
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA ruthenium water oxidation catalyst based on a carboxamide ligand†
Wangchuk
Rabten
a,
Torbjörn
Åkermark
a,
Markus D.
Kärkäs
*a,
Hong
Chen
bc,
Junliang
Sun
bd,
Pher G.
Andersson
*a and
Björn
Åkermark
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: markus.karkas@su.se; pher.andersson@su.se; bjorn.akermark@su.se
bBerzelii Centre EXSELENT on Porous Materials, Department of Materials and Environmental Chemistry, Stockholm University, SE-10691, Stockholm, Sweden
cFaculty of Material Science and Chemistry, China University of Geosciences, 430074, Wuhan, China
dCollege of Chemistry and Molecular Engineering, Peking University, 100871, Beijing, China
First published on 1st February 2016
Abstract
Herein is presented a single-site Ru complex bearing a carboxamide-based ligand that efficiently manages to carry out the four-electron oxidation of H2O. The incorporation of the negatively charged ligand framework significantly lowered the redox potentials of the Ru complex, allowing H2O oxidation to be driven by the mild oxidant [Ru(bpy)3]3+. This work highlights that the inclusion of amide moieties into metal complexes thus offers access to highly active H2O oxidation catalysts.
In efforts towards developing a sustainable and clean energy resource, extensive research has been devoted towards splitting of H2O into O2 and H2 (eqn (1)).1 Here, the design of robust artificial water oxidation catalysts (WOCs) appears to be the stumbling block in attempts to develop systems for the generation of solar fuels. Several research groups are therefore pursuing the construction of robust artificial WOCs based on Ru,2,3 Mn,4 Fe,5 Cu6 and Co.7
| 2H2O → 4H+ + 4e− + O2 | (1) |
In order to develop more efficient WOCs, single-site Ru complexes where the metal is coordinated to a negatively charged ligand have been studied. The use of such ligand frameworks offers the possibility of stabilizing the metal center at a highly oxidized state by electron-donation to the metal center, which results in lowering of the redox potentials.8 The resulting high-valent metal species are important catalytic intermediates during the oxidation of H2O and may be key to accessing highly active and robust WOCs.9 Recent studies focusing on the incorporation of negatively charged ligands have shown that such complexes can result in Ru WOCs with sufficiently low redox potentials to allow H2O oxidation to be driven by light.10
We have recently reported on the unexpected formation of the single-site Ru complex 2 bearing a mixed pyridinecarboxylate ligand (1) (Fig. 1). Complex 2 was found to have a sufficiently low redox potential to allow H2O oxidation to be driven by the mild single-electron oxidant [Ru(bpy)3]3+ (bpy = 2,2′-bipyridine).11 It was also markedly more active than the corresponding dicarboxylate complex.12 The incorporation of the amide moiety into WOCs thus seemed to create a suitable ligand framework for producing robust catalysts. It was therefore reasoned that replacing the carboxylate unit in ligand 1 by an additional amide moiety, to give the dicarboxamide ligand 3, could potentially offer access to an even more active catalyst. Indeed, the substitution of ligand 1 with the dicarboxamide ligand 3 (H4pdca = 2,6-pyridine-dicarboxamide), resulted in a more active Ru-based WOC (4, Fig. 1). When using the mild one-electron oxidant [Ru(bpy)3]3+ at neutral pH, the designed Ru complex 4 managed to reach turnover numbers (TONs) close to 400 and turnover frequencies (TOFs) of ∼1.6 s−1, which is almost a two-fold increase compared to Ru complex 2 housing the mixed carboxylate–amide ligand 1.
