
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards stable single-atom catalysts: strong binding of atomically dispersed transition metals on the surface of nanostructured ceria†
Alberto
Figueroba
a,
Gábor
Kovács
a,
Albert
Bruix
b and
Konstantin M.
Neyman
 *ac
*ac
aDepartament de Química Física & Institut de Química Teòrica i Computacional, Universitat de Barcelona, C/Martí i Franquès 1, 08028 Barcelona, Spain
bInterdisciplinary Nanoscience Center (iNANO) and Department of Physics and Astronomy, Aarhus University, DK-8000 Aarhus C, Denmark
cInstitució Catalana de Recerca i Estudis Avançats (ICREA), 08010 Barcelona, Spain. E-mail: konstantin.neyman@icrea.cat
First published on 1st March 2016
Abstract
The interaction of a series of different transition metal atoms with nanoparticulate CeO2 has been studied by means of density-functional calculations. Recently, we demonstrated the ability of sites exposed on {100} nanofacets of CeO2 to very strongly anchor atomic Pt, making the formed species exceptionally efficient single-atom anode catalysts for proton-exchange membrane fuel cells. Herein, we analyzed the capacity of these surface sites to accommodate all other group VIII–XI transition metal atoms M = Fe, Ru, Os, Co, Rh, Ir, Ni, Pd, Cu, Ag, and Au. The interaction of the M atoms with {100} nanofacets of ceria leads to oxidation of the former and such interaction is calculated to be stronger than the binding of the atoms in the corresponding metal nanoparticles. Comparing the stability of metal–metal and metal-oxide bonds allows one to establish which metals would more strongly resist agglomeration and hence allows the proposal of promising candidates for the design of single-atom catalysts. Indeed, the remarkable stability of these adsorption complexes (particularly for Pt, Pd, Ni, Fe, Co, and Os) strongly suggests that atomically dispersed transition metals anchored as cations on {100} facets of nanostructured ceria are stable against agglomeration into metal particles. Therefore, these sites appear to be of immediate relevance to the preparation of stable catalysts featuring the highest possible metal efficiency in nanocatalysis.
1. Introduction
Reducing the amount of precious noble metals used in catalytic materials is one of the challenges in catalysis research.1 The ever-increasing demand from the automobile industry along with limited supply has a critical effect on the price of such metals, which is an obstacle for the large-scale implementation of applications that require noble-metal-based catalysts.2–4 It is well known, for example, that the very high cost of platinum is one of the stringent factors limiting the wider use of fuel cell technology.3,4Two strategies are commonly followed to cope with the challenge of reducing the content of precious metals in catalysts. The first strategy involves the partial or complete replacement of the precious metal by less-expensive materials. Following it, numerous alternatives have been proposed that can substantially decrease the cost of the catalyst.5–7 However, the resulting catalytic performance is often inferior to that of the analogous noble-metal systems. The other strategy focuses on the more efficient utilization of the noble metal rather than its substitution. This approach aims to maximize the specific catalytic performance of the noble-metal phase, i.e. its per-atom activity. The latter has been customarily achieved by finely dispersing the metal on supports – a paradigm of nanocatalysis.8,9
The limiting case of metal dispersion corresponds to catalytic systems with atomic metal species on the surface of the support, denoted as single-atom catalysts (SACs).10–12 Notably, non-reducible metal-oxide supports exposing regular surfaces, such as MgO(100),13 adsorb atomic transition metal species too weakly to prevent their clustering. A similar situation takes place on the most stable (111) surface of CeO2, a reducible oxide support widely used in catalysis.14 This indicates that the formation of transition metal SACs that are sufficiently stable to counteract metal–metal bond formation upon sintering requires special strongly binding sites on the supports.
Various composites featuring atomically dispersed noble metals have been reported to be catalytically active for different reactions. For instance, cationic Pt and Au species on nanostructured ceria15 and more inert oxides, such as zeolites and silica,16,17 were found to catalyze the water-gas shift reaction at low temperatures. Materials formed by Pd cations anchored on alumina18 and Pt cations on FeOx (ref. 19) were reported to be active towards CO oxidation. High catalytic hydrogenation activity has also been attributed to such SACs, as for instance Pd atoms anchored to cavities of mesoporous graphitic carbon nitride20 and FeOx-supported Pt.21 These and analogous systems represent a promising new generation of cost-effective catalytic materials. Most publications point to oxidized states of the supported noble-metal atoms as the active species in these catalysts. It should be noted, however, that the structure of SAC materials can change under the often harsh catalytic conditions, leading to partial loss of the specific activity of the metal due to its sintering or bulk diffusion. These phenomena can strongly reduce the number of active metal sites exposed to reactants. Thus, it is essential that the support provides sufficient concentration of surface sites that can anchor the metal atoms strongly enough to prevent agglomeration and bulk diffusion. To this end, {100} nanofacets of ceria nanoparticles (NPs) have been shown to be remarkably efficient in anchoring single Pt atoms as Pt2+ cations.22,23 Density-functional calculations combined with X-ray photoemission spectroscopy (XPS) experiments were used to determine that Pt2+ cations adsorbed on such sites are efficiently protected from reduction, aggregation or diffusion into the bulk, up to high temperatures.22 This specific nanofacet site thus fulfills the stability requirements for a SAC. Importantly, this surface site consisting of four O atoms in a square-planar geometry (below referred to as an O4 site), is not unique to ceria NPs. This structural motif can also be found on extended CeO2(100) surfaces24 and on the step edges of low-energy CeO2(111) islands.25
The extraordinary stability of Pt atoms on CeO2 {100} nanofacets suggests that these sites may also strongly bind other transition metal atoms. To assess this peculiar binding propensity important for preparing stable SACs, we have investigated the interaction of these sites with transition metal atoms M = Fe, Ru, Os, Co, Rh, Ir, Ni, Pd, Cu, Ag, and Au in groups VIII–XI of the periodic table. Our results reveal that the O4 sites on {100} facets of ceria NPs strongly bind and oxidize single atoms of all studied metals. Comparison of the adsorption energies of the M1 species with the binding energies of the corresponding atoms in metal NPs indicates high energetic stability of the anchored M1 species against agglomeration. The oxide support acts in the anchoring according to coordination chemistry principles – as a polydentate ligand formed by surface oxygen anions.26 This explains how the adsorption bonds are as strong as metal–ligand bonds in common transition metal complexes. Findings of the present study suggest general guidelines for the preparation of enduring transition metal SACs.
