large because of the exchange of a water molecule as a sixth ligand with the imidazole moiety. Indeed, an energy difference between CH3–Co(corrin)–OH2 and CH3–Co(corrin)–Im was estimated at the BP86/SV(P) level of theory to be −4.2 kcal mol−1. The value includes the effect of a dielectric constant of 74.8, which is a standard value of aqueous solution. The computed energy difference is in a good agreement with the experimentally determined equilibrium constant for the ligand exchange of the water molecule with imidazole (K = 4.52 × 102, ΔG = −RTln(K) = –3.6 kcal mol−1).68 In contrast, the Co–C bond formation is smoothly promoted in the protein environment since the solvent water molecules is not accessible to the proximal site and the histidine residue occupies at the coordination site of the cobalt atom.
- In contrast to the His93 residue in the myoglobin matrix, imidazole does not serve as a suitable axial ligand because the addition of imidazole (1 mM) to the CoII(TDHC) aqueous solution (0.05 mM) does not produce spectral changes. The binding constant of CoII(TDHC) for apomyoglobin was determined by UV-vis spectral changes in the titration experiment to be 20 μM−1 in 0.1 M potassium phosphate buffer (pH 7.0) at 25 °C (Fig. S13 in the ESI†).
- The chemical shifts of methyl groups of Nε2-methylated histidine and of Nδ1-methylated histidine were determined to be 36.20 ppm and 33.89 ppm, respectively, in D2O/CD3CN co-solvent (9:1, v/v) containing 1% TFA at 25 °C.
- In this study, we focus on the two pathways because activation barrier for reductive elimination pathway is much higher than that for the concerted pathway.24 In methionine synthase, a radical mechanism is one of the considerable pathways of the transmethylation from methylcobalamin to deprotonated homocysteine.18,69 However, the nucleophilic mechanism is more likely than a radical mechanism in our system because the radical mechanism requires the significantly uphill electron transfer (ET) from His64 to the methylated complex to form a diradical species. The relative energy of the intermediate generated by ET was estimated to be 102.9 kcal mol−1 at the BP86/6-31G(d) level of theory by following equation.
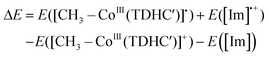 where [CH3–CoIII(TDHC′)]˙ and [Im]˙+ are one electron-reduced methylated complex and imidazole radical cation, respectively. The energy difference is significantly larger than the energy barrier of the stepwise pathway, indicating that the nucleophilic mechanism is more plausible than the radical mechanism.
where [CH3–CoIII(TDHC′)]˙ and [Im]˙+ are one electron-reduced methylated complex and imidazole radical cation, respectively. The energy difference is significantly larger than the energy barrier of the stepwise pathway, indicating that the nucleophilic mechanism is more plausible than the radical mechanism.
- The truncated model of Co(TDHC) and 4-ethylimidazole as a model of distal and proximal histidine residues, His64 and His93, were used for DFT calculations (Fig. S12 in the ESI†).
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
-
T. Hayashi, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, ch. 23, vol. 5, pp. 1–69 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1986, 84, 4524–4529 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- K. P. Jensen and U. Ryde, J. Phys. Chem. A, 2003, 107, 7539–7545 CrossRef CAS.
- C. Rovira, X. Biarnés and K. Kunc, Inorg. Chem., 2004, 43, 6628–6632 CrossRef CAS PubMed.
- C. Rovira and P. M. Kozlowski, J. Phys. Chem. B, 2007, 111, 3251–3257 CrossRef CAS PubMed.
- N. Dölker, A. Morreale and F. Maseras, J. Biol. Inorg. Chem., 2005, 10, 509–517 CrossRef PubMed.
- J. Kuta, S. Patchkovskii, M. Z. Zgierski and P. M. Kozlowski, J. Comput. Chem., 2006, 27, 1429–1437 CrossRef CAS PubMed.
- P. M. Kozlowski, T. Kamachi, T. Toraya and K. Yoshizawa, Angew. Chem., Int. Ed., 2007, 46, 980–983 CrossRef CAS PubMed.
- P. M. Kozlowski, T. Kamachi, T. Toraya and K. Yoshizawa, J. Biol. Inorg. Chem., 2011, 17, 293–300 CrossRef PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- G. C. Hayward, H. A. Hill, J. M. Pratt, N. J. Vanston and R. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1965, 6485–6493 CAS.
- N. Kumar, M. Jaworska, P. Lodowski, M. Kumar and P. M. Kozlowski, J. Phys. Chem. B, 2011, 115, 6722–6731 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: 10.1039/c5dt04109k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a