
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic, structural, and computational investigations of N-alkyl benzo-2,1,3-selenadiazolium iodides and their supramolecular aggregates†
Lucia M.
Lee
,
Victoria B.
Corless
,
Michael
Tran
,
Hilary
Jenkins
,
James F.
Britten
and
Ignacio
Vargas-Baca
*
Department of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, ON, Canada L8S 4M1. E-mail: vargas@chemistry.mcmaster.ca
First published on 6th January 2016
Abstract
Despite their versatility, the application of telluradiazoles as supramolecular building blocks is considerably constrained by their sensitivity to moisture. Albeit more robust, their selenium analogues form weaker supramolecular interactions. These, however, are enhanced when one nitrogen atom is bonded to an alkyl group. Here we investigate general methods for the synthesis of such derivatives. Methyl, iso-propyl and tert-butyl benzo-2,1,3-selenadiazolium cations were prepared by direct alkylation or cyclo-condensation of the alkyl-phenylenediamine with selenous acid. While the former reaction only proceeds with the primary and tertiary alkyl iodides, the latter is very efficient. Difficulties reported in earlier literature are attributable to the formation of adducts of benzoselenadiazole with its alkylated cations and side reactions initiated by aerobic oxidation of iodide. However, the cations themselves are resilient to oxidation and stable in acidic to neutral aqueous medium. X-ray crystallography was used in the identification and characterization of the following compounds: [C6H4N2(R)Se]+X−, (R = CH(CH3)2, C(CH3)3; X = I−, I3−], [C6H4N2(CH3)Se]+I−, and [C6H4N2Se][C6H4N2(CH3)Se]2I2. Formation of Se⋯N secondary bonding interactions (chalcogen bonds) was only observed in the last structure as anion binding to selenium is a strong competitor. The relative strengths of those forces and the structural preferences they enforce were assessed with DFT-D3 calculations supplemented by AIM analysis of the electron density.
Introduction
While hydrogen bonding and the coordination of metal ions are preponderant in supramolecular chemistry, other interactions are receiving increasing interest in this field. A notable example is halogen bonding,1–3 a special case of the phenomenon termed secondary bonding4 that is recurrent in the structures of compounds of the heavy p-block elements. The potential of secondary bonding in supramolecular chemistry is well exemplified by the derivatives of the 1,2,5-telluradiazole ring (1, Chart 1). Their molecules usually associate by two antiparallel Te⋯N secondary bonding interactions forming the [Te–N]2 supramolecular synthon,5 a virtual four-membered ring (Scheme 1, E = Te).6 In the absence of substantial steric hindrance, each molecule can simultaneously form the [Te–N]2 supramolecular synthon twice building in this way supramolecular ribbons.7 The driving force for association of these molecules is strong enough to overcome moderate steric repulsion by allowing structural distortion of the ribbons. This effect induces properties such as chromotropism8 and second-harmonic generation9 which are of interest for practical applications. In addition to the self-association of 1,2,5-telluradiazoles through Te⋯N interactions, other studies have also evaluated their affinity to Lewis bases in both solid10 and solution.11–13 In spite of their versatility, prospects of widespread application of telluradiazoles are limited by their sensitivity to moisture.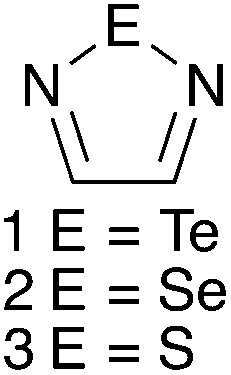 | ||
| Chart 1 | ||
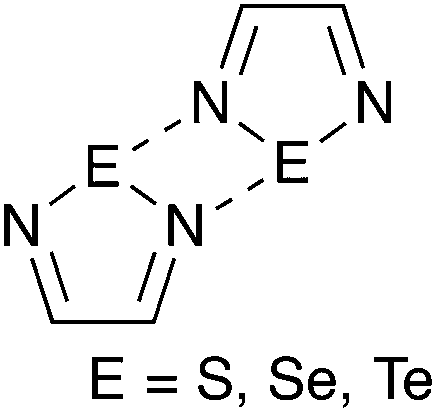 | ||
| Scheme 1 | ||
The lighter members of the chalcogenadiazole family (2, 3, Chart 1) are much more robust but SBIs centred on S and Se atoms are weaker than those formed by Te14 and instances of supramolecular association are less frequent in such cases. However, crystallographic evidence suggests that attachment of Lewis acid (a transition metal ion15,16 or a borane17), on one nitrogen of the heterocycle strengthens the secondary interactions made by Se and Te, shortening the corresponding distances. Such effect is also strong in the cations formed by N-alkylation and provides an explanation for the peculiar trend in reduction potentials of the N-methyl-benzo-2,1,3-chalcogenadiazolium cations in acetonitrile.18 Such evidence of molecular association in solution through SBIs is consistent with the observation of [M2H]+ supramolecular aggregates in the mass spectrum of benzo-2,1,3-telluradiazoles19 as well as calculated interaction energies and AIM parameters.12,18
