
New approaches from nanomedicine for treating leishmaniasis
Víctor
Gutiérrez
a,
Amedea B.
Seabra
b,
Rosa M.
Reguera
c,
Jayant
Khandare
d and
Marcelo
Calderón
*a
aFreie Universität Berlin, Institute for Chemistry and Biochemistry, Takustrasse 3, 14195 Berlin, Germany. E-mail: marcelo.calderon@fu-berlin.de; Fax: +49-30-838459368; Tel: +49-30-83859368
bExact and Earth Sciences Department, Universidade Federal de São Paulo, Diadema, São Paulo, Brazil
cDepartamento de Ciencias Biomédicas, Universidad de León, León, Spain
dMaharashtra Institute of Pharmacy, MIT Campus, Paud Road, Kothrud, Pune 411038, India
First published on 21st October 2015
Abstract
Leishmaniasis, a vector-borne disease caused by obligate intramacrophage protozoa, threatens 350 million people in 98 countries around the world. There are already 12 million infected people worldwide and two million new cases occur annually. Leishmaniasis has three main clinical presentations: cutaneous (CL), mucosal (ML), and visceral (VL). It is considered an opportunistic, infectious disease and the HIV-leishmaniasis correlation is well known. Antimonial compounds are used as first-line treatment drugs, but their toxicity, which can be extremely high, leads to a number of undesirable side effects and resultant failure of the patients to adhere to treatment. There is also a reported increase in Leishmania sp. resistance to these drugs. Nanotechnology has emerged as an attractive alternative because of its improved bioavailability and lower toxicity, and other characteristics that help to relieve the burden of this disease. In this review we will present some of the recent advances in the nanotechnological research regarding the treatment of leishmaniasis. The preclinical results regarding the approaches for a biomedical treatment of the disease have been encouraging, but further efforts will still be necessary for this therapy to have greater clinical applicability in humans.
 Víctor Gutiérrez | Victor Gutierrez is a physician from Universidad de la Sabana in Colombia. He has worked in the last 8 years in the fields of General medicine, Internal Medicine, Infectious diseases and Tropical medicine in different countries of Latinamerica such as Colombia, Brasil and Argentina. Recently he has received a Tropical medicine's Diploma from Charité University in Berlin Germany. He joined Prof. Marcelo Calderón's team at the Institute of Chemistry and Biochemistry in the Free University of Berlin in 2013. Currently he works in the Internal Medicine and Nephrologie department at the Südharz Clinic in Nordhausen, Germany. |
 Amedea B. Seabra | Amedea B. Seabra holds a diploma degree in Chemistry from University of São Paulo (USP). She finished her PhD at Chemistry Institute, State University of Campinas (UNICAMP) in 2006. Thereafter, she worked as a Research Fellow at UNICAMP (2006–2008), and she took postdoctoral studies at the Chemistry and Biochemistry Department, Concordia University in Montreal, Canada (2008–2010). Since 2011 she is associated professor at Federal University of São Paulo (UNIFESP). Her present research is focused on the preparation, characterization and evaluation of the cytotoxicity of nitric oxide-releasing nanomaterials for biomedical applications. |
 Rosa M. Reguera | Dr Rosa M. Reguera is professor of Biomedical Sciences at the University of Leon (Spain). She received her PhD in Veterinary Medicine in 1996 with honors. She spent a post-doctoral training in Washington University at St Louis in 2003. In addition to teaching, the scientific career of Dr Reguera is focused on searching therapeutic targets of parasitic protozoa (Leishmania). For the last five years her lab has been focused on the creation of fluorescent clones from different leishmania strains in order to develop a HTS platform devoted to seek specific, efficient and safe drugs against Leishmania. |
 Jayant Khandare | Jayant Khandare is Professor in Pharmaceutics at Maharashtra Inst. of Pharmacy, Pune. His research interests are at the interface of macromolecular chemistry, super-hydrophobic surfaces, targeted drug delivery, 3D cancer cell scaffolds for enhanced cellular interactions. His recent research on multicomponent nanosystem for capturing ‘Circulating Tumor cells (CTCs)’ has been published in Adv. Healthcare Mat. 2013, as cover page, inside. While, he published his work in ‘Small’ 2012 on enhancing surface interactions with colon cancer cells using transferrin-conjugated 3D nanostructured substrates. His research start-up, Actorius Innovations and Research (AIR) is involved in designing multi-components for cancer diagnostics. |
 Marcelo Calderón | Marcelo Calderón is an Assistant Professor for Macromolecular and Organic Chemistry at the Department of Biology, Chemistry, and Pharmacy from the Freie Universität Berlin, Germany. Dr Calderón received his PhD in organic chemistry in 2007 from the National University of Córdoba, Argentina, under the supervision of Prof. Miriam Strumia. In 2007 he joined the Research Group of Prof. Rainer Haag at the Freie Universitat Berlin for his Postdoctoral work, where he pursued the development of polymer–drug conjugates for the passive targeted delivery of drugs, genes, and imaging probes. Dr Calderón was the recipient of the Arthur K. Doolittle Award in 2010 (American Chemical Society, Polymer Materials: Science and Engineering Division), the Cesar Milstein Fellowship in 2011 (Ministry of Science, Technology and Productive Innovation, Argentina), and the NanoMatFutur Grant for Young Scientists from the German Ministry of Science in 2012 (BMBF). |