2. Computational details
The adsorption of single atoms of 11 different transition metals on the {100} O4 site of a ceria NP has been investigated by means of periodic spin-polarized density-functional calculations. The generalized gradient approach (GGA) corrected with the on-site Coulomb interaction Hubbard term U (GGA+U) has been used.27–29 Common exchange-correlation potentials suffer from an incomplete cancellation of the self-interaction error, thus leading to a poor description of strongly correlated systems. The U-correction scheme offers a computationally feasible improvement, which enables an appropriate treatment of transition and rare-earth metal oxides featuring localized electrons in d and f orbitals, respectively. The GGA+U approach has been widely used to describe ceria-based systems, whose Ce 4f states can be occupied upon reduction.30 However, there is no unique U value allowing simultaneous reproduction of all properties of oxidized and reduced ceria.31 For example, calculations with larger values of U predict band gaps in better agreement with experiment, but overestimate interatomic distances in ceria. Following previous studies of ceria NPs,32–35 present calculations have been performed using the PW91 (ref. 36) exchange-correlation potential corrected with a value of U = 4 eV (referred to as the PW91+4 scheme). The effect of using different U values on the calculated properties was assessed (see section 4).Calculations have been carried out using the VASP code,37–39 representing the valence states in plane-wave basis sets with a cut-off of 415 eV for the kinetic energy. The core–valence interaction has been described through the projector augmented wave method.40 Only the Γ-point has been used to sample the reciprocal space. The electron density was self-consistently converged with a 10−4 eV total energy threshold and all geometric structures were optimized until forces acting on each atom became smaller than 0.02 eV Å−1. Some adsorbed metal atoms can exhibit different oxidation states. This has been explored and the resulting oxidation states of the adsorbed M1 species have been determined by the analysis of the localized magnetic moments on Ce cations. Bader charge analysis41 has also been performed.
3. M1–CeO2 and Mn models
A cuboctahedral Ce40O80 NP (Fig. 1) has been chosen as a representative model of nanostructured CeO2, previously shown to be capable of reproducing various experimental observations22,32,35 (effect of the NP size on the calculated observations is briefly addressed in section 4). The structure of this NP resulted from a global optimization using interionic potential and density-functional calculations.33,34 The Ce40O80 NP retains the cubic fluorite-type crystallinity of bulk CeO2 and exposes small O-terminated {111} and {100} facets. The latter corresponds to the polar (100) surface, less stable than the (111) surface.42 Yet, ceria nanosystems with abundant [100] terminations have also been prepared, ranging from nanocrystals with extended (100) terraces24 to nanocubes exposing only {100} facets.43 The size of the periodically repeated unit cell was chosen to keep distances between NPs in neighboring cells ≥700 pm to avoid significant spurious interactions of the NPs.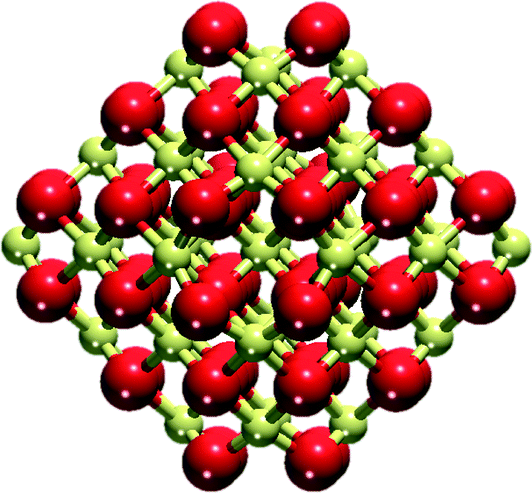 | ||
| Fig. 1 Cuboctahedral Ce40O80 nanoparticle model of nanostructured ceria. Yellow and red spheres represent Ce4+ cations and O2− anions, respectively. | ||
The adsorption energies, Ead, of a metal atom M on the ceria NP were calculated as Ead = E(M1–Ce40O80) − E(M1)–E(Ce40O80), where E(M1) is the total ground-state energy of an isolated metal atom and E(M1–Ce40O80) and E(Ce40O80) are the total energies of the ceria NP with and without the adsorbed metal atom, respectively. Negative adsorption energy values correspond to stabilizing interactions with respect to the separated M1 and Ce40O80 fragments. In order to compare the strength of this interaction with that of M1 agglomeration into metal particles Mn, we have calculated the binding energy, Ead79, of an edge M atom in the M79 model (a bulk cut of the fcc crystal, Fig. 2) as Ead79 = E(M79) − E(M1) − E(M78). Similarly, the propensity of the anchored M1 species to agglomerate can be assessed by comparing the Ead values with the experimental metal cohesive energies (see Table S1†).