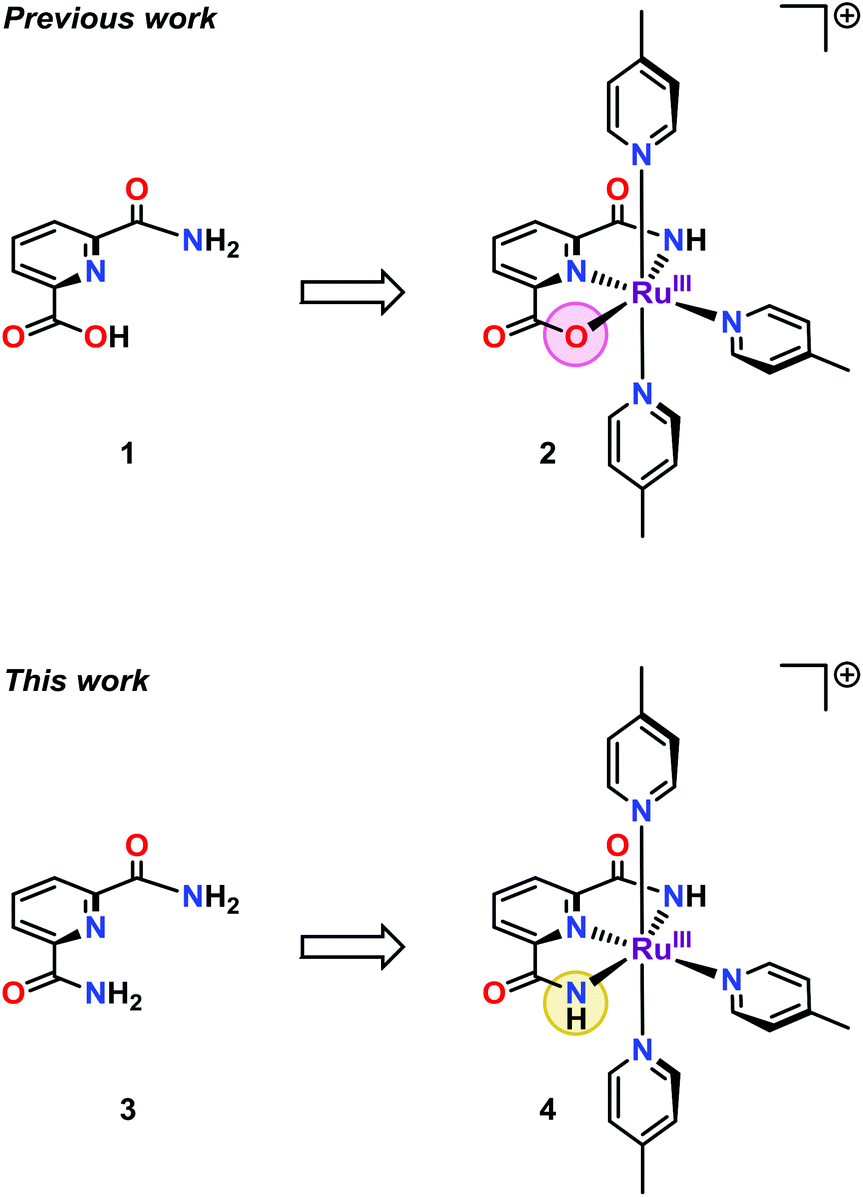 | ||
| Fig. 1 Structures of the previously reported single-site Ru complex 2 containing ligand 1 and Ru complex 4 based on the dicarboxamide ligand 2,6-pyridine-dicarboxamide (3, H4pdca). | ||
Ru complex 4, [Ru(H2pdca)(pic)3]+, was synthesized from the commercially available dicarboxamide ligand 3 (H4pdca = 2,6-pyridine-dicarboxamide) by refluxing a solution of ligand 3, Ru(DMSO)Cl2 and Et3N overnight. To this solution was added 4-picoline and the resulting solution was further refluxed for 48 h. This afforded Ru complex 4 as an orange solid in 25% yield. Complex 4 was characterized by 1H NMR, high-resolution mass spectrometry (HRMS), X-ray crystallography, elemental analysis and UV-vis spectroscopy to confirm the structure of the single-site Ru complex 4.