1 Introduction
Leishmaniasis is a deadly infectious disease, caused by the parasitic protozoan Leishmania, which is transmitted to mammals by the bite of a phlebotomine sandfly vector.1,2 There are three major forms of the infection: cutaneous (CL), mucosal (ML), and visceral leishmaniasis (VL), also called kala-azar. Cutaneous manifestations can be subdivided into localized, diffuse, leishmaniasis recidivans, and post-kala-azar dermal leishmaniasis. Leishmaniasis is caused by 20 different species that belong to the genus Leishmania, including the L. donovani complex with 2 species (L. donovani, L. infantum also known as L. infantum in America); L. mexicana complex with 3 main species (L. mexicana, L. amazonensis, and L. venezuelensis); L. tropica; L. major; L. aethiopica; and the subgenus Viannia (V) with 4 main species L. (V.) braziliensis, L. (V.) guyanensis, L. (V.) panamensis, and L. (V.) peruviana. The different species are morphologically indistinguishable, but they can be differentiated by isoenzyme analysis, molecular methods, or monoclonal antibodies.1 Leishmaniasis threatens 350 million people in 98 countries around the world.2 There are already 12 million infected people worldwide and two million new cases occur annually. Leishmaniasis is also an important opportunistic infection in HIV patients, which is potentially fatal, even when treated appropriately. HIV infection can increase the risk of VL's development by 10–100 fold in endemic areas.3 The Leishmania parasite exists in two life forms: an elongated, flagellated promastigote in the midgut of the sandfly, and a small, rounded, and non-motile form called amastigote in macrophages and other antigen presenting cells such as dendritic cells and neutrophils.4,5 Once the sandfly bites the host in order to take a meal, the parasites invade the local phagocytic host cells. Then the promastigotes transform into amastigotes and multiply by simple division inside the phagolysosomes of the resident macrophages. After a local dissemination, the distant macrophages get infected. The parasite, host, and other factors affect whether the infection becomes symptomatic and whether CL or VL results. Sandflies become infected by ingesting infected cells during blood meals1 (Fig. 1). Amastigotes living inside the macrophage's phagolysosomes represent the main target of antileishmanial treatment, but they are not an easy target because there are major structural barriers that antileishmanial drugs have to overcome. | ||
| Fig. 1 (1) The female sandfly transmits the protozoan by infecting itself with the Leishmania amastigotes contained in the blood of the human or mammalian host. (2) The parasite continues its development inside the sandfly, where amastigotes transform into the promastigote form by binary fission. (3) Promastigotes migrate to the pharynx and buccal cavity of the sandfly. (4) Sandfly bites its host and injects the promastigotes into the blood stream. (5–7) Once in the host, the promastigotes are phagocytized by neutrophils and then by macrophages of the reticuloendothelial system where they transform into the amastigote form. (8) Amastigote forms also multiply by binary fission, which leads to the cell's rupture and liberation of these forms into the circulation. Free amastigotes invade fresh cells. Some of them are taken up by the sandfly during its blood meal. | ||
Effective vaccines against this disease are still under development and the available drugs can be quite toxic and costly, and there may be some parasitic resistance.6 During the last seven decades the chemotherapy for leishmaniasis has been dependent on antimonial compounds. The old-fashioned pentavalent antimonium-based (SbV+) drugs (Glucantime and Pentostam) were developed and introduced as antileishmanials during the 1950s.7 These drugs have several flaws, including the need of repeated parenteral administration, the occurrence of many undesirable side effects (including cardiotoxicity and pancreatitis), as well as resistance from over usage.8 Drug combinations of antimonials with allopurinol (a xanthine oxidase inhibitor) and the antibiotic paromomycin very much improve the curative outcome.9
In a second instance, the macrolide antifungal agent amphotericin B (AmB) is a good alternative to the former compounds. This fungicide, which is formulated as a deoxycholate salt or, even better, is delivered by liposomes (AmBisome), has resulted in a good therapy against visceral leishmaniasis, but it is extremely expensive for developing and poor countries and requires intravenous (i.v.) administration.10 A promising alternative to (SbV+) is the alkylphosphocholine derivative miltefosine (Impavido) that was firstly synthesized as an antineoplasic drug. This compound is the only oral drug prescribed against visceral leishmaniasis.11,12 However, despite its proved efficacy, miltefosine should not be administered to pregnant women due to their teratogenic effects. Finally, clinical studies performed with the broad spectrum and low cost aminoglycoside antibiotic paromomycin (Humatin) have shown that the latter has similar efficacy with AmB but with fewer side effects, and has therefore been approved for the treatment of visceral leishmaniasis in India.13 Many other compounds are considered second line drugs for leishmaniasis including the aromatic diamidine pentamidine or the antifungal azole fluconazole as well others with different stages of approval status. This suggests that we still need newer drugs and delivery conditions. Fig. 2 discloses the chemical structures of some of the typically used drugs.