 | ||
| Fig. 2 Model M79 NP used for estimating the binding energy of atoms in metal particles. The darker green M atom in the edge position was removed for calculating its binding energy to the remaining M78 species. | ||
4. Results and discussion
We performed a comparative study of the adsorption of 3d (Fe, Co, Ni, Cu), 4d (Ru, Rh, Pd, Ag) and 5d (Os, Ir, Pt,22 Au) metal atoms on O4 sites of {100} nanofacets of the Ce40O80 NP. This is not the only site on the NP where metal atoms can be anchored, but the adsorption of Pt on other NP sites was calculated to be much weaker than on the O4 site22,23 and similar in strength to the adsorption on CeO2(111) surfaces.44 Since this site preference is expected for the adsorption of all metal atoms under scrutiny, we focused on interactions with the O4 site.The optimized structures for the M1–Ce40O80 systems are shown in Fig. 3. Via the analysis of spin moments, we were able to quantify the charge transfer taking place upon atom deposition on the {100} nanofacet. Such charge transfer results in the oxidation of the M1 adsorbate with the concomitant reduction of a certain number of Ce4+ cations to Ce3+. The number of reduced Ce cations depends on the M atom and equals its oxidation state. Corner Ce4+ cations in the Ce40O80 NP are the easiest to reduce due to their lower coordination number and an accordingly less destabilizing electrostatic environment.34,45 These corner Ce4+ cations accepted electrons from M atoms, which were concomitantly oxidized to +1 or +2 oxidation states. In oxidation states higher than +2, a search for the most stable locations of the additionally formed Ce3+ cations has not been performed in view of a very high number of possible configurations. The location of the Ce3+ cations in less stable positions of the NP could induce a destabilization of the adsorption by up to 40 kJ mol−1 per Ce3+ cation.34 Yet, this difference does not affect the upcoming discussion of the adsorption energy values of M atoms. Note that the appearance of Ce3+ cations (larger than Ce4+) significantly elongates the corresponding Ce–O distances, to 228 pm from 213 pm in the pristine CeO2 NP (see Fig. S1 and S2†).
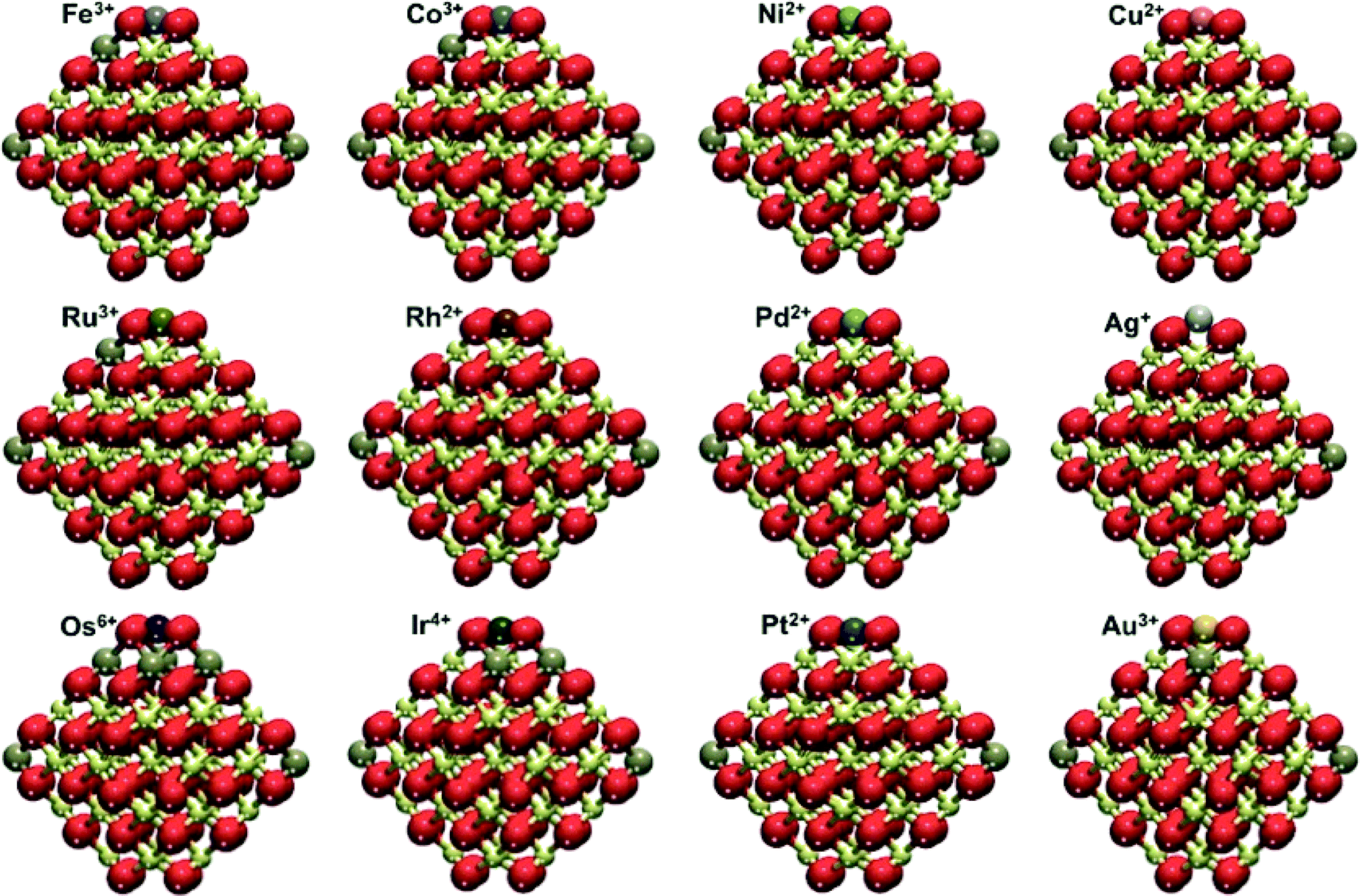 | ||
| Fig. 3 Overview of the M1–Ce40O80 structures calculated for the adsorption of different transition metal atoms (M) on the {100} facet of the Ce40O80 nanoparticle. Yellow, brown and red spheres represent Ce4+, Ce3+ and O2− ions, respectively. See selected calculated interatomic distances in Fig. S1 and S2.† | ||
Herein, we discuss the oxidation states for each adsorbed metal atom and the M–O coordination modes (see for details Fig. S1 and S2†). We also comment on how our results compare to pertinent experimental data.