Single crystals of X-ray diffraction quality were obtained from an aqueous-methanolic solution. The crystal structure of Ru complex 4 is depicted in Fig. 2. The structure reveals that the RuIII center is located in a slightly distorted [RuN6] octahedral configuration. The electron-rich dicarboxamide ligand scaffold 3 thus stabilizes the Ru center and makes it possible to isolate the complex at the RuIII state. In the equatorial plane, three positions are occupied by the three nitrogen atoms from the tridentate 2,6-pyridine-dicarboxamide ligand 3. The fourth position in the equatorial plane and the two axial positions are occupied by 4-picoline ligands.
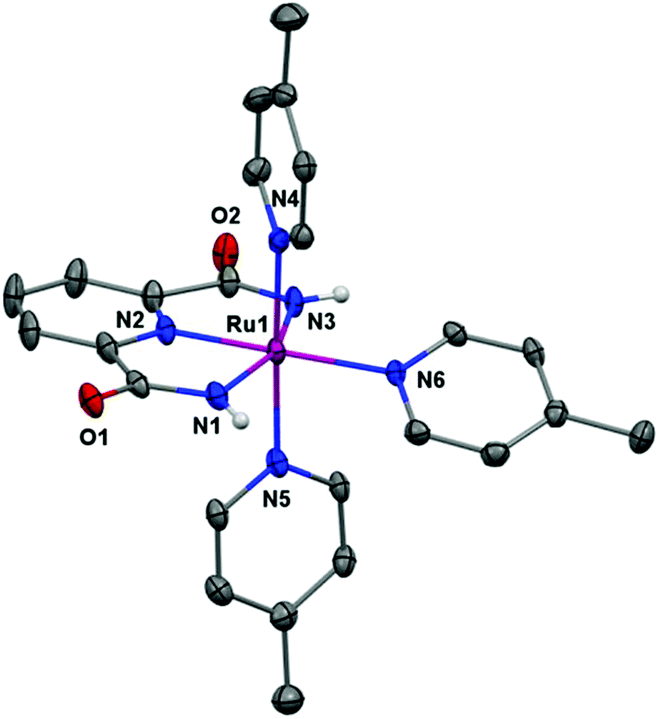 | ||
| Fig. 2 X-ray crystal structure of the single-site Ru complex 4 at the 50% probability level. Hydrogen atoms (except the N–H) and PF6 have been omitted for clarity. | ||
A comparison of the crystal structure of the previously reported RuIII complex 2,11 shows that the bond angle N(1)–Ru(1)–N(3) in Ru complex 4 is close to that found in complex 2, 158.98° and 160.0°, respectively (see Tables 1, S2 and S3†). The Ru–N(pic) distances are all ∼2.10 Å, which is longer than in the related RuII dicarboxylate complex [Ru(pdc)(pic)3]2+ (H2pdc = 2,6-pyridinedicarboxylic acid).12 All the Ru–N bond distances are in general slightly longer in Ru complex 4 than those found for complex 2 (see Table S3†).
| Bond lengths | |||
| Ru(1)–N(1) | 2.032(2) | Ru(1)–N(4) | 2.110(2) |
| Ru(1)–N(2) | 1.973(2) | Ru(1)–N(5) | 2.098(2) |
| Ru(1)–N(3) | 2.033(2) | Ru(1)–N(6) | 2.117(2) |
| Bond angles | |||
| N(1)–Ru(1)–N(2) | 79.50(8) | N(2)–Ru(1)–N(6) | 178.27(7) |
| N(1)–Ru(1)–N(3) | 158.98(7) | N(3)–Ru(1)–N(4) | 93.30(7) |
| N(1)–Ru(1)–N(4) | 88.20(7) | N(3)–Ru(1)–N(5) | 88.10(7) |
| N(1)–Ru(1)–N(5) | 91.18(7) | N(3)–Ru(1)–N(6) | 98.76(7) |
| N(1)–Ru(1)–N(6) | 102.18(7) | N(4)–Ru(1)–N(5) | 177.64(7) |
| N(2)–Ru(1)–N(3) | 79.55(8) | N(4)–Ru(1)–N(6) | 90.96(7) |
| N(2)–Ru(1)–N(4) | 89.48(7) | N(5)–Ru(1)–N(6) | 86.95(7) |
| N(2)–Ru(1)–N(5) | 92.64(7) | ||
HRMS analysis of Ru complex 4 in aqueous methanol/acetonitrile solutions showed a major peak at m/z 544.1172 with a distinct isotopic pattern (Fig. S3†), which can be assigned to [RuIII(H2pdca)(pic)3]+ (4). Additionally, a peak at m/z 492.0854 could be observed (Fig. S5†), which corresponds to the acetonitrile-containing Ru-species [RuIII(H2pdca)(pic)2(MeCN)]+. In this species, one of the picoline ligands has been displaced for a solvent acetonitrile molecule. However, for the Ru complex to become catalytically active it is important to have access to the corresponding aqua complex. Formation of this aqua complex allows for proton-coupled electron transfer (PCET) and for the synchronous removal of protons and electrons, thus avoiding charge accumulation and high-energy intermediates.13 The picoline-aqua ligand displacement was therefore studied by HRMS. Upon dissolution of Ru complex 4 in aqueous solutions, a peak at m/z 469.0683 appeared (Fig. S4†). This peak corresponds to the RuIII-aqua species [RuIII(H2pdca)(pic)2(OH2)]+, which shows that ligand displacement occurs in aqueous solutions to generate the catalytically important Ru-aqua species.