 | ||
| Fig. 2 Chemical structures of drugs typically used for the treatment of leishmaniasis. | ||
Despite the efforts of scientific community, compounds included in preclinical studies do not move forward to development. Current scenario of first and second-line drugs against leishmaniasis obligates to strengthen interaction between researchers developing new in vivo models and experts in nanomaterials. This is particularly important because leishmaniasis is a complex of diseases with different clinical manifestations as a consequence of the anatomical distribution of parasites. These different locations might be a challenge that requires joint efforts of multidisciplinary teams, that should include chemists that design new drug and delivery systems that are able to accumulate at specific locations (spleen, liver, bone morrow, dermis) after administration. However, the final market of these products will be the poorest among the developing countries. Following World Health Organization (WHO) recommendations, future therapy in visceral leishmaniasis should be self-administrated by patients, without the need of specialized professionals or infrastructures, meanwhile topical and photoactivatable therapies would be eligible for cutaneous and mucocutaneous leishmaniasis.
The commonest and currently used animal models include Balb/c mice and Syrian golden hamster (primary test) and dogs (secondary test). Mice are used in acute infections that examine the activity of the drug against the liver but not the spleen infection. By its part, Syrian golden hamster infection provides a simultaneous progression of disease in liver and spleen, which develops into a chronic non-cure infection more similar to human VL. Finally, during phase II and III in clinical trials the most experimental animal in used is the dog. Drugs or vaccine candidates are evaluated after a short period of time post-treatment, using spleen and liver impression smears, which involve the undesired euthanasia of animals. Although there is not a consensual approach of in vivo drug evaluation, the most used assays do not consider the assessment of similar parasitic load in experimental animals before starting the treatment. In addition, another disadvantage is that appraisal of the disease for longer periods of time after the end of treatment, involves performing repeated tissue biopsies by high qualify experts and increasing the number of slaughtered animals.
These drawbacks are avoided by the use of in vivo real-time imaging systems that allow the acquisition of images at infection sites after inoculating the animals with genetically modified parasites expressing reporters. These systems have several advantages, namely, (a) eliminate the repeated biopsies, the consuming time smear preparations and microscope analyses, (b) allow treatment of animals with similar parasite loads, (c) facilitate appraisal of infection for longer periods of times after the end of treatment, (d) tackle individual evaluation through all the period and (e) reduce the number of experimental animals. In addition, these methods give access to periodic data of other biomedical parameters from blood and urine that might inform about toxicity at particular organs such as liver or kidney. Despite the great need of novel diagnosis tools, almost no examples of concepts based on nanomedicine have been reported related to leishmaniasis. Combining new drugs, vaccines, or diagnostic probes with appropriate delivery systems seems to be a promising approach to give response the WHO requirements against clinical leishmaniasis in future.
The major challenge in the treatment of leishmaniasis is the fact that the parasite infects the macrophage, therefore traditional antileishmanial drugs face difficulties to penetrate inside the macrophages to kill the parasite.14 Recently, the combination of antileishmanial drugs with nanocarriers has been emerging as promising approach in the treatment of leishmania. These nanocarriers have the ability to penetrate into macrophages, release the drug inside the cell, leading to a local high concentration of the therapeutic, and ultimately killing the protozoa. In this sense, the main strategy in the treatment of leishmania is to target the drugs directly to macrophages by using nanocarriers, which have the ability to overcome biological barriers.15,16 Moreover, the use of nanocarriers would permit to reduce the drug toxicity, enhance treatment efficacy, improve selectivity, modulate the drug pharmacokinetics, increase drug solubilization, protect the drug from degradation, and promote a sustained drug release directly in the target site.16 Another advantage is the possibility to design the nanocarriers to carry more than one drug, allowing the combination therapy that might have a synergist effect.17 Nanocarriers allow surface modification that should be further investigated to increase parasite selectivity.
Nanoparticles like liposomes, polymers, and nanospheres have proven to be very important for drug delivery as nanocarriers. This technology may prove to be superior to current treatment including reduced cost of drug, improved bioavailability, and lower drug toxicity, which definitely enhances the patient's adherence to treatment. The use of nanotechnology based formulations for the treatment of leishmaniasis has shown promising results, however a higher attention of the scientific community should be paid to such neglected diseases.18,19 This review revises the recently reported research concerning nanotechnology against leishmaniasis in its three clinical forms, with special emphasis on the utilization of liposomes, polymers, metal nanoparticles, carbon-based materials, and nitric oxide (NO) releasing nanoparticles.