Group VIII metals
Adsorbed atoms of group VIII feature different oxidation states, +3 – Fe and Ru, and +6 – Os. Two Ce3+ cations are located in the corner positions, whereas additional f electrons are found in other surface Ce positions close to the occupied {100} site. Single M atoms bind to oxygen atoms of the O4 site in a square-planar fashion. The nearest M–O distances are 184–187 pm for Fe, 196–199 pm for Ru and 184–185 pm for Os (see Fig. S1†), much shorter than the ca. 225 pm distance between the O atoms and the center of the O4 moiety in the pristine Ce40O80 NP.22 A similar coordination mode of Fe was detected by Mössbauer spectroscopy for Fe3+ cations inserted into the lattice of ceria–zirconia catalysts.46 In turn, the self-dispersion of Ru metal powder on ceria in the form of small particles and single atoms in a highly oxidized state was demonstrated to be very stable by X-ray absorption near edge structure (XANES) and Raman spectroscopic techniques.47Group IX metals
Atoms of this group manifested more diverse oxidation states. Co is the only metal atom in this group that features a +3 state. Despite the well-known stability of the +3 state for Rh, we calculated it to be adsorbed only in the +2 state. Similar to group VIII, only the 5d element Ir undergoes further oxidation to the +4 state. Just as for adsorbed group VIII atoms, Ce3+ cations are formed in NP corners and close to the O4 site occupied by M. Co and Ir are bound in a square-planar coordination mode with M–O distances 185–186 pm for Co and 192–197 pm for Ir. Differently, Rh2+ appears to prefer linear coordination with two short (198 pm) and two long distances (208 pm). Such group IX cationic species have been identified experimentally on ceria-based materials. Co3+ cations were detected using temperature-programmed reduction (TPR) and XPS experiments in ceria–zirconia catalysts.48 Rh2+ cations were evidenced on ceria–zirconia mixed oxides by Fourier transform infrared (FTIR) spectroscopy.49 Finally, surface Ir4+ species were identified in ceria by TPR and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS).50Group X metals
Group X atoms Ni and Pd are calculated to be adsorbed similarly to Pt as +2 cations, coordinated in a square-planar mode, very characteristic of d8 metal centers of this group. The interatomic M–O distances are 189 pm for Ni and 205 pm for both Pd and Pt. The same oxidation state was recently reported for Ni2+ on defect sites of the CeO2(111) surface.51 It was shown that the interaction of Pd with ceria can lead to the formation of Pd–O–Ce surface superstructures featuring Pd2+ cations and active in catalytic combustion of methane.52 There is experimental evidence that Pd2+ cations in the square-planar coordination by oxygen are highly stable in PdOx–CeO2 solid solutions.53 The presence of atomic platinum as Pt2+ cations on ceria surfaces has been documented by several theoretical and experimental studies. Formation of these species was often related to the exposure of (100)-terminated facets.22,23,54–56The present GGA+U data depend on the chosen U value. In particular, larger U values stabilize the presence of localized f electrons favoring processes that involve the reduction of Ce4+ to Ce3+. To benchmark this dependence we calculated Ead with different U values for a single Pd atom adsorbed on the Ce40O80 NP (see Fig. 4). The use of U = 3 eV decreases Ead by 53 kJ mol−1 compared to that obtained with U = 4 eV chosen in this work. The use of a larger U value of 5 eV has an opposite effect on the Ead increasing its magnitude by 54 kJ mol−1. A similar trend was found for the binding of supported atomic Pt species on ceria.22,57 In general, the usage of smaller, i.e. less interfering U values seems to be preferable. Yet, U < 4 eV values often do not allow complete localization of 4f electrons on the Ce3+ cations. The U = 4 eV employed throughout this work seems to be an adequate compromise providing a physically correct description of localized Ce 4f states and minimizing spurious stabilization of reduced Ce3+ species. One can also question, to what extent the O4 site on the NP Ce40O80 represents the corresponding sites on other ceria nanostructures with similar surface terminations. Data in Fig. 4 shed light on this issue as well. Indeed, enlargement of the NP model to Ce80O160 (ref. 33 and 34) results in the strengthening of Pd–O4 interactions by ca. 30 kJ mol−1, implying that on larger ceria species transition metal atoms should be bound to the O4 sites at least as strong as on the Ce40O80 model used in this study.