N-Alkyl substituted selenadiazolium cationic rings could therefore be a convenient alternative to telluradiazoles in supramolecular architecture, provided that they are reasonably stable and the preparation methods are flexible enough to accommodate multiple variations in the structure. The most straightforward synthetic method would be the direct alkylation of the heterocycle in 4 (Chart 2). However, early reports of this approach describe very inefficient reactions plagued by multiple by-products with most common alkylating reagents.20,21 Clean and efficient alkylations have been reported rather recently using very reactive agents such as Me–OS(O)2–R′ (R′ = OMe, C6H2(NO2)2-2,4),20 F3C–SO3–CH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 or (CH3)3O+BF4−;22 but these methods are applicable with only a handful of alkyl groups. Alternatively, the alkyl group could be attached to nitrogen before formation of the heterocycle. This approach was actually reported in an early publication20,21 but its products were not structurally authenticated. A third route to N-alkyl selenadiazolium cations, also limited in flexibility, consists of the spontaneous de-alkylation of one nitrogen atom during the reaction of N,N′-bis(tert-butyl)-diazabutadiene with SeCl4.23
18 or (CH3)3O+BF4−;22 but these methods are applicable with only a handful of alkyl groups. Alternatively, the alkyl group could be attached to nitrogen before formation of the heterocycle. This approach was actually reported in an early publication20,21 but its products were not structurally authenticated. A third route to N-alkyl selenadiazolium cations, also limited in flexibility, consists of the spontaneous de-alkylation of one nitrogen atom during the reaction of N,N′-bis(tert-butyl)-diazabutadiene with SeCl4.23
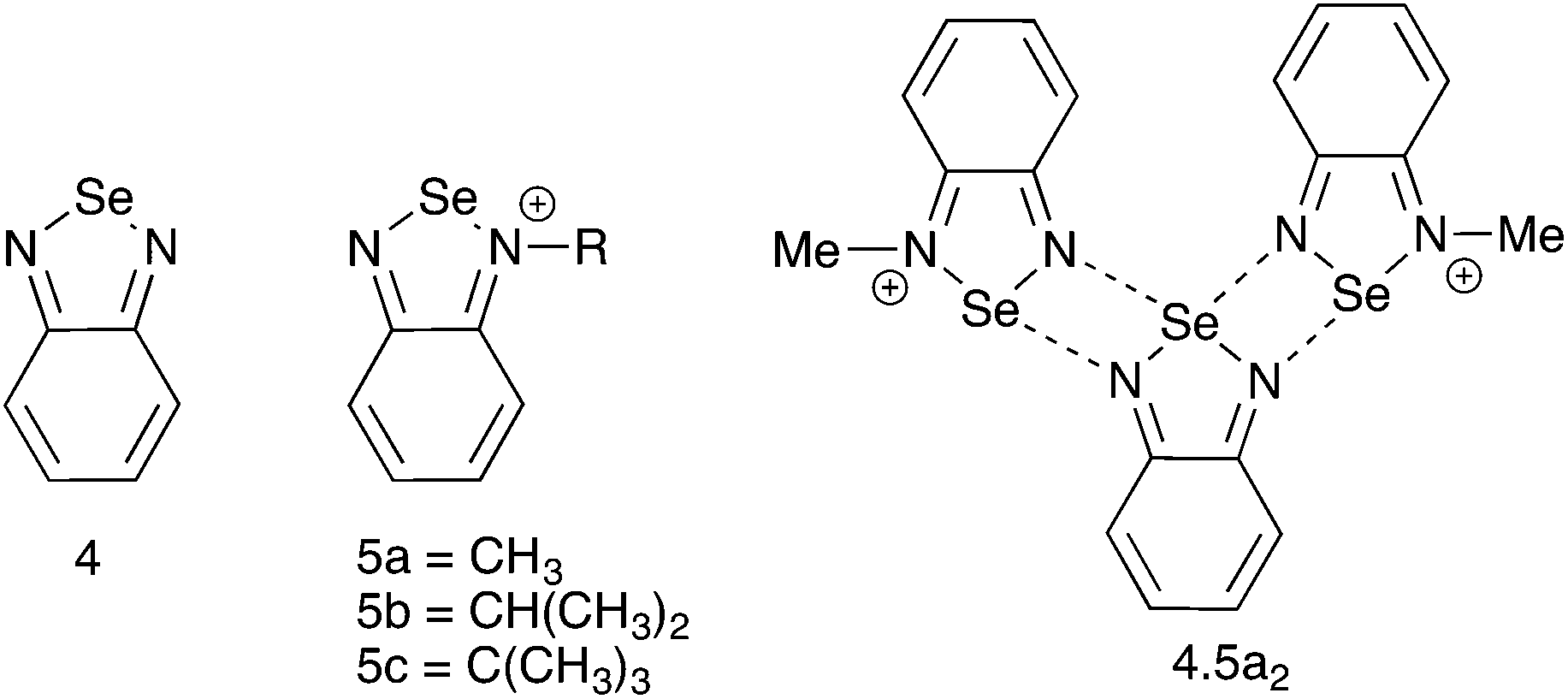 | ||
| Chart 2 | ||
As we are interested in using the N-alkylated selenadiazolium cations as supramolecular building blocks, in this report we compare the two most flexible synthetic methods for the preparation of the derivatives with primary, secondary and tertiary alkyl groups (5, Chart 2). We examine the crystal structures of their iodide salts, the affinity of the cations for halide anions, and spectroscopic properties useful for the characterization of these species in solution.
Experimental
Materials and general procedures
Selenium dioxide, iodomethane were used as received from commercial suppliers. The N-alkyl 1,2-phenylenediamines were prepared from the reaction of the corresponding commerically available primary amines with 1-fluoro-2-nitrobenzene, followed by reduction with H2 catalyzed by Pd/C.24–26 Benzo-2,1,3-selenadiazole was prepared by the reaction of 1,2-phenylenediamine with selenium dioxide according to literature.27 Anhydrous-grade solvents were used without further purification.Instrumentation
1H, 13C and 77Se NMR spectra were recorded on Bruker 200 and Bruker 600 spectrometers at ambient temperature. Chemical shifts are reported in δ values (parts per million) with respect to the resonances of tetramethylsilane for 1H and 13C and with respect to the line of Me2Se for the chemical shift of 77Se. In the last case, spectra were obtained using 1H–77Se HMBC unless otherwise stated. IR spectra were recorded on polyethylene pellets on a Nicolet 6700 FT-IR spectrometer with a resolution of 4 cm−1. Raman spectra were obtained with a Renishaw inVia spectrometer exciting at 785 nm, 30 mW, averaging 10 × 10 s scans. Low- and high-resolution electrospray spectral analyses were performed using Micromass GCT spectrometer and a Micromass Quattro Ultima for electron ionization spectra. In each case, the sample was introduced into the ionization chamber in a shortened borosilicate glass capillary on a probe rod. The samples were then heated and the temperature range that displayed the most intense parent ion peaks was used for spectral acquisition. A 70 eV electron stream ionized the sample and the positively charged ions were identified by a time-of-flight detector. A Cary 50 UV-vis spectrometer and its Cary Win-UV software package were used to acquire the spectra. Additional processing was carried out using the GramsAI suite (version 8.0). Melting points were measured on a Thomas-Hoover melting point apparatus and are reported uncorrected. Elemental analysis was performed by the Science Centre of London Metropolitan University, 29 Hornsey Road, London, UK, N7 7DD.All single crystal X-ray diffraction samples were mounted with paratone oil at the tip of a glass fibre installed on a MiTeGen goniometer head, and kept under a cold stream of nitrogen while on the diffractometer. Data were collected at 100 K on a Bruker APEX2 diffractometer, using Mo Kα radiation (λ = 0.71073 Å) and outfitted with an Oxford cryostream low-temperature accessory. Ω and ϕ scans were collected in 0.5° steps with a crystal to detector distance of 4.954 cm. The preliminary unit cell parameters were determined using a minimum of 50 frames from three different orientations, and final cell refinement after integration in SAINT. Data were corrected for absorption and scaled using face-indices as well as redundant data in SADABS. Crystal structures were solved using SHELXL and most structures were refined by full-matrix least square of all F2 values with the WinGX package.
Syntheses
 ]+), 1529.7 (C6H4N2
]+), 1529.7 (C6H4N2 ) ppm. IR (cm−1): 3602 (m), 2854(b), 2341 (m), 2150 (vw), 2018 (w), 1982 (w), 1949 (w), 1812 (w), 1600 (w), 1529 (m), 1515 (m), 1464 (sh), 1134 (vw), 1044 (m), 905 (vw), 730 (sh) 720 (sh). Mp. 148–150 °C (d). E.A. % calcd for C20H18I2N6Se3: C 28.83, H 2.18, N 10.09, found C 28.72, H 2.09, N 9.98.