2 Liposomes and nanoemulsions
Liposomes are small artificial vesicles of spherical shape that can be created from cholesterol and natural non-toxic phospholipids. Due to their size, biocompatibility, hydrophobic/hydrophilic balance, stability, and flexibility to load various molecules as cargo, liposomes are currently used as drug delivery systems. Of all the nanomedicines implemented for antileishmaniasis treatment, liposomes are perhaps the most studied and therefore have the largest number of anticipated clinical applications nowadays. The best example is AmBisome®, a liposomal formulation of AmB which has proven to be successful against leishmaniasis. In 1981, New et al. examined the effects of liposomal AmB (L-AmB) (Fig. 3)20 using the leishmania model and reported that L-AmB had a lower toxicity than AmB itself and that treatment with a higher dose of L-AmB could be feasible. During the 80s several trials showed that L-AmB had a lower toxicity than AmB in host animals and thus could be administered at higher doses.21–23 A clinical trial performed by Lopez Berestein et al. in 1985 in cancer patients with confirmed fungal infection showed higher tolerance with L-AmB than with AmB.24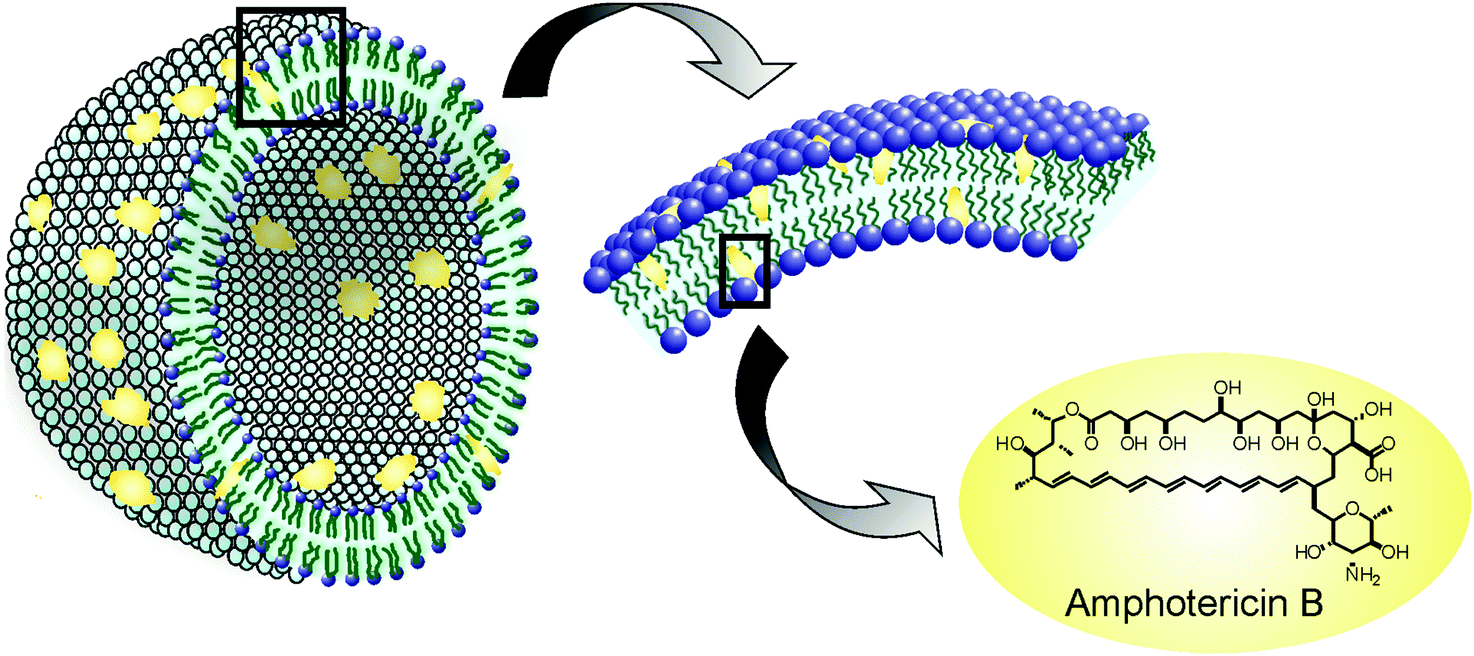 | ||
| Fig. 3 Schematic representation of liposomal formulation of amphotericin B. | ||
In 1987, Szoka et al. prepared small unilamellar vesicles (SUV) containing sterol and explored the effects of component substances of liposome and size of the particle on the expression of toxicity.25 They concluded that the sterol including L-AmB was less toxic than without sterol. The authors also reported that smaller liposomes are less toxic than larger liposomes, when sterol was integrated. Based on these findings, NeXtar Inc. succeeded in formulating the SUV type L-AmB.26 Studies published between 1998 and 2005 focused on phase II dose optimization, mainly in the Mediterranean region of Europe and South Asia, and confirmed earlier studies’ results showing that total doses of 18–20 mg kg−1 appeared effective, at least in non-immuno compromised individuals.26 Also the use of conventional liposomes with antileishmanial drugs proved to be associated with an important reduction in their toxicity profile.27 AmBiosome's market launch was in 1990 and then in August 1997 FDA approved its use for the treatment of patients with VL, Aspergillosis, Candidiasis, and/or Cryptococcal infections refractory or intolerant to AmB.20 However, its use continues to be restricted in several areas due to its high cost.
In 2011 Roychoudhury et al. investigated the efficacy of sodium stibogluconate (SSG) in phosphatidylcholine stearylamine-bearing liposomes (PC-SA-SSG), PC-cholesterol liposomes (PC-Chol-SSG), and free AmB against SSG-resistant L. donovani strains in 8-week infected BALB/c mice.28 Therapy with a single dose of PC-SA-SSG was effective in curing mice infected with two differentially originated SSG-unresponsive parasite strains at significantly higher levels than AmB, unlike free and PC-Chol-SSG. Successful therapy correlated also with a complete suppression of disease-promoting interleukin-10 (IL-10) and transforming growth factor beta (TGF-β), upregulation of T helper cells (Th1) cytokines, and expression of macrophage microbicidal. When administered as PC-SA-SSG versus free SSG, due to the elevated accumulation of SSG in intracellular parasites irrespective of SSG-resistance, a cure happened as a result of increased drug retention and improved therapy.