The electrochemical properties of Ru complex 4 were subsequently studied in aqueous solution at neutral pH by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Under neutral pH, the cyclic voltammogram of complex 4 shows a rapid increase of current at 1.21 V vs. NHE, which is due to the catalytic oxidation of H2O (Fig. S7†). The electrochemistry of Ru complex 4 was further analyzed by DPV under neutral conditions (Fig. S8†). DPV of complex 4 revealed three redox peaks at 0.18, 0.94 and 1.15 V vs. NHE (see Table 2). Based on previous work with Ru complex 2, these events can be assigned to the RuIII/RuII, RuIV/RuIII and RuV/RuIV redox couples, respectively. It should be noted that the corresponding redox potentials for the related Ru complex 2 were found to occur at 0.35, 0.72 and 0.92 V vs. NHE. From the obtained electrochemical data it is obvious that the substitution of the carboxylate moiety in Ru complex 2 for an amide unit to form Ru complex 4 alters the electrochemical properties of the Ru complex. However, the change is not straightforward because the RuIII/RuII redox couple is decreased as might be expected, while the higher potentials are increased.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Redox couple | E 1/2 (V vs. NHE) |
|---|---|
| a Electrochemical measurements were performed in an aqueous phosphate buffer solution (0.1 M, pH 7.2). All potentials were obtained from DPV and are reported vs. NHE. Conditions: scan rate 0.1 V s−1, glassy carbon disk as the working electrode, a platinum spiral as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. Potentials were converted to NHE by using the [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple as a standard (E1/2 = 1.26 V vs. NHE). | |
| RuIII/RuII | 0.18 |
| RuIV/RuIII | 0.94 |
| RuV/RuIV | 1.15 |
| E onset | 1.21 |
The low onset potential of 1.21 V vs. NHE of Ru complex 4 suggests that the complex is able to mediate catalytic H2O oxidation when driven by the mild single-electron oxidant [Ru(bpy)3]3+. To evaluate Ru complex 4 as a molecular WOC, an aqueous solution (phosphate buffer; 0.1 M, pH 7.2) containing complex 4 was added to the chemical oxidant [Ru(bpy)3]3+. The gaseous products were subsequently analyzed in real-time by mass spectrometry. Indeed, O2 evolution was immediately triggered upon the addition of an aqueous solution containing Ru complex 4 to the oxidant (Fig. 3). The O2 evolution dependence on pH was also investigated and it could be shown that pH 6.0 and 7.2 afforded the highest catalytic activity (see Fig. S13 and Table S1†). Background experiments were also carried out to verify that complex 4 is necessary for maintaining catalytic activity. In the absence of Ru complex 4, spontaneous decomposition of the [Ru(bpy)3]3+ oxidant occurs without any detectable formation of O2, highlighting that complex 4 is essential for oxidizing H2O. It should be noted that the low O2 evolution yields, i.e. conversion yields from oxidant to O2, depicted in Table 3 are due to competing pathways in which the chemical oxidant [Ru(bpy)3]3+ is spontaneously decomposed, thus resulting in unproductive reaction pathways without any evolution of O2.