) ppm. IR (cm−1): 3602 (m), 2854(b), 2341 (m), 2150 (vw), 2018 (w), 1982 (w), 1949 (w), 1812 (w), 1600 (w), 1529 (m), 1515 (m), 1464 (sh), 1134 (vw), 1044 (m), 905 (vw), 730 (sh) 720 (sh). Mp. 148–150 °C (d). E.A. % calcd for C20H18I2N6Se3: C 28.83, H 2.18, N 10.09, found C 28.72, H 2.09, N 9.98.
Computational methods
All the structures considered in this study were fully optimized using the ADF DFT package (versions 2013.01–2014.01).28,29 The Adiabatic Local Density Approximation (ALDA) was used for the exchange–correlation kernel30,31 and the differentiated static LDA expression was used with the Vosko–Wilk–Nusair parameterization.32,33 Calculation of model geometries was gradient-corrected with the exchange and correlation functionals of Perdew, Burke and Ernzerhof (PBE)34 and applying the zeroth-order regular approximation (ZORA)34–38 formalism with the specially adapted triple-ζ all-electron plus one-polarization-function basis sets. The contribution of dispersion was modelled with Grimme's correction.39 Analytical frequency calculations were performed to ensure that each geometry was at an energy minimum.40,41 The TD-DFT calculation was performed from the optimized geometry of [C6H4N(NCH3)Se]+ using the statistical average of potentials model for exchange and correlation (SAOP)42–44 and a basis set of quadruple-zeta quality plus polarization.Results and discussion
Syntheses by direct alkylation
Although benzo-2,1,3-selenadiazole is a weak base, it is readily protonated by strong acids.45 For this reason, the purported inefficiency20,21 of its reaction with primary alkyl-iodides is unusual and merited reinvestigation using a more reactive tertiary iodide. Initial attempts at bench-top alkylation with (CH3)3CI in a hot toluene solution yielded a complex mixture, as made evident by the 1H NMR spectrum. Slow evaporation of the solution yielded a mixture of crystals with multiple morphologies; three of which were identified by single-crystal X-ray diffraction: [5c][I3], [4]2[4-H]2[I][I3] and [4]2(C6H4(NH2)2H+)2][I]2. The first species does contain the N-tert-butyl benzo-selenadiazolium cation (5c), which demonstrates that the alkylation reaction does proceed. However, the presence of the triiodide anion, plus protonated phenylenediamine and benzoselenadiazole point to the actual problem with this experiment: the iodide ion is likely oxidized by atmospheric oxygen to I3− which then halogenates the solvent or benzoselenadiazole to generate HI. Crystals of [5c][I] were obtained when the reaction was carried out under an atmosphere of nitrogen, albeit in low yield. Similarly, the alkylation reaction proceeded with neat CH3I in anaerobic conditions, however, in this case the alkylation is incomplete due to the formation of the crystalline 4[5a]2[I]2, which was identified by X-ray diffraction. The salt [5a][I] was obtained from 4[5a]2[I]2 by recrystallization from dilute solutions. Interestingly, iso-propyl iodide was completely unreactive towards benzoselenadiazole under the same conditions. While the reaction of tert-butyl iodide is likely to proceed by a SN1 mechanism, it is likely that the primary and secondary iodides prefer a SN2 mechanism but steric hindrance prevents the reaction of the iso-propyl halide.Condensation of selenous acid with N-alkylated phenylenediamines
Even in aqueous medium, benzoselenadiazole is readily formed by the reaction of H2SeO3 and phenylenediamine. N-substituted ortho-diamino benzenes were reported to undergo an analogous reaction in glacial acetic acid producing the benzoselenadiazolium cations, which are isolated by precipitation with sodium halide.46,47 This procedure works as expected but we found more convenient to carry out the reaction in a mixture of ethanol and trifluoroacetic acid in order to avoid using the less volatile acetic acid. Structural characterization by X-ray diffraction did confirm the identity of the products. We also found that exposure of the cations in solution to the atmosphere resulted in iodide oxidation and crystallization of the corresponding triiodide salts, which was verified by the strong scattering band at 111 cm−1 in the Raman spectrum48 or crystallographic analysis.X-ray crystallography