An interesting study by Perez et al. analyzed the in vitro antileishmanial activity of liposomes with different deformability properties and loaded with the photosensitizer zinc phthalocyanine (ZnPcAL).29 They compared two liposomal systems, one of them built with soybean phosphatidylcholine, sodium cholate, total polar archaeolipids (TPAs), and other one with an ultradeformable character, lacking of TPAs. They found that the photodynamic liposomes were innocuous against promastigotes, however a low concentration (0.01 μM ZnPc and 7.6 μM phospholipids) irradiated at a very low-energy density (0.2 J cm−2) eliminated L. braziliensis amastigotes from J774 macrophages (Fig. 4), without reducing the viability of the host cells, HaCaT keratinocytes, and bone marrow-derived dendritic cells. Interestingly, they found that the only liposomes containing TPAs were captured by macrophages, leading to 2.5-fold increased intracellular delivery of ZnPc as compared to the ultradeformable liposomes (UDL).
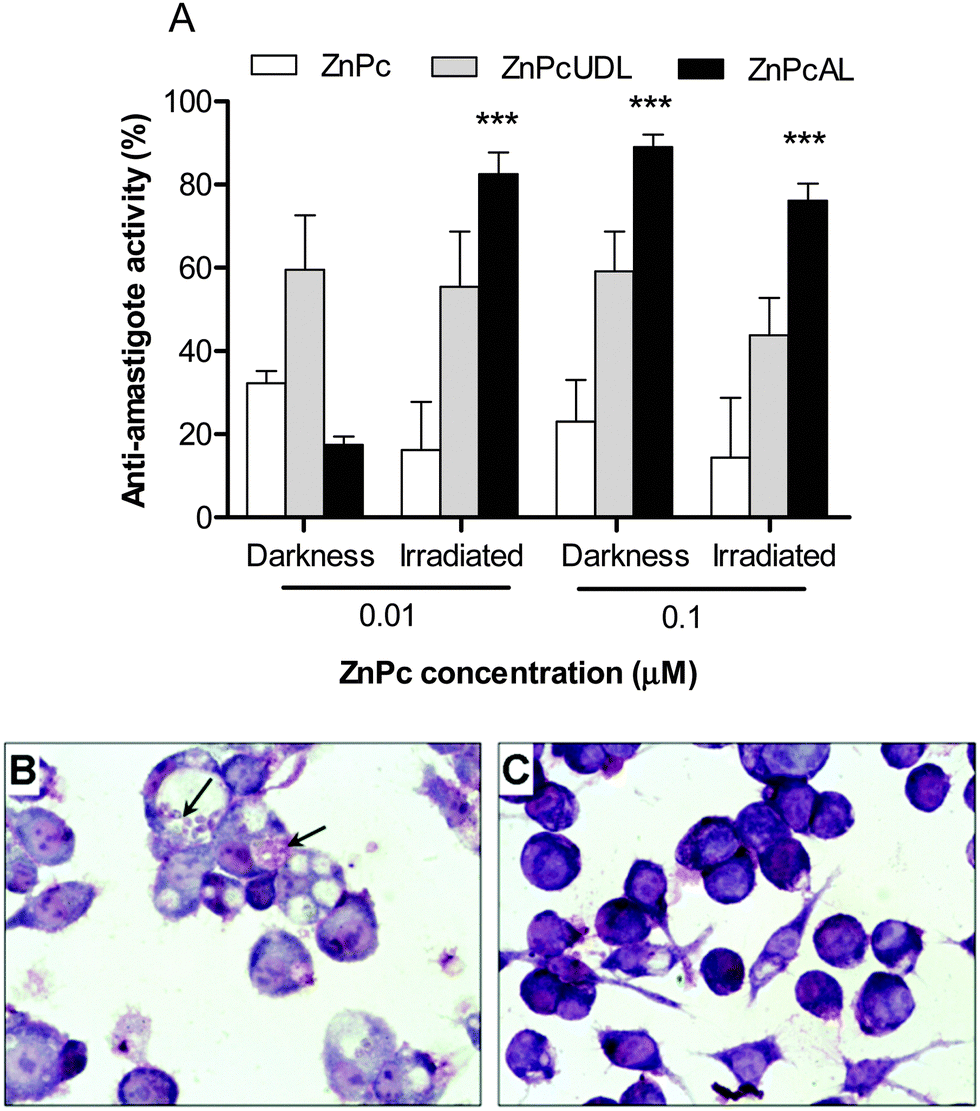 | ||
| Fig. 4 (A) Antiamastigote efficacy of ZnPc (free or liposomal) in darkness or after irradiation. (B, C) Optical microscopy of infected J774 cells incubated with free ZnPc (B) or ZnPc on UDL (C). Reprinted with modifications from ref. 29. Copyright 2014, Dove Press. | ||
A layer-by-layer method to prepare nanocapsules (NCs) with a nanoemulsion core loaded with doxorubicin (NCs-DOX), which was further grafted with phosphatidylserine (PS) in order to enhance the cellular uptake, was reported by Kansal et al. in 2012.30 The authors compared PS-NCs-DOX with non-PS-coated NCs-DOX for their potential ability to target L. donovani