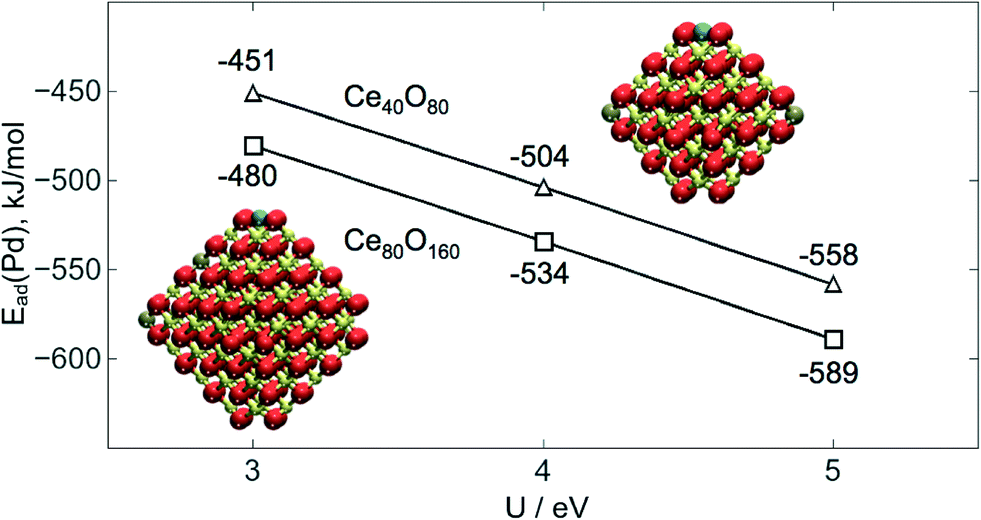 | ||
| Fig. 4 Calculated adsorption energies Ead(Pd) of single Pd atoms on the O4 site of Ce40O80 and Ce80O160 NPs as a function of the U value used with the PW91 functional. | ||
Group XI metals
Finally, adsorbed atoms of group XI, like those of group IX, exhibit several oxidation states. Moreover, each adsorbed group XI element can be in different oxidation states. The presence of Cu as Cu2+ ions in the ceria lattice was recently reported.58 We calculated Cu+ to be coordinated in a distorted linear fashion with two short (189 pm) and two long Cu–O distances (231 and 262 pm). Cu2+ adsorbs in a square-planar coordination mode with four equal Cu–O distances (199 pm). The Cu2+ state is found to be by 54 kJ mol−1 more stable than the Cu+ state (Table 1). Atomic Ag can be adsorbed as either Ag+ or Ag3+, the latter was calculated to be somewhat more stable. The not very usual Ag3+ forms four Ag–O distances of 205–206 pm, in line with its square-planar coordination in oxoargentates.59 Ag+ is in a distorted square-planar coordination, where the metal atom is 90 pm above the O4 plane. This distortion elongates Ag–O distances to 239–241 pm. Atomic Au can also be adsorbed as Au+ and Au3+, the latter being more stable by 70 kJ mol−1 (Table 1). Au, similar to the other presently investigated 5d metals, forms the highest oxidation state (+3) within the group. A linear coordination of Au+ cations is preferred, with two short (206 pm) and two long distances (274 and 276 pm), while Au3+ prefers square-planar coordination (Au–O distances are 204–205 pm). Ag adsorbed as single atoms on microporous hollandite manganese oxide was found by XANES to feature the +1 oxidation state.60 Meanwhile, gold can be stabilized both as Au3+ and Au+ surface cations in ceria-supported gold catalysts.61| M | E ad, kJ mol−1 | E ad79, kJ mol−1 | E ad79 − Ead, kJ mol−1 | q, au |
|---|---|---|---|---|
| Group VIII | ||||
| Fe3+ | −785 | −630 | 155 | 1.50 |
| Ru3+ | −812 | −729 | 83 | 1.46 |
| Os6+ | −978 | −844 | 134 | 2.49 |
| Group IX | ||||
| Co3+ | −709 | −548 | 161 | 1.29 |
| Rh2+ | −678 | −612 | 66 | 1.07 |
| Ir4+ | −830 | −821 | 9 | 1.54 |
| Group X | ||||
| Ni2+ | −678 | −519 | 159 | 1.06 |
| Pd2+ | −504 | −377 | 127 | 0.91 |
| Pt2+ | −678 | −548 | 130 | 0.93 |
| Group XI | ||||
| Cu+ | −412 | 0.73 | ||
| Cu2+ | −466 | −371 | 95 | 1.03 |
| Ag+ | −277 | −263 | 14 | 0.63 |
| Ag3+ | −251 | 1.06 | ||
| Au+ | −264 | 0.41 | ||
| Au3+ | −334 | −310 | 24 | 1.08 |
The adsorption energies in Table 1 reveal that the {100} nanofacet of ceria can strongly anchor not only atomic Pt, but other transition metal atoms as well. Furthermore, atoms of all considered transition metals are oxidized upon adsorption on the O4 site. The Bader charges of adsorbed M1 species reflect the formal oxidation states only qualitatively, being notably smaller than the latter and manifesting significant covalence in the M–O interactions. Despite the simplicity of the presently considered O4 sites exposed by the Ce40O80 model NP, they are expected to be representative for a variety of experimental coordination environments of different transition metal cations. In particular, we are confident that our model NP allows us to quantify the strength of the M–O(–Ce) bonds formed by oxidized metal atoms. To assess whether these metal centers are resistant to agglomeration and sintering processes, which is a crucial issue in the design of metal-efficient SACs, one can compare the binding energies of transition metal atoms on the ceria NP surface and in the corresponding M79 NPs (Table 1).
For all M atoms under scrutiny, the adsorption energy Ead on the ceria NP is larger in magnitude (more negative) than the binding energy Ead79 of an edge atom in the M79 NPs. This indicates that the metal dispersion as single atoms on the O4 sites of ceria nanostructures is energetically favored over the formation of metallic particles. Therefore, very stable adsorption complexes on the nanoparticulate oxide support should resist sintering processes, especially for metals featuring substantial energy differences ΔE = Ead79 − Ead (Table 1). The ΔE value defines the propensity of anchored metal atoms to form particles: the larger ΔE, the less prone the metal center is to sintering. We predict Fe3+, Os6+, Co3+ and group X metals to be particularly resistant to agglomeration in oxidative media. For group X metals, the extraordinary stability of the square-planar coordination for d8 metal centers explains the particularly strong interaction with the ceria NP. Small ΔE values calculated for cationic Ag, Au, and Ir species suggest that these complexes are less resistant to sintering and might form metallic particles more readily. In order to further stabilize these metals as single atoms one should explore other supporting materials. Notably, the O4 sites of CeO2 nanostructures (not limited solely to Ce40O80 NPs) bind metal atoms notably more strongly than the extended CeO2(111) surface. For instance, the adsorption of Cu, Ag, and Au atoms on the latter surface resulted in PW91+3 Ead values of −179, −96, and −69 kJ mol−1, respectively.62 This strongly suggests that the adsorption complexes of M1 on {100} facets of CeO2 NPs are substantially more resistant to agglomeration processes than on the CeO2(111) surface. It should be noted that since Pt was found to adsorb on both such surface and other non-{100} sites of the ceria NP with comparably low Ead,22 other metals are expected to behave similarly.