Crystallographic and refinement data for the salts of the three cations are presented in Table 1; selected distances and angles in each crystal structure are provided in Table 2.| 4[5a]2[I]2 | [5a][I] | [5b][I3] | [5b][I] | [5c][I3] | [5c][I] | |
|---|---|---|---|---|---|---|
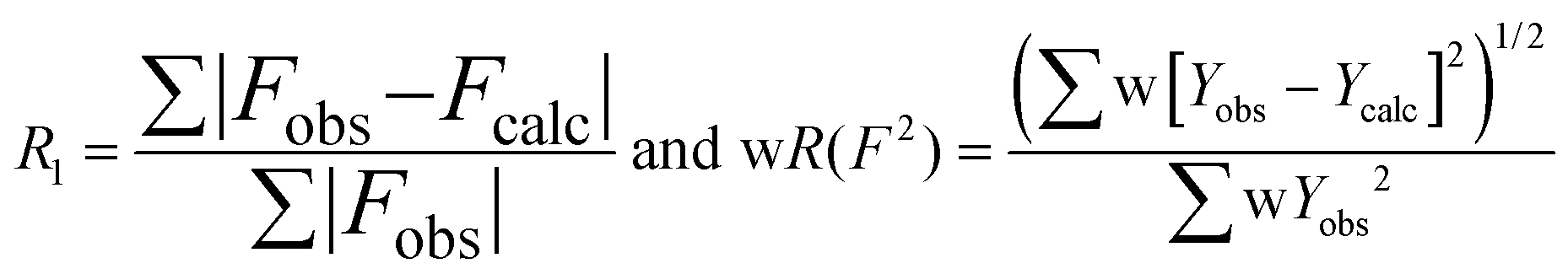 . . |
||||||
| Chemical formula | C20H18I2N6Se3 | C7H7N2SeI | C9H11N2SeI3 | C9H11N2SeI | C10H13N2SeI3 | C10H13N2SeI |
| CCDC | 1424372 | 1424371 | 1424373 | 1424374 | 1424370 | 1434655 |
| Formula mass | 833.08 | 325.01 | 606.86 | 353.06 | 620.9 | 367.08 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| a, Å | 15.986(1) | 6.471(1) | 11.317(2) | 16.084(1) | 7.668(1) | 11.668(1) |
| b, Å | 11.671(1) | 13.216(2) | 9.818(2) | 10.525(1) | 10.014(2) | 7.699 (1) |
| c, Å | 12.743 (1) | 10.474(2) | 13.569(2) | 6.597(1) | 20.949(4) | 13.253 (1) |
| α, ° | 90 | 90 | 90 | 90 | 90 | 90 |
| b, ° | 92.566(3) | 92.426(4) | 105.047(3) | 90 | 95.631(3) | 90 |
| γ, ° | 90 | 90 | 90 | 90 | 90 | 90 |
| Unit cell volume, Å3 | 2375.2(2) | 894.9(3) | 1456.0(4) | 1116.8(2) | 1600.7(5) | 1190.6(1) |
| Temperature, K | 100.15 | 100.15 | 100.15 | 100.15 | 100.15 | 296 |
| Space group | C12/c1 | P21/n | P121/n1 | Pna21 | P21/c | Pnma |
| Z | 4 | 4 | 4 | 4 | 4 | 4 |
| No. of reflections measured | 2700 | 12698 |
19082 |
16547 |
8761 | 26960 |
| No. independent reflections | 2213 | 2521 | 4908 | 3306 | 4094 | 3066 |
| Final R1 values (I > 2σ(I)) | 0.0404 | 0.0199 | 0.0261 | 0.0229 | 0.0381 | 0.0331 |
| Final wR(F2) values (I > 2σ(I)) | 0.0685 | 0.0385 | 0.0447 | 0.0419 | 0.0759 | 0.0828 |
| Final R1 values (all data) | 0.0550 | 0.0262 | 0.0399 | 0.0276 | 0.0682 | 0.0413 |
| Final wR(F2) values (all data) | 0.0733 | 0.0404 | 0.0481 | 0.0434 | 0.0861 | 0.0847 |
| Compound | 4[5a]2[I]2 | [5a][I] | 5b[I3] | [5b][I] | [5b][I3] | 5c·I |
|---|---|---|---|---|---|---|
| Se–N1 | 1.856(5) | 1.785(2) | 1.766(3) | 1.774(3) | 1.742(5) | 1.749(2) |
| Se–N2 | 1.777(5) | 1.875(2) | 1.857(3) | 1.881(2) | 1.841(4) | 1.864(2) |
| N1–C1 | 1.339(7) | 1.327(3) | 1.333(4) | 1.333(4) | 1.349(8) | 1.343(3) |
| N2–C6 | 1.336(7) | 1.329(3) | 1.338(4) | 1.338(4) | 1.347(6) | 1.339(3) |
| C1–C6 | 1.443(7) | 1.440(3) | 1.444(4) | 1.439(4) | 1.414(7) | 1.442(4) |
| C1–C2 | 1.432(8) | 1.431(3) | 1.355(5) | 1.428(6) | 1.414(7) | 1.421(4) |
| C2–C3 | 1.366(8) | 1.351(4) | 1.426(5) | 1.355(6) | 1.342(9) | 1.361(3) |
| C3–C4 | 1.428(8) | 1.431(4) | 1.426(5) | 1.360(4) | 1.400(9) | 1.417(4) |
| C4–C5 | 1.371(8) | 1.359(3) | 1.353(4) | 1.427(5) | 1.345(9) | 1.368(4) |
| C5–C6 | 1.428(8) | 1.422(3) | 1.422(5) | 1.425(5) | 1.418(1) | 1.421(3) |
| N1–Se–N2 | 88.7(2) | 88.16(9) | 89.6(1) | 88.6(2) | 90.4(4) | 90.2(1) |
| Se–N1–C1 | 111.3(4) | 111.2(2) | 110.7(2) | 111.0(3) | 109.7(7) | 110.3(2) |
| Se–N2–C6 | 111.6(4) | 111.4(1) | 110.6(2) | 111.0(3) | 109.8(7) | 110.2(2) |
| Se–N2–C7 | 124.0(4) | 124.9(2) | 124.4(2) | 124.7(3) | 122.4(6) | 122.4(2) |
| C6–N2–C7 | 124.4(5) | 123.7(2) | 125.1(3) | 124.3(3) | 127.8(8) | 127.5(2) |
The crystal structure of 4[5a]2[I]2 consists of a molecule of 4 bound to two 5a, conforming a pseudo-trimer assembled by two asymmetric [Se–N]2 supramolecular synthons (Fig. 1) with asymmetric SBI distances of 2.573(4) and 2.937(1) Å. This aggregate is analogous to the product formed by the reaction of benzoselenadiazole with [(CH3)3O][BF4],22 in which the Se⋯N and Se⋯F secondary bonding distances are 2.573(2) and 2.966(6) Å and 2.970(4) Å respectively. In this arrangement, the molecules of 4 and 5a are nearly coplanar with a deviation angle of 5.06°. This structure also features Se–I short distances at 3.528(1) Å and 3.831(1) Å. Both iodides sit out of the average plane of the aggregate forming a virtual four-membered ring (i.e. the [Se I]2 supramolecular synthon) with the Se atoms of the 5a molecules of two neighbouring pseudotrimers.
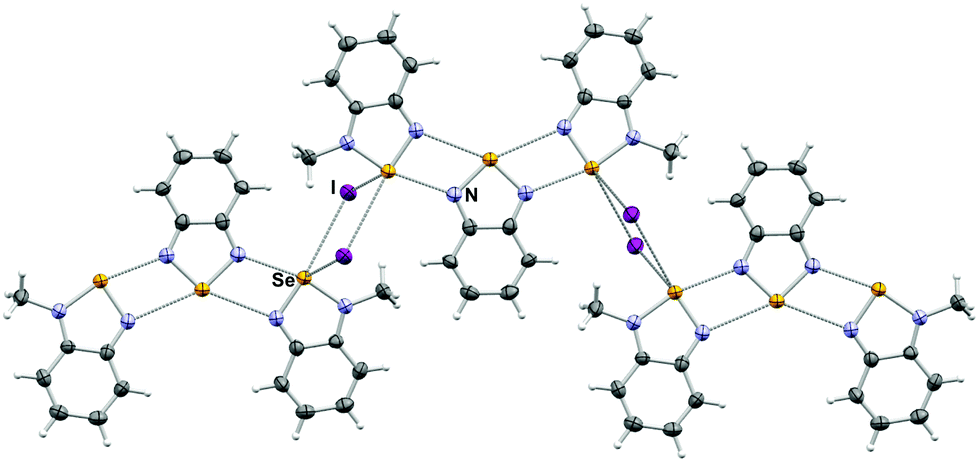 | ||
| Fig. 1 Arrangements of molecules and ions in the crystal 4[5a]2[I]2. Selected distances: Se1⋯N1: 2.573(4) Å and 2.937(1) Å. | ||
In clear contrast with the structures of the pseudo-trimers,22 and the dimers observed for [5a][CF3SO3]18 and [C2H2N(NC(CH3)3)Se][GaCl4],49 the crystal structures of the iodide and triiodide salts of the alkylated cations 5a–c feature no association of the heterocycles to each other. In all cases, strong Se–anion interactions prevent formation of the Se⋯N SBIs (Fig. 2 and 3).