parasites, which are known to cause VL. Cellular uptake by J774A.1 macrophages, intracellular localization, in vivo pharmacokinetics, and organ distribution studies were performed. In vivo antileishmanial activity of free DOX, NCs-DOX, and PS-NCs-DOX was tested against VL in Leishmania donovani-infected hamsters. Flow cytometric revealed 1.75-fold enhanced uptake of PS-NCs-DOX in J774A.1 macrophage cell lines when compared with NCs-DOX. In vivo organ distribution studies in hamsters demonstrated a significantly higher extent of accumulation of PS-NCs-DOX compared with NCs-DOX particularly in liver and spleen. There was a significant improvement in the antileishmanial activity with PS-NCs-DOX than with NCs-DOX. PS-NCs-DOX showed 85.23% inhibition of the splenic parasitic burden, whereas NCs-DOX and free DOX showed 72.88% and 42.85% parasite inhibition, respectively, in Leishmania-infected hamsters. The parasite inhibition with blank PS-NCs and NCs was 8.73% and 13.8%, respectively (Fig. 5).30
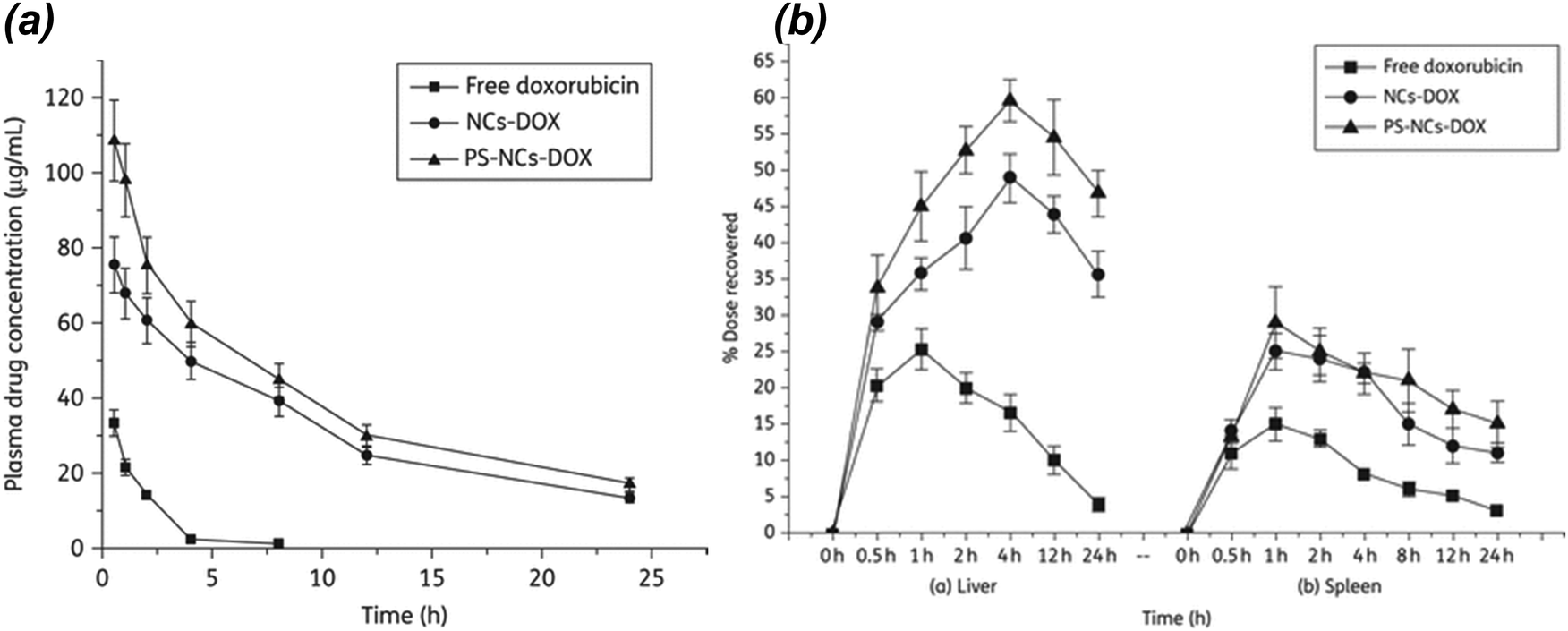 | ||
| Fig. 5 (a) Plasma doxorubicin concentrations profiles of different formulations administered intravenously in Wistar rats. (b) Effect of NCs-DOX and PS-NCs-DOX on hepatic and splenic uptake of doxorubicin. PS-NCs-DOX show significantly enhanced uptake in comparison with NCs-DOX (P < 0.05). Reprinted with permission from ref. 30. Copyright 2012, Oxford Journals. | ||
3 Polymeric nanoparticles