Clear trends in calculated adsorption energies emerge along the rows and groups of the periodic table (Fig. 5). Both Ead and Ead79 generally decrease in magnitude, when moving from the left to the right of the period. This indicates that metals with less occupied d bands form stronger metal–metal bonds and also bind more strongly to the oxide support. For 4d and 5d metals, such decreases in the Ead and Ead79 are quite monotonous, with more pronounced differences for Au. The trend along the period for 3d metals is less linear and both Ead and Ead79 values are crossed with those of the 4d and 5d metals. These trends also indicate that, except for Au, 5d metals form the strongest bonds with the O4 site of the ceria NP, whereas 4d metals form the weakest bonds, with the exception of Ru.
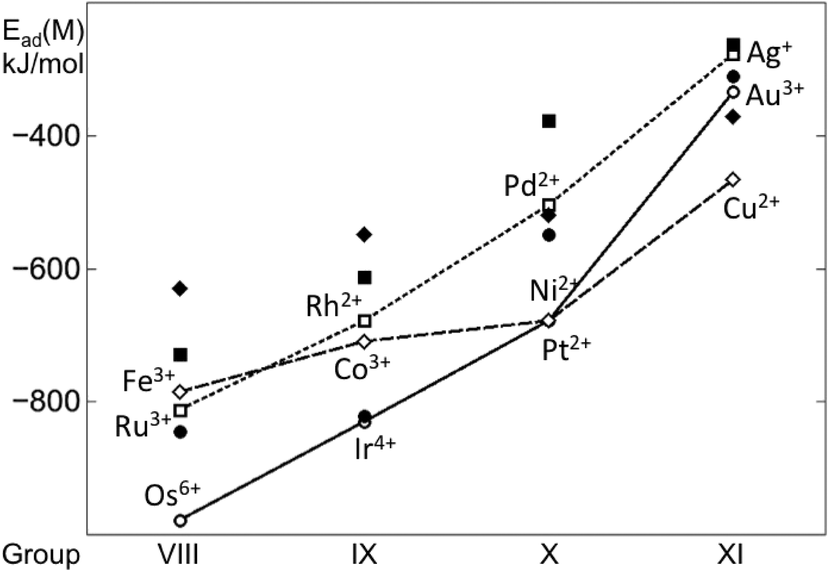 | ||
| Fig. 5 Adsorption energies of single transition metal atoms (M) on the {100} facet of the Ce40O80 NP (empty points, connected as a guide) and binding energies of these atoms in edge positions of the M79 NP depicted in Fig. 2 (filled points). Results for 3d, 4d and 5d metals are shown by diamonds, squares, and circles, respectively. | ||
The agglomeration of the transition metal content into the corresponding oxide phase under an oxidative environment would also lead to the destruction of the single-atom sites. Similar to sintering into metal NPs, the formation of the oxide phase would lead to less exposed metal atoms, with different properties and local environment. The propensity of the single-atom sites to such restructuring can be assessed from the experimental standard heats of formation (ΔHf) for the most stable oxide phases of the metals under study (see Table S2†). In general, 3d metals are more prone to the formation of oxides than 4d and 5d metals, and the oxides of metals situated at the beginning of the period are more stable than those at the end of the period.63 Comparing Ead and ΔHf values for each studied transition metal one expects generally high resistance against decomposition via the formation of a metal oxide phase except for Fe, which is quite susceptible to oxide formation.
All results of the present study correspond to stoichiometric ceria NPs, which are mostly relevant to conditions of ultra-high vacuum or very low oxygen pressure.23 Yet, under ambient atmosphere and oxidative catalytic conditions ceria NPs can be stabilized by an excess of oxygen.64–66 There, a variety of oxygen-containing surface species is expected to create a pool of additional adsorption sites capable of stabilizing oxidized transition metal atoms as potential SACs. We expect the binding properties of such sites to be quite similar to those of the sites on the {100} O-terminated facets of the stoichiometric NPs. This assumption is supported by the finding that oxygen atoms of the {100} O4 sites are loosely bound to the ceria NPs,33,34 which is reminiscent to the binding of the species adsorbed on the NP faces under excess O2. Therefore, the present theoretical prediction that surface oxygen sites of nanostructured ceria are able to make diverse single-atom metal catalysts resistant to sintering probably can also be generalized to different experimental conditions.
The above results on the extreme stability of supported single metal atoms are expected to provide a guideline establishing suitable candidates for the design of SACs. This work opens a way to examine the catalytic function of the proposed materials individually for each reaction of interest. Co-sputtering of metals under an oxygen atmosphere, allowing the preparation of nanocomposites of atomically dispersed Pt on ceria,22,67 can also be used to disperse other metals, the atoms of which are strongly bound to the square-planar O4 sites. This has been demonstrated for model catalysts prepared according to the guidelines provided by the present calculations and comparing the stability and reactivity of Pt2+, Pd2+, and Ni2+ species on nanostructured ceria.68 Interestingly, well-characterized steps on extended CeO2(111) surfaces25,69 also appear to efficiently anchor Pt2+ by forming PtO4 moieties, indicating that the preparation of ceria surfaces with very abundant steps also facilitates metal dispersion in the form of atoms.70 In addition, the detailed structural data calculated in this work (e.g. metal coordination and metal–oxygen distances, see Fig. S1 and S2†) provide a benchmark for the characterization of atomically dispersed metal sites in SACs supported on CeO2 NPs. For example, Pt coordination and Pt–O bond length measured by means of extended X-ray absorption fine structure (EXAFS)54 experiments fully agree with the calculated structure of Pt2+ on the {100} sites of the ceria NPs.22,23 Another implication of the present findings for nanocatalysis is related to the ability of some M1–ceria SACs to undergo agglomeration and re-dispersion cycles under certain reaction conditions, forming metal clusters during the reaction and re-dispersing to the M1–ceria state upon termination of the reaction.71 The estimated stability ΔE = Ead79 − Ead of the M1–ceria materials with respect to the agglomeration in metal clusters (Table 1) controls that clusters remain small enough (possibly, sub-nano) and can readily transform back into single-atom species.