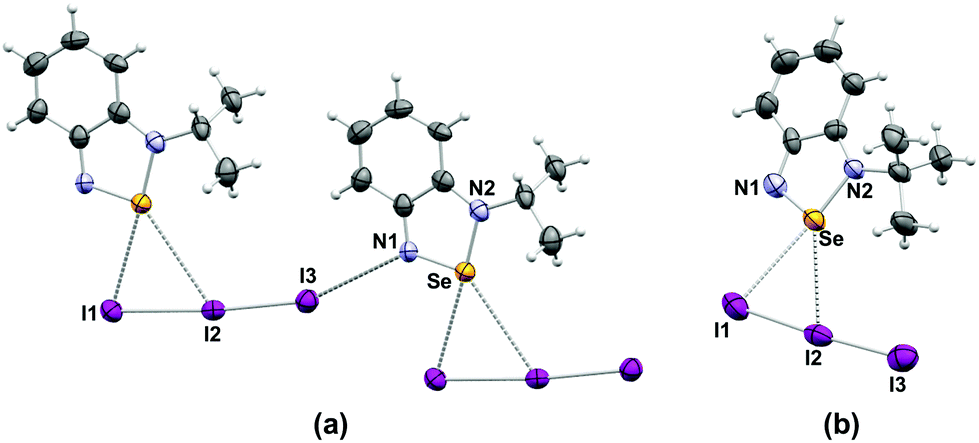 | ||
| Fig. 2 Crystal structures of (a) [5b][I3] and (b) [5c][I3]. Selected bond distances (a) Se⋯I1: 3.249(1) Å, Se⋯I2: 3.626(5) Å; (b) Se⋯I1: 3.309(1) Å, Se⋯I2: 3.727(1) Å. | ||
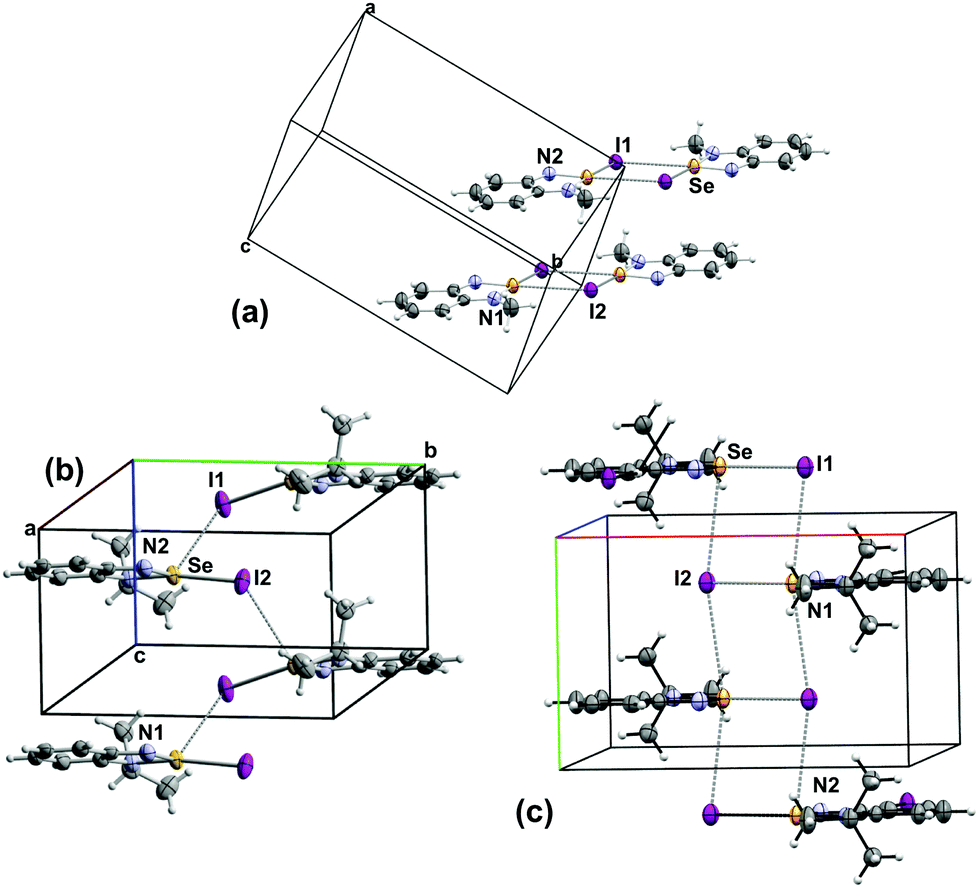 | ||
| Fig. 3 Packing arrangement in the crystal structures of the iodide salts of (a) 5a (b) 5b and (c) 5c. | ||
The triiodide anions in the crystals of [5b][I3] and [5c][I3] engage in two interactions with the selenium atom. The shortest (3.249(1), 3.309(1) Å, respectively) is with the terminal iodine atom I1; the contact to the middle I2 atom is longer (3.626(1), 3.727(1) Å, respectively), cf. the sum of van der Waals radii 3.88 Å. In the case of [5b][I3] the terminal iodine atom I3 is in close proximity (3.491(3) Å, cf. the sum of van der Waals radii 3.53 Å) to the nitrogen atom of a neighbouring 5b cation, the nearly linear geometry of this arrangement is indicative of a halogen bond. Consequently, the I2–I3 bond is longer (2.879(1) Å) in [5b][I3] than in [5c][I3] (2.829(1)].
The arrangement of the ions and the Se⋯I interactions in the [5a–c][I] lattices are influenced the size of the alkyl group (Fig. 3). The small methyl group in [5a][I] confines the Se⋯I interactions to the plane of the cation forming iodide-bridged dimers that conform the [Se I]2 supramolecular synthon with short (3.178 (1) Å) and long (3.610 (1) Å) SBIs. Similar binding of anions to Se has been observed in related species. For example, a virtual four-membered ring consisting of Se and Cl atoms is formed in the crystal structure of [C2H2NN(H)Se][Cl] (2.900(1) Å and (3.075 (1) Å).45 The structure of [C2H2N(NC(CH3)3)Se][Cl] also features a planar selenadiazole ring with one Se–Cl bond distance of 2.605 (1) Å.23 In contrast, the GaCl4– salt of the same selenadiazole heterocycle does not display any Se–Cl short interactions, instead it contains the [Se–N]2 supramolecular synthon.23 The larger alkyl groups favour interlayer interactions (eminently electrostatic) in [Se I]∞ supramolecular chains. The Se–I distances are 3.043 (1) Å and 3.696 (1) Å in the structure of [5b][I] and 3.147 (1) and 3.880 (1) Å in [5c][I].