To date, linear poly(ethylene glycol) (PEG), poly(lactide-co-glycolide) (PLGA), polysaccharide based, and amino acid based polymers have been successfully implicated, particularly to deliver bioactives for the treatment of a series of diseases through the systemic circulation. More recently, advanced polymeric architectures such as dendritic polymers have been introduced and are now being evaluated for their safety and ability to deliver therapeutic agents. A growing volume of literature indicates that an array of structurally diverse nanostructures of different sizes, e.g., dendritic polymers, prodrug conjugates, nanospheres, polyplexes, nanogels, polymeric micelles, etc., are being developed for diagnostics or treatment related purposes (Fig. 6).31,32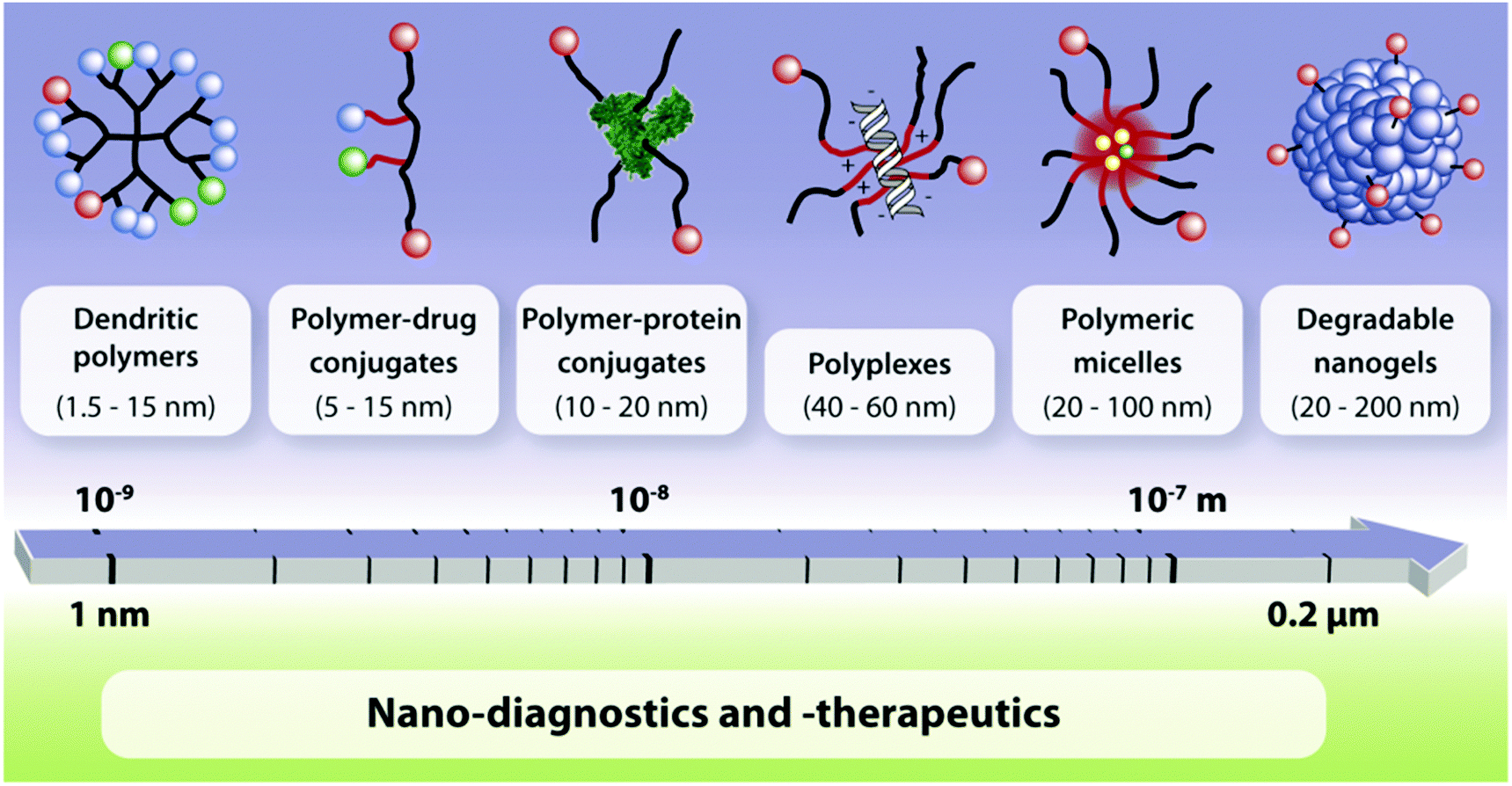 | ||
| Fig. 6 Multifunctional polymeric nanocarriers according to different chemical compositions and size.31 | ||
Polymeric nanocarriers possess advanced physicochemical properties that improve bioavailability, biodegradability, enhance cellular dynamics, and control targetability in drug delivery.33 In polymer-based drug delivery systems, a drug is either non-covalently encapsulated in the interior of the polymer or covalently conjugated to form a macromolecular prodrug. In the encapsulation approach the release can be triggered by structural change within the polymeric scaffold, i.e. backbone degradation, cleavage of shell, charging of functional groups, etc., while in the macromolecular prodrug approach, the mechanism of release involves the splitting of the linker between the polymer and the bioactive agent.34 These characteristics are crucial to obtain an intracellular delivery and sustained release of drugs at a therapeutically relevant level.35 Therefore, the development that the field of polymeric nanocarriers is currently experiencing could be of great use for the treatment of leishmaniasis.