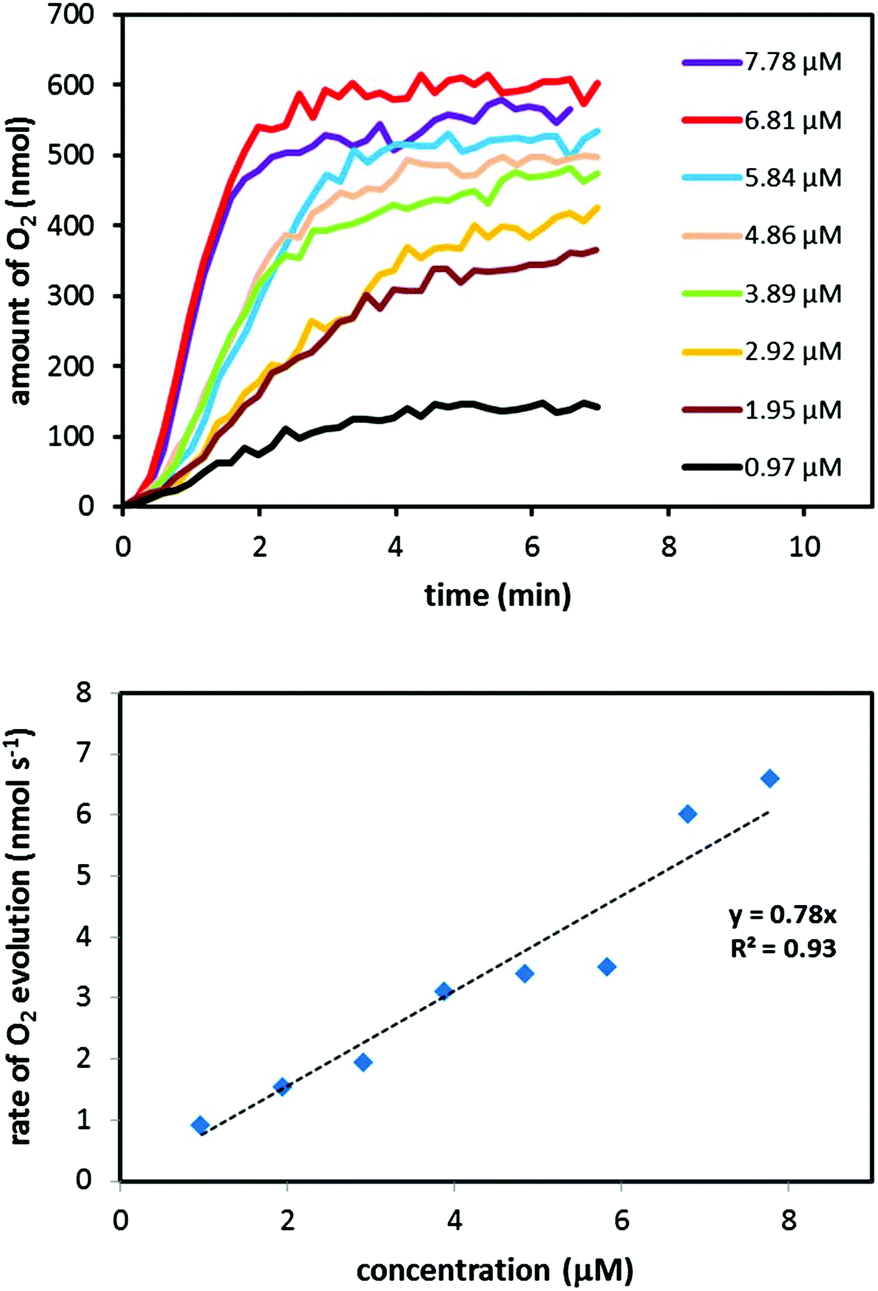 | ||
| Fig. 3 (upper) Plots of O2 evolution versus time at various concentrations of Ru complex 4. Reaction conditions: an aqueous phosphate buffer solution (0.1 M, pH 7.2, 0.50 mL) containing Ru complex 4 was added to the oxidant [Ru(bpy)3](PF6)3 (3.6 mg, 3.6 μmol). (lower) Initial rate of O2 evolution plotted as a function of the concentration of Ru complex 4. Rates of O2 evolution were calculated from the slopes of linearly fitted O2 evolution plots in the period of 30–90 s. | ||
a
| Catalyst concentration (μM) | TONb (nmol O2/nmol cat.) | Yield of O2 (4·amount O2/amount oxidant) |
|---|---|---|
| a Reaction conditions: an aqueous phosphate buffer solution (0.1 M, pH 7.2, 0.50 mL) containing Ru complex 4 was added to the oxidant [Ru(bpy)3](PF6)3 (3.6 mg, 3.6 μmol). b Turnover numbers (TONs) were calculated from moles of produced O2/moles of catalyst. | ||
| 7.78 | 149 | 64.4% |
| 6.81 | 181 | 68.5% |
| 5.84 | 183 | 59.4% |
| 4.86 | 214 | 57.9% |
| 3.89 | 269 | 58.2% |
| 2.92 | 305 | 49.4% |
| 1.95 | 388 | 41.9% |
| 0.97 | 319 | 17.2% |
The catalytic activity of Ru complex 4 was compared with that of the previously developed [Ru(bpb)(pic)2]+ complex (H2bpb = N,N′-1,2-phenylene-bis(2-pyridine-carboxamide)), based on a tetradentate bisamide ligand scaffold, where O2 evolution halted after ∼200 TONs because of the formation of the catalytically inactive mono-carbonyl complex [Ru(bpb)(CO)(OH2)].8c By contrast, such species do not appear to interfere with Ru complexes 2 and 4. It is also important to compare the catalytic efficiencies of Ru complexes 2 and 4. Ru complex 4 is able to generate a TON of ∼400 and a TOF of ∼1.6 s−1, while complex 211 is less efficient and produces a TON of ∼200 and a TOF of ∼1.32 s−1. Although Ru complexes 2 and 4 have a high structural resemblance and share almost the same tridentate ligand framework, there is a difference in catalytic activity. This