Spectroscopy in solution
The iodide salts of 5a–c are stable in air. The compounds are slightly soluble in water at room temperature, their solubility decreases with the size of the R group. In fact, it is possible to recrystallize 5a from hot water. Aqueous solutions of the cations have a characteristic red-brown colour, their UV-vis absorption spectrum displays prominent maxima in the range of 330–343 nm and a weaker band between 440–460 nm. These spectra are in excellent agreement with the TD-DFT calculation for the 5a, which attributes the bands to the LUMO ← HOMO and LUMO ← HOMO−1 excitations (Fig. 4). The UV-vis spectra do not change from neutral to acidic pH, but hydrolysis ensues in basic medium. Given the modest solubility of the 5a–c iodides, their 77Se NMR chemical shifts are best obtained through 1H–77Se heteronuclear multiple bond correlation (HMBC) experiments. The 77Se δ values (1455–1490 ppm) appear at slightly lower frequency than the 1515 ppm reported for [C6H4N(NCH3)Se][CF3SO3] in acetonitrile.18 For the pseudo-trimer 4[5a]2[I]2, two resonances were observed with frequencies that correspond to those of the constituting neutral and cationic molecules, suggesting that the aggregate dissociates in solution.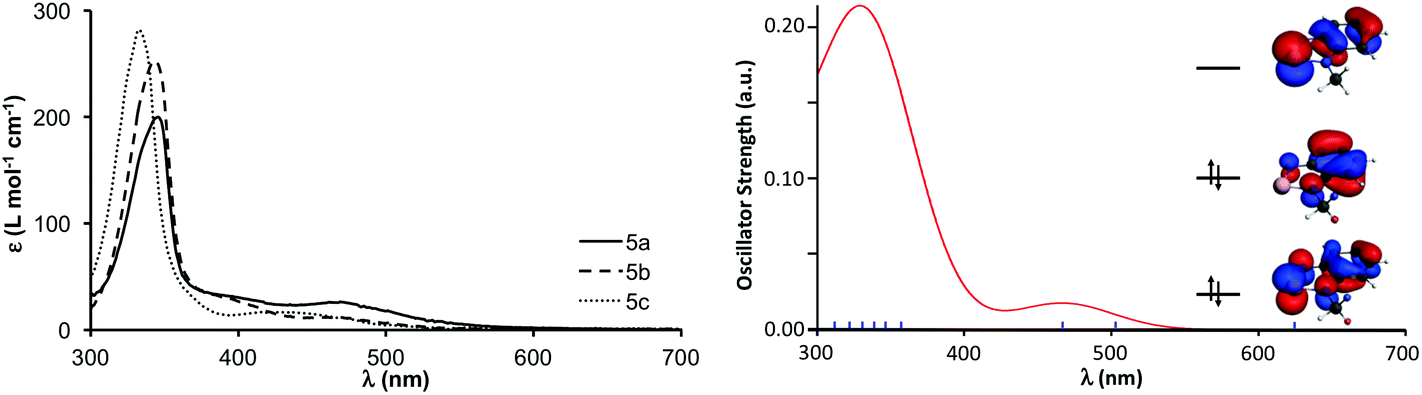 | ||
| Fig. 4 Experimental (left) and calculated (right) UV-vis spectra of [5a][I]. Frontier orbitals shown as an inset in the calculated spectrum. | ||
DFT calculations
As the crystal structures show there is competition between the formation of the [Se–N]2 supramolecular synthon and binding of the anion to selenium, the relative strengths of these interactions were evaluated using DFT and were extended to include the chloride and bromide salts. For computational expediency, GGA calculations were performed with the PBE functional, supplemented by dispersion and relativistic corrections. Model structures were optimized from the crystallographic coordinates and expanded to include hypothetical isomers and the analogues with chloride and bromide anions. Whenever an experimental structure was available, bond lengths were reproduced within 0.01 Å, the largest deviations correspond to the intramolecular Se–N distances. Selenium-centred secondary bond distances were reproduced within 0.01 Å to nitrogen and 0.50 Å to iodide. The large deviation in the latter case is likely due to the additional interactions of the anion in the lattices. Interaction energies were evaluated using the transition-state method50 that partitions the energy of interaction between two molecules or fragments in a hypothetical process in which the constituting units are first calculated individually, brought to their equilibrium positions without orbital mixing, and finally the electron density is relaxed by the interaction of fragment orbitals. The change from the first to the second step is regarded as the total steric interaction that results from the sum of the electrostatic (EElstat) and Pauli-repulsion (EPauli). The third step gives the orbital interaction energy (EOrb). Dispersion is treated as a separate contribution (EDisp). The total interaction energy is given by the sum of all contributions (eqn (1)).| EInteraction = EElstat + EPauli + EOrb + EDisp | (1) |
Three partitioning schemes (Fig. 5, i–iii) were used to evaluate the secondary interactions within the pseudo-trimer 4[5a]2[I]2 the results are summarized in Table 3. In this analysis the average interaction energy of the [Se–N]2 supramolecular synthon was 56.4 kJ mol−1. Formation of the interactions in iii and ii could be regarded as steps in the assembly of the pseudotrimer and the calculations show that the interaction energies are nearly additive.
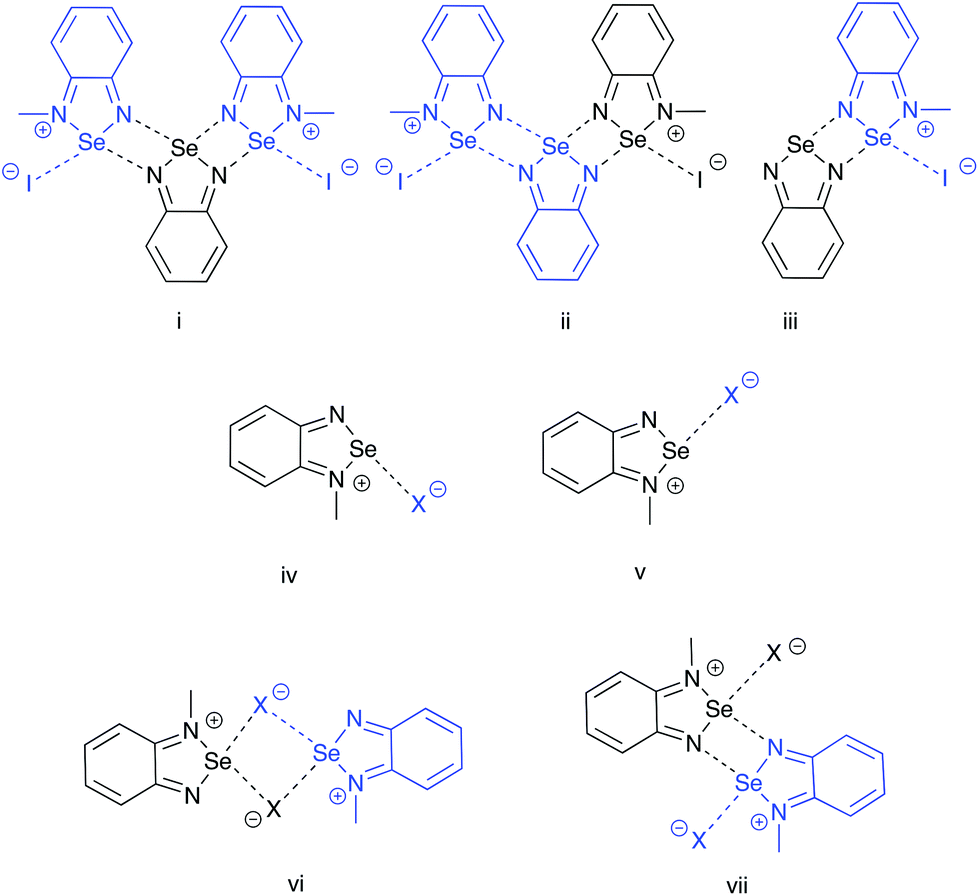 | ||
| Fig. 5 Partition schemes used in the DFT-D3 fragment interaction analyses. Each fragment (region) is identified with a distinct colour. | ||
| Partition scheme | E Int | E Elstat | E Pauli | E Orb | E Disp |
|---|---|---|---|---|---|
| i | −112.05 | −294.18 | 416.15 | −203.76 | −30.26 |
| ii | −55.54 | −145.38 | 209.63 | −102.26 | −17.54 |
| iii | −57.65 | −147.58 | 208.39 | −103.30 | −15.16 |
In the case of the [5a][X] (X = Cl, Br, I) ion pairs, two geometries were considered, cis and trans to the alkylated nitrogen atom (Fig. 5, iv and v). The results, summarized in Table 4, indicate that in all three cases the trans structure is preferred. This is consistent with the results of previous calculations of the electrostatic potential maps of the borane adducts of chalcogenadiazoles which showed that the most prominent σ hole on the chalcogen is opposite to the substituted nitrogen.12
| Partition scheme | X | E Int | E Elstat | E Pauli | E Orb | E Disp |
|---|---|---|---|---|---|---|
| iv | Cl | −520.8 | −545.8 | 284.0 | −255.2 | −3.8 |
| Br | −492.8 | −512.0 | 263.2 | −239.6 | −4.4 | |
| I | −461.4 | −470.7 | 232.7 | −217.7 | −5.7 | |
| v | Cl | −543.1 | −558.9 | 375.5 | −357.2 | −2.5 |
| Br | −516.1 | −521.0 | 345.1 | −337.3 | −2.9 | |
| I | −487.5 | −475.8 | 310.8 | −319.1 | −3.4 |