5. Summary
The interaction of 11 different transition metal atoms M = Fe, Ru, Os, Co, Rh, Ir, Ni, Pd, Cu, Ag, Au with the representative model Ce40O80 NP has been studied by means of GGA+U density-functional calculations. We have shown that adsorption sites of the ceria {100} nanofacets can very effectively anchor all considered metal atoms in the form of Mn+ cations. The oxidation of the M centers takes place with the accompanying reduction of Ce4+ cations to Ce3+. Calculated data indicate that higher oxidation states are favored by transition metals in later periods and in groups more to the left in the periodic table. The deposition of the M atoms leads to a significant structural reconstruction of the oxygen atoms constituting the adsorption site, which coordinate differently to the Mn+ cation depending on its oxidation state. Remarkably, the adsorption for every M1–ceria system studied is stronger than the binding of the corresponding M atom in a metal NP M79. These adsorption energy differences are particularly large for the group X metals (Ni, Pd, and Pt) and for Fe, Co, and Os. This suggests that, especially for these metals, CeO2-based materials exposing O4 sites such as those on {100} nanofacets could provide suitable architectures for preparing single-metal atom catalysts with very high resistance to sintering. In general, the present calculated results are expected to be helpful in the preparation and atomic-level characterization of efficient single-atom catalysts, the limiting case in nanocatalysis featuring the smallest active species.Acknowledgements
This work was financially supported by the European Community (FP7-NMP.2012.1.1-1 project ChipCAT, reference no. 310191), Spanish MINECO (grants CTQ2012-34969, CTQ2015-64618-R) and Generalitat de Catalunya (grants 2014SGR97 and XRQTC). The authors acknowledge a support by the COST Action CM1104 “Reducible oxide chemistry, structure and functions”. Computer resources, technical expertise and assistance were provided by the Red Española de Supercomputación. We are grateful to Dr. Hristiyan Aleksandrov for his comments on the manuscript. AB acknowledges support from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013) /Marie Curie Actions/grant no. 626764 (Nano-DeSign).References
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433–442 CrossRef CAS.
- J. Tollefson, Nature, 2007, 450, 334–335 CrossRef CAS PubMed.
- W. Vielstich, A. Lamm and H. A. Gasteiger, Handbook of Fuel Cells: Fundamentals, Technology, Applications, Wiley, Chichester, 2003 Search PubMed.
- W. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef PubMed.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- W. Yang, T.-P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- C. T. Campbell, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef CAS PubMed.
- R. Schlögl and S. B. Abd Hamid, Angew. Chem., Int. Ed., 2004, 43, 1628–1637 CrossRef PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos, Acc. Chem. Res., 2014, 47, 783–792 CrossRef CAS PubMed.
- K. M. Neyman, C. Inntam, V. A. Nasluzov, R. Kosarev and N. Rösch, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 823–828 CrossRef CAS.
- A. Bruix, F. Nazari, K. M. Neyman and F. Illas, J. Chem. Phys., 2011, 135, 244708 CrossRef PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- Y. P. Zhai, D. Pierre, R. Si, W. L. Deng, P. Ferrin, A. U. Nilekar, G. W. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. H. Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková, J. Mysliveček, R. Fiala, M. Václavů, K. C. Prince, S. Bruyère, V. Potin, F. Illas, V. Matolín, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS PubMed.
- H. A. Aleksandrov, K. M. Neyman and G. N. Vayssilov, Phys. Chem. Chem. Phys., 2015, 17, 14551–14560 RSC.
- Y. Pan, N. Nilius, C. Stiehler, H.-J. Freund, J. Goniakowski and C. Noguera, Adv. Mater. Interfaces, 2014, 1, 1400404 Search PubMed.
- S. M. Kozlov, F. Viñes, N. Nilius, S. Shaikhutdinov and K. M. Neyman, J. Phys. Chem. Lett., 2012, 3, 1956–1961 CrossRef CAS.
- A. Hu, K. M. Neyman, M. Staufer, T. Belling, B. C. Gates and N. Rösch, J. Am. Chem. Soc., 1999, 121, 4522–4523 CrossRef CAS.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767–808 CrossRef CAS.
- V. I. Anisimov, I. V. Solovyev, M. A. Korotin, M. T. Czyzyk and G. A. Sawatzky, Phys. Rev. B: Condens. Matter, 1993, 48, 16929–16934 CrossRef CAS.
- I. V. Solovyev, P. H. Dederichs and V. I. Anisimov, Phys. Rev. B: Condens. Matter, 1994, 50, 16861–16871 CrossRef CAS.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- C. Loschen, J. Carrasco, K. M. Neyman and F. Illas, Phys. Rev. B: Condens. Matter, 2007, 75, 035115 CrossRef.
- G. N. Vayssilov, A. Migani and K. Neyman, J. Phys. Chem. C, 2011, 115, 16081–16086 CAS.
- A. Migani, G. N. Vayssilov, S. T. Bromley, F. Illas and K. M. Neyman, Chem. Commun., 2010, 46, 5936–5938 RSC.