effect is not clear but could originate from the labile carboxylate unit that exists in the ligand scaffold in Ru complex 2. It has previously been found that high-valent Ru-oxo species are prone to undergo extrusion of CO2 with subsequent cleavage of the (hetero)aryl–carbonyl bond.14 This sort of Ru-catalyzed decarboxylation would hence be regarded as an unfavorable reaction pathway that limits the catalytic activity of Ru complex 2. The small structural change when changing the carboxylate unit in complex 2 to an amide moiety in complex 4 apparently has a significant impact on the catalytic activity. Realizing these small, but fundamental, structural variations in artificial WOCs could thus be of value for the development of more robust WOCs.
To conclude, through a rational molecular design, a highly active WOC has been developed based on a carboxamide ligand scaffold. The designed Ru complex 4 was found to have a low overpotential for H2O oxidation, which is attractive, and thus enabled H2O oxidation to be driven by the mild chemical oxidant [Ru(bpy)3]3+. It could be established that when driven by [Ru(bpy)3]3+, Ru complex 4 generated a TON close to 400, which is almost a two-fold increase compared to Ru complex 2 that is based on the mixed carboxylate–amide ligand 1. These findings are intriguing and it is believed that the results presented herein will contribute to further development of Ru-based WOCs for creating sustainable H2O splitting devices.
Acknowledgements
Financial support from the Swedish Research Council (621-2013-4872 and 348-2014-6070), the Knut and Alice Wallenberg Foundation and the Carl Trygger Foundation is gratefully acknowledged. We would also like to thank Beamline I19 at Diamond Light Source, UK for crystal data collection.Notes and references
- (a) M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100 CrossRef PubMed; (b) W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15 RSC.