The dimerization energies the [5a][X] ion pairs were evaluated in two possible geometries (Fig. 5, vi and vii), the results are compiled in Table 5. While dimer vi is formed from the cis (iv) structures and contains the [Se–N]2 supramolecular synthon, the trans (v) models form the [Se X]2 supramolecular synthon in dimer vii. The energy of interaction is more negative in latter case, which indicates that the structure of the dimer of the methyl cation experimentally observed in the crystal of [5a][I] salt is favoured by the greater stability of the trans geometry of the ion pair. The results also show that the dimerization of the ion pairs [5a][I] is stronger than the interaction of the ion pair with the neutral molecule of 4; however, the overall sum of interactions favours the formation of the pseudo-trimer.
| Partition scheme | X | E Int | E Elstat | E Pauli | E Orb | E Disp |
|---|---|---|---|---|---|---|
| vi | Cl | −61.2 | −60.5 | 51.4 | −39.6 | −12.3 |
| Br | −64.3 | −68.2 | 66.23 | −48.6 | −13.7 | |
| I | −63.7 | −75.2 | 87.1 | −61.0 | −14.7 | |
| vii | Cl | −57.2 | −172.6 | 242.3 | −110.7 | −16.2 |
| Br | −65.4 | 265.1 | −187.8 | −124.8 | −17.8 | |
| I | −71.6 | 298.5 | −206.8 | −144.0 | −19.3 |
The pseudo-trimer structures 4[5a]2[X]2 (X = I, BF4) are especially interesting because of their two distinct Se⋯N SBI distances. In order to assess the relative strength of each SBI, a bond-energy decomposition analysis was performed using the extended-transition-state method from the natural orbitals for chemical valence (ETS-NOCV).51 In this method the orbital component to the total bonding energy between fragments is calculated as in eqn (2).
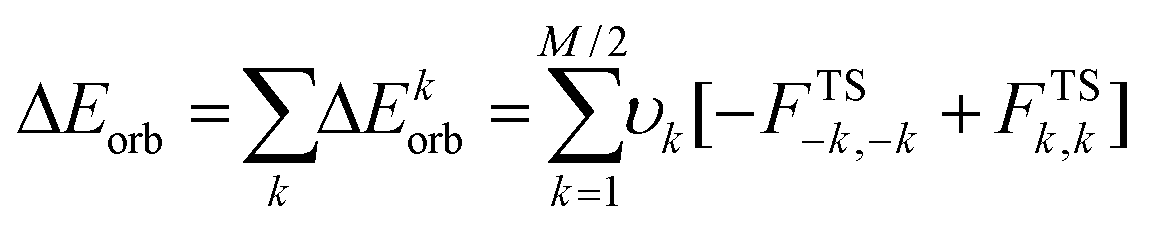 | (2) |
The terms −FTS−k,−k and FTSk,k are diagonal transition-state Kohn–Sham matrix elements corresponding to NOCV's with eigenvalues −υk and υk respectively. This particular orbital interaction term provides both a qualitative and quantitative picture of the electronic density reorganization in the [Se–N]2 supramolecular synthons. Each of the fragment interaction calculations (i–iii) shows that the shorter Se⋯N SBI, that opposite to the quaternary nitrogen, is stronger (−13.0 to −16.9 kcal mol−1) than the longer (−2.4 to −7.5 kcal mol−1).
The Se⋯N interactions in 4[5a]2[I]2 were also examined through Bader's theory of atoms in molecules (AIM).52 Previous studies12 applied AIM to the examination the Lewis acidity and basicity of 1–3; their results highlighted the ambiphilic character of the chalcogenadiazoles molecules and showed that the attachment of a Lewis acid to one nitrogen of the heterocycle strengthens the [E–N]2 supramolecular synthon formed by the other nitrogen. The calculated electron density (ρ(r)) and its Laplacian (∇2ρ(r)) at the bond critical points (BCPs) of 4[5a]2[I]2 are given in Table 6. Although the difference of Se⋯N distances is small, the AIM parameters suggest that they are very different in strength as there is indication that the energy of interaction scales linearly with the BCP density.
| AIM parameter at BCP | Se⋯N | Se⋯I | |
|---|---|---|---|
| Long | Short | ||
| ρ (e Å−3 × 102) | 3.81 | 8.24 | 3.69 |
| ∇2ρ (e Å−5 × 102) | 2.38 | 6.67 | 4.67 |
For comparison, the corresponding BCP parameters of the Se⋯I interaction in the pseudo-trimer are included in Table 6. It is interesting that while the BCP electron density is smaller, the its Laplacian is intermediate between the values for the Se⋯N SBIs. The comparison was further extended by calculating the BCP parameters of the dimers of [5a][X] (X = Cl, Br, I) in the two possible conformations considered under partition schemes vi and vii. The results are compiled in Table 7. All these values suggest that the Se⋯X and Se⋯N interactions are of similar strength. In some cases, the calculated BCP parameters approach those of the Te⋯N SBI in the neutral dimer of 1,2,5-telluradiazole (4.20 e Å−3 × 10−2 and 10.9 e Å−5 × 10−2).12
Conclusions
As these investigations show, the most convenient method for the preparation of N-alkyl benzoselenadiazolium cations is the condensation of selenous acid with N-substituted phenylenediamines. Such intermediates can be conveniently obtained from o-fluoronitrobenzene and primary amines. As a wide variety of aromatic diamines are commercially available or readily prepared, this approach will certainly enable the preparation of a diversity of polycations that will be useful as building blocks for supramolecular structures, including species capable of forming large supramolecular aggregates. However, the N-alkyl benzoselenadiazolium cations have a strong affinity for halide anions, therefore the anions must be replaced for non-coordinating ions in order to enable the self-assembly of the building blocks through Se⋯N supramolecular interactions. In contrast to the telluradiazoles, and as a great advantage, the cations are not only stable in air but also in neutral to acidic aqueous media.Acknowledgements
The financial support of the Natural Science Engineering Research Council of Canada (NSERC, DG-IVB, PGSD-LML) and the Ontario Graduate Scholarship program (LML) is gratefully acknowledged. Portions of this work were made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: http://www.sharcnet.ca) and Compute/Calcul Canada.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- G. R. Desiraju, Chem. Commun., 1997, 1475–1482 RSC.