- A. Migani, G. N. Vayssilov, S. T. Bromley, F. Illas and K. M. Neyman, J. Mater. Chem., 2010, 20, 10535–10546 RSC.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244–13249 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöch, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- R. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, USA, 1994 Search PubMed.
- M. M. Branda, R. M. Ferullo, M. Causà and F. Illas, J. Phys. Chem. C, 2011, 115, 3716–3721 CAS.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- A. Bruix, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2010, 114, 14202–14207 CAS.
- M. A. Sk, S. M. Kozlov, K. H. Lim, A. Migani and K. M. Neyman, J. Mater. Chem. A, 2014, 2, 18329–18338 CAS.
- R. Nedyalkova, D. Niznansky and A.-C. Roger, Catal. Commun., 2009, 10, 1875–1880 CrossRef CAS.
- A. Satsuma, M. Yanagihara, J. Ohyama and K. Shimizu, Catal. Today, 2013, 201, 62–67 CrossRef CAS.
- S. S.-Y. Lin, D. H. Kim, M. H. Engelhard and S. Y. Ha, J. Catal., 2010, 273, 229–235 CrossRef CAS.
- M. Haneda, K. Shinoda, A. Nagane, O. Houshito, H. Takagi, Y. Nakahara, K. Hiroe, T. Fujitani and H. Hamada, J. Catal., 2008, 259, 223–231 CrossRef CAS.
- Y. Huang, A. Wang, L. Li, X. Wang, D. Su and T. Zhang, J. Catal., 2008, 255, 144–152 CrossRef CAS.
- J. Carrasco, D. López-Durán, Z. Liu, T. Duchoň, J. Evans, S. D. Senanayake, E. J. Crumlin, V. Matolín, J. A. Rodríguez and M. V. Ganduglia-Pirovano, Angew. Chem., Int. Ed., 2015, 54, 3917–3921 CrossRef CAS PubMed.
- S. Colussi, A. Gayen, M. Farnesi Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Angew. Chem., Int. Ed., 2009, 48, 8481–8484 CrossRef CAS PubMed.
- R. V. Gulyaev, T. Y. Kardash, S. E. Malykhin, O. A. Stonkus, A. S. Ivanova and A. I. Boronin, Phys. Chem. Chem. Phys., 2014, 16, 13523–13539 RSC.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- T. Wu, X. Pan, Y. Zhang, Z. Miao, B. Zhang, J. Li and X. Yang, J. Phys. Chem. Lett., 2014, 5, 2479–2483 CrossRef CAS PubMed.
- Y. Gao, W. Wang, S. Chang and W. Huang, ChemCatChem, 2013, 5, 3610–3620 CrossRef CAS.
- A. Bruix, A. Migani, G. N. Vayssilov, K. M. Neyman, J. Libuda and F. Illas, Phys. Chem. Chem. Phys., 2011, 13, 11384–11392 RSC.
- J. S. Elias, M. Risch, L. Giordano, A. N. Mansour and Y. Shao-Horn, J. Am. Chem. Soc., 2014, 136, 17193–17200 CrossRef CAS PubMed.
- H. Müller-Buschbaum, Z. Anorg. Allg. Chem., 2004, 630, 2125–2175 CrossRef.
- P. Hu, Z. Huang, Z. Amghouz, M. Makkee, F. Xu, F. Kapteijn, A. Dikhtiarenko, Y. Chen, X. Gu and X. Tang, Angew. Chem., Int. Ed., 2014, 53, 3418–3421 CrossRef CAS PubMed.
- A. M. Venezia, G. Pantaleo, A. Longo, G. Di Carlo, M. P. Casaletto, F. L. Liotta and G. Deganello, J. Phys. Chem. B, 2005, 109, 2821–2827 CrossRef CAS PubMed.
- M. M. Branda, N. C. Hernández, J. F. Sanz and F. Illas, J. Phys. Chem. C, 2010, 114, 1934–1941 CAS.
- D. F. Shriver and P. W. Atkins, Inorganic Chemistry, Oxford University Press, 3rd edn, 1999 Search PubMed.
- G. Preda, A. Migani, K. M. Neyman, S. T. Bromley, F. Illas and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 5817–5822 CAS.
- J. Kullgren, K. Hermansson and P. Broqvist, J. Phys. Chem. Lett., 2013, 4, 604–608 CrossRef CAS PubMed.
- X. Huang and M. Beck, Chem. Mater., 2015, 27, 2965–2972 CrossRef.
- R. Fiala, A. Figueroba, A. Bruix, M. Václavů, A. Rednyk, I. Khalakhan, M. Vorokhta, J. Lavková, F. Illas, V. Potin, I. Matolínová, K. M. Neyman and V. Matolín, Appl. Catal., B, 2016 DOI:10.1016/j.apcatb.2016.02.036.
- A. Neitzel, A. Figueroba, Y. Lykhach, T. Skála, M. Vorokhta, N. Tsud, S. Mehl, K. Sevciková, K. C. Prince, K. M. Neyman, V. Matolín and J. Libuda, submitted.
- N. Nilius, S. M. Kozlov, J.-F. Jerratsch, M. Baron, X. Shao, F. Viñes, S. Shaikhutdinov, K. M. Neyman and H.-J. Freund, ACS Nano, 2012, 6, 1126–1133 CrossRef CAS PubMed.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- M. Hatanaka, N. Takahashi, N. Takahashi, T. Tanabe, Y. Nagai, A. Suda and H. Shinjoh, J. Catal., 2009, 266, 182–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected calculated structural data of M1–Ce40O80 models, experimental cohesive energies of the considered metals M and heats of formation of their most stable oxides. See DOI: 10.1039/c6cy00294c |
| This journal is © The Royal Society of Chemistry 2016 |