- For recent examples of single-site ruthenium-based water oxidation catalysts, see: (a) M. R. Norris, J. J. Concepcion, Z. Fang, J. L. Templeton and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 13580 CrossRef CAS PubMed; (b) C. J. Richmond, R. Matheu, A. Poater, L. Falivene, J. Benet-Buchholz, X. Sala, L. Cavallo and A. Llobet, Chem. – Eur. J., 2014, 20, 17282 CrossRef CAS PubMed; (c) L. Wang, L. Duan, Y. Wang, M. S. G. Ahlquist and L. Sun, Chem. Commun., 2014, 50, 12947 RSC; (d) T.-T. Li, W.-L. Zhao, Y. Chen, F.-M. Li, C.-J. Wang, Y.-H. Tian and W.-F. Fu, Chem. – Eur. J., 2014, 20, 13957 CrossRef CAS PubMed; (e) Y. Liu, S.-M. Ng, S.-M. Yiu, W. W. Y. Lam, X.-G. Wei, K.-C. Lau and T.-C. Lau, Angew. Chem., Int. Ed., 2014, 53, 14468 CrossRef CAS PubMed; (f) J. T. Muckerman, M. Kowalczyk, Y. M. Badiei, D. E. Polyansky, J. J. Concepcion, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2014, 53, 6904 CrossRef CAS PubMed; (g) Y. Wang and M. S. G. Ahlquist, Phys. Chem. Chem. Phys., 2014, 16, 11182 RSC; M. D. Kärkäs, R.-Z. Liao, T. M. Laine, T. Åkermark, S. Ghanem, P. E. M. Siegbahn and B. Åkermark, Catal. Sci. Technol., 2016 10.1039/C5CY01704A; (h) M. D. Kärkäs, R.-Z. Liao, T. M. Laine, T. Åkermark, S. Ghanem, P. E. M. Siegbahn and B. Åkermark, Catal. Sci. Technol., 2016 10.1039/C5CY01704A.
- For recent examples of dinuclear ruthenium-based water oxidation catalysts, see: (a) Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 8934 CrossRef CAS PubMed; (b) S. Neudeck, S. Maji, I. López, S. Meyer, F. Meyer and A. Llobet, J. Am. Chem. Soc., 2014, 136, 24 CrossRef CAS PubMed; (c) H. Isobe, K. Tanaka, J.-R. Shen and K. Yamaguchi, Inorg. Chem., 2014, 53, 3973 CrossRef CAS PubMed; (d) T. M. Laine, M. D. Kärkäs, R.-Z. Liao, T. Åkermark, B.-L. Lee, E. A. Karlsson, P. E. M. Siegbahn and B. Åkermark, Chem. Commun., 2015, 51, 1862 RSC; (e) T. M. Laine, M. D. Kärkäs, R.-Z. Liao, P. E. M. Siegbahn and B. Åkermark, Chem. – Eur. J., 2015, 21, 10039 CrossRef CAS PubMed.
- For examples of manganese-based water oxidation catalysts, see: (a) E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715 CrossRef CAS PubMed; (b) K. J. Young, B. J. Brennan, R. Tagore and G. W. Brudvig, Acc. Chem. Res., 2015, 48, 567 CAS; (c) W. A. A. Arafa, M. D. Kärkäs, B.-L. Lee, T. Åkermark, R.-Z. Liao, H.-M. Berends, J. Messinger, P. E. M. Siegbahn and B. Åkermark, Phys. Chem. Chem. Phys., 2014, 16, 11950 RSC; (d) K. Yamamoto, S. Nakazawa and T. Imaoka, Mol. Cryst. Liq. Cryst., 2002, 379, 407 CrossRef CAS; (e) E. A. Karlsson, B.-L. Lee, R.-Z. Liao, T. Åkermark, M. D. Kärkäs, V. Saavedra Becerril, P. E. M. Siegbahn, X. Zou, M. Abrahamsson and B. Åkermark, ChemPlusChem, 2014, 79, 936 CrossRef CAS; (f) L. Ma, Q. Wang, W.-L. Man, H.-K. Kwong, C.-C. Ko and T.-C. Lau, Angew. Chem., Int. Ed., 2015, 54, 5246 CrossRef CAS PubMed; (g) R.-Z. Liao, M. D. Kärkäs, B.-L. Lee, B. Åkermark and P. E. M. Siegbahn, Inorg. Chem., 2015, 54, 342 CrossRef CAS PubMed.