- A. Nangia and G. R. Desiraju, Top. Curr. Chem., 1999, 198, 57–95 CrossRef.
- A. F. Cozzolino, P. J. W. Elder and I. Vargas Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef CAS.
- A. F. Cozzolino, J. F. Britten and I. Vargas Baca, Cryst. Growth Des., 2006, 6, 181–186 CAS.
- A. F. Cozzolino, P. S. Whitfield and I. Vargas Baca, J. Am. Chem. Soc., 2010, 132, 17265–17270 CrossRef CAS PubMed.
- A. F. Cozzolino and I. Vargas Baca, Cryst. Growth Des., 2011, 11, 668–677 CAS.
- N. A. Semenov, A. V. Lonchakov, N. A. Pushkarevsky, E. A. Suturina, V. V. Korolev, E. Lork, V. G. Vasiliev, S. N. Konchenko, J. Beckmann, N. P. Gritsan and A. V. Zibarev, Organometallics, 2014, 33, 4302–4314 CrossRef CAS.
- G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS PubMed.
- A. F. Cozzolino, P. J. W. Elder, L. M. Lee and I. Vargas Baca, Can. J. Chem., 2013, 91, 338–347 CrossRef CAS.
- P. Singh, S. Sharma, H. B. Singh and R. J. Butcher, Proc. Natl. Acad. Sci., India, Sect. A, 2014, 84, 269–280 CrossRef CAS.
- A. F. Cozzolino, I. Vargas Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef CAS PubMed.
- C. J. Milios, P. V. Ioannou, C. P. Raptopoulou and G. S. Papaefstathiou, Polyhedron, 2009, 28, 3199–3202 CrossRef CAS.
- G. Mukherjee, P. Singh, C. Ganguri, S. Sharma, H. B. Singh, N. Goel, U. P. Singh and R. J. Butcher, Inorg. Chem., 2012, 51, 8128–8140 CrossRef CAS PubMed.
- A. F. Cozzolino, A. D. Bain, S. Hanhan and I. Vargas Baca, Chem. Commun., 2009, 4043–4045 RSC.
- M. Risto, R. W. Reed, C. M. Robertson, R. Oilunkaniemi, R. S. Laitinen and R. T. Oakley, Chem. Commun., 2008, 3278–3280 RSC.
- A. F. Cozzolino, G. Dimopoulos-Italiano, L. M. Lee and I. Vargas Baca, Eur. J. Inorg. Chem., 2013, 2013, 2751–2756 CrossRef CAS.
- A. J. Nunn and J. T. Ralph, J. Chem. Soc., 1965, 6769–6777 RSC.
- A. J. Nunn and J. T. Ralph, J. Chem. Soc. C, 1966, 1568–1570 RSC.
- G. Berionni, B. Pegot, J. Marrot and R. Goumont, CrystEngComm, 2009, 11, 986–988 RSC.
- J. L. Dutton, J. J. Tindale, M. C. Jennings and P. J. Ragogna, Chem. Commun., 2006, 2474–2476 RSC.
- P. Zhang, E. A. Terefenko, J. Bray, D. Deecher, A. Fensome, J. Harrison, C. Kim, E. Koury, L. Mark, C. C. McComas, C. A. Mugford, E. J. Trybulski, A. T. Vu, G. T. Whiteside and P. E. Mahaney, J. Med. Chem., 2009, 52, 5703–5711 CrossRef CAS PubMed.
- M. Chakrabarty, S. Karmakar, R. Mukherjee, S. Arima and Y. Harigaya, Monatsh. Chem., 2008, 140, 375–380 CrossRef.
- Y. Jahng and M. A. F. M. Rahman, Synth. Commun., 2006, 36, 1213–1220 CrossRef.
- I. W. Harvey, M. D. McFarlane, D. J. Moody and D. M. Smith, J. Chem. Soc., Perkin Trans. 1, 1988, 1939–1943 RSC.
- G. te Velde and F. M. Bickelhaupt, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- S. Van Gisbergen, J. Snijders and E. Baerends, Phys. Rev. Lett., 1997, 78, 3097–3100 CrossRef CAS.
- S. J. A. van Gisbergen, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1998, 109, 10644–10656 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. Van Lenthe and E. Baerends, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943 CrossRef CAS.
- E. Van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505 CrossRef CAS.
- E. Van Lenthe and R. Van Leeuwen, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- L. Fan and T. Ziegler, J. Chem. Phys., 1992, 96, 9005–9012 CrossRef CAS.
- L. Fan and T. Ziegler, J. Phys. Chem., 1992, 96, 6937–6941 CrossRef CAS.
- O. V. Gritsenko, P. R. T. Schipper and E. J. Baerends, Chem. Phys. Lett., 1999, 302, 199–207 CrossRef CAS.
- O. V. Gritsenko, P. R. T. Schipper and E. J. Baerends, Int. J. Quantum Chem., 2000, 76, 407–419 CrossRef CAS.
- P. Schipper and O. V. Gritsenko, J. Chem. Phys., 2000, 112(13), 1344–1352 CrossRef CAS.
- L. M. Lee, P. J. W. Elder, A. F. Cozzolino, Q. Yang and I. Vargas Baca, Main Group Chem., 2010, 9, 117–133 CAS.
- G. I. Eremeeva, B. K. Strelets and L. S. Efros, Khim. Geterotsikl. Soedin., 1975, 276–277 CAS.
- G. I. Eremeeva, Y. I. Akulin, T. N. Timofeeva, B. K. Strelets and L. S. Efros, Khim. Geterotsikl. Soedin., 1982, 1129–1130 CAS.
- L. Andrews, E. S. Prochaska and A. Loewenschuss, Inorg. Chem., 1980, 19, 463–465 CrossRef CAS.
- J. L. Dutton and P. J. Ragogna, Inorg. Chem., 2009, 48, 1722–1730 CrossRef CAS PubMed.
- T. Ziegler and A. Rauk, Theor. Chem. Acc., 1977, 46, 1–10 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details for [4]2[4-H]2][I][I3] and [4]2(C6H4(NH2)2H+)2][I]2; Cartesian coordinates of all calculated structures. CCDC 1424370–1424374 and 1434655. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04314j |
| This journal is © The Royal Society of Chemistry 2016 |