- For examples of iron-based water oxidation catalysts, see: (a) F. Acuña-Parés, M. Costas, J. M. Luis and J. Lloret-Fillol, Inorg. Chem., 2014, 53, 5474 CrossRef PubMed; (b) C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273 CrossRef CAS PubMed; (c) R.-Z. Liao, X.-C. Li and P. E. M. Siegbahn, Eur. J. Inorg. Chem., 2014, 728 CrossRef CAS; (d) M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531 CrossRef CAS PubMed; (e) A. R. Parent, T. Nakazono, S. Lin, S. Utsunomiya and K. Sakai, Dalton Trans., 2014, 43, 12501 RSC.
- For examples of copper-based water oxidation catalysts, see: (a) S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed; (b) D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689 CrossRef CAS PubMed; (c) S. G. Winikoff and C. J. Cramer, Catal. Sci. Technol., 2014, 4, 2484 RSC.
- For examples of cobalt-based water oxidation catalysts, see: (a) H.-Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499 CrossRef CAS PubMed; (b) X. Zhou, F. Li, H. Li, B. Zhang, F. Yu and L. Sun, ChemSusChem, 2014, 7, 2453 CrossRef CAS PubMed; (c) M. Chen, S.-M. Ng, S.-M. Yiu, K.-C. Lau, R. J. Zeng and T.-C. Lau, Chem. Commun., 2014, 50, 14956 RSC; (d) H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268 CrossRef CAS PubMed; (e) I. Siewert and J. Gałęzowska, Chem. – Eur. J., 2015, 21, 2780 CrossRef CAS PubMed; (f) N. Morlanés, K. S. Joya, K. Takanabe and V. Rodionov, Eur. J. Inorg. Chem., 2015, 49 CrossRef; (g) B. Das, A. Orthaber, S. Ott and A. Thapper, Chem. Commun., 2015, 51, 13074 RSC.
- (a) M. D. Kärkäs, E. V. Johnston, E. A. Karlsson, B.-L. Lee, T. Åkermark, M. Shariatgorji, L. Ilag, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Chem. – Eur. J., 2011, 17, 7953 CrossRef PubMed; (b) L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397 CrossRef CAS PubMed; (c) M. D. Kärkäs, T. Åkermark, H. Chen, J. Sun and B. Åkermark, Angew. Chem., Int. Ed., 2013, 52, 4189 CrossRef PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863 CrossRef PubMed.
- (a) Y. Xu, L. Duan, L. Tong, B. Åkermark and L. Sun, Chem. Commun., 2010, 46, 6506 RSC; (b) M. D. Kärkäs, T. Åkermark, E. V. Johnston, S. R. Karim, T. M. Laine, B.-L. Lee, T. Åkermark, T. Privalov and B. Åkermark, Angew. Chem., Int. Ed., 2012, 51, 11589 CrossRef PubMed.
- W. Rabten, M. D. Kärkäs, T. Åkermark, H. Chen, R.-Z. Liao, F. Tinnis, J. Sun, P. E. M. Siegbahn, P. G. Andersson and B. Åkermark, Inorg. Chem., 2015, 54, 4611 CrossRef CAS PubMed.
- L. Duan, Y. Xu, M. Gorlov, L. Tong, S. Andersson and L. Sun, Chem. – Eur. J., 2010, 16, 4659 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2010, 43, 1019 CrossRef CAS PubMed.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. CCDC 1420348. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00327c |
| This journal is © The Royal Society of Chemistry 2016 |