In 2013 Costa Lima et al. developed PLGA-based nanospheres (NS) containing AmB with suitable physicochemical properties and anti-parasitic activity for VL therapy.18 BALB/c mice infected with stationary phase promastigotes received i.v. injection of a single-shot treatment of drug-free PLGA-NS, AmBisome®, and a single or three consecutive daily doses of AmB-PLGA-NS. Results showed significant in vitro and in vivo AmB-PLGA-NS efficacy and preferential accumulation in the visceral organs. In addition, an immune-modulatory effect was observed in mice treated with AmB-PLGA-NS, correlating with improved treatment efficacy. The in vitro cytotoxic response of the T-lymphocytes, which was accomplished using a LIVE/DEAD cell mediated cytotoxicity kit, revealed that AmB-PLGA-NS efficacy against VL infection was strictly due to the action of CD8+ but not CD4+ T lymphocytes. The authors could demonstrate a crucial role for CD8+ cytotoxic T lymphocytes in the efficacy of AmB-PLGA-NS.
Andrographolide (AG) is a diterpenoid lactone extracted from the leaves of the Indian medicinal plant Andrographis paniculata that has been shown to be a potent antileishmanial agent with low cytotoxicity. Roy et al. studied its efficacy by loading it into 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 poly(DL-lactide-co-glycolic acid) nanoparticles (AGnp) stabilized by polyvinyl alcohol (PVA).36 Antileishmanial activity in Albino mice macrophages was found to be significant for the nanoparticle preparation with 4% PVA (IC50 34 μM) in about one-fourth of the dosage of the pure compound AG (IC50 160 μM). The authors then concluded this compound could provide an effective low-cost chemotherapy of leishmaniasis acting through an alternative mechanism of leishmaniasis conventional therapy.
:50 poly(DL-lactide-co-glycolic acid) nanoparticles (AGnp) stabilized by polyvinyl alcohol (PVA).36 Antileishmanial activity in Albino mice macrophages was found to be significant for the nanoparticle preparation with 4% PVA (IC50 34 μM) in about one-fourth of the dosage of the pure compound AG (IC50 160 μM). The authors then concluded this compound could provide an effective low-cost chemotherapy of leishmaniasis acting through an alternative mechanism of leishmaniasis conventional therapy.
A step further was realized by Mondal et al. by studying AG's behavior against resistance.35 The authors designed AG nanoparticles with P-gp efflux inhibitor vitamin E D-a-tocopheryl polyethyleneglycol succinate (TPGS). AGnps stabilized by vitamin E TPGS were delivered into macrophage cells infested with sensitive and drug resistant amastigotes of L. donovani parasites. Antileishmanial activity was found to be significant for AGnp with TPGS in about one-tenth of the dosage of the free AG and one-third of the dosage of the AGnp without TPGS. Another important aspect was cytotoxicity of AGnp, which was found to be significantly less with or without TPGS than standard antileishmanial chemotherapeutics like AmB, paromomycin, or sodium stibogluconate.
A possible pathway to deliver compounds to the parasite which is found within a parasitophorous vacuole (PV) in the macrophages could be through polymer–drug conjugates. These conjugates are taken into cells by endocytosis and then trafficked through endosomes to lysosomes. The PV has a lot of similarities to late endosomes/lysosomes and multiple vacuole trafficking pathways can intersect with Leishmania PV. This could mean that it is likely that the polymer–drug conjugates can also be trafficked to this compartment.37 In 2012 Nicoletti et al. decided to investigate polymer–drug conjugates based on N-(2-hydroxypropyl)methacrylamide (HPMA). GlyPheLeuGly (GFLG) was chosen as a linker from the polymer to the drug, in this case AmB. This combination reports a high antileishmanial in vitro and in vivo activity. When alendronate was added to this backbone, however, there was not much improvement in the antileishmanial activity than with HPMA–GFLG–AmB alone.37
Kóczán et al. showed in 2002 that a branched polypeptide–methotrexate conjugate with a polycationic carrier could increase the effect of methotrexate (MTX) upon L. donovani infection in mice, which led to an important antileishmanial activity.38 They concluded that the covalent bond between carrier and this drug is crucial for in vivo and in vitro activities. In 2003 Nan et al. described an antileishmanial activity using HPMA–drug conjugates for the treatment of VL. Conjugates of HPMA copolymer with NPC1161, an 8-aminoquinoline analog with antileishmanial activity, containing N-acetylmannose-amine (ManN) in the side chains, were synthesized and characterized in vitro and in vivo (Fig. 7).39 ManN was conjugated to target the macrophages of the reticuloendothelial system (RES). When compared to nontargeted conjugates in mice, targeted conjugates were significantly more effective against L. donovani amastigotes and also showed a higher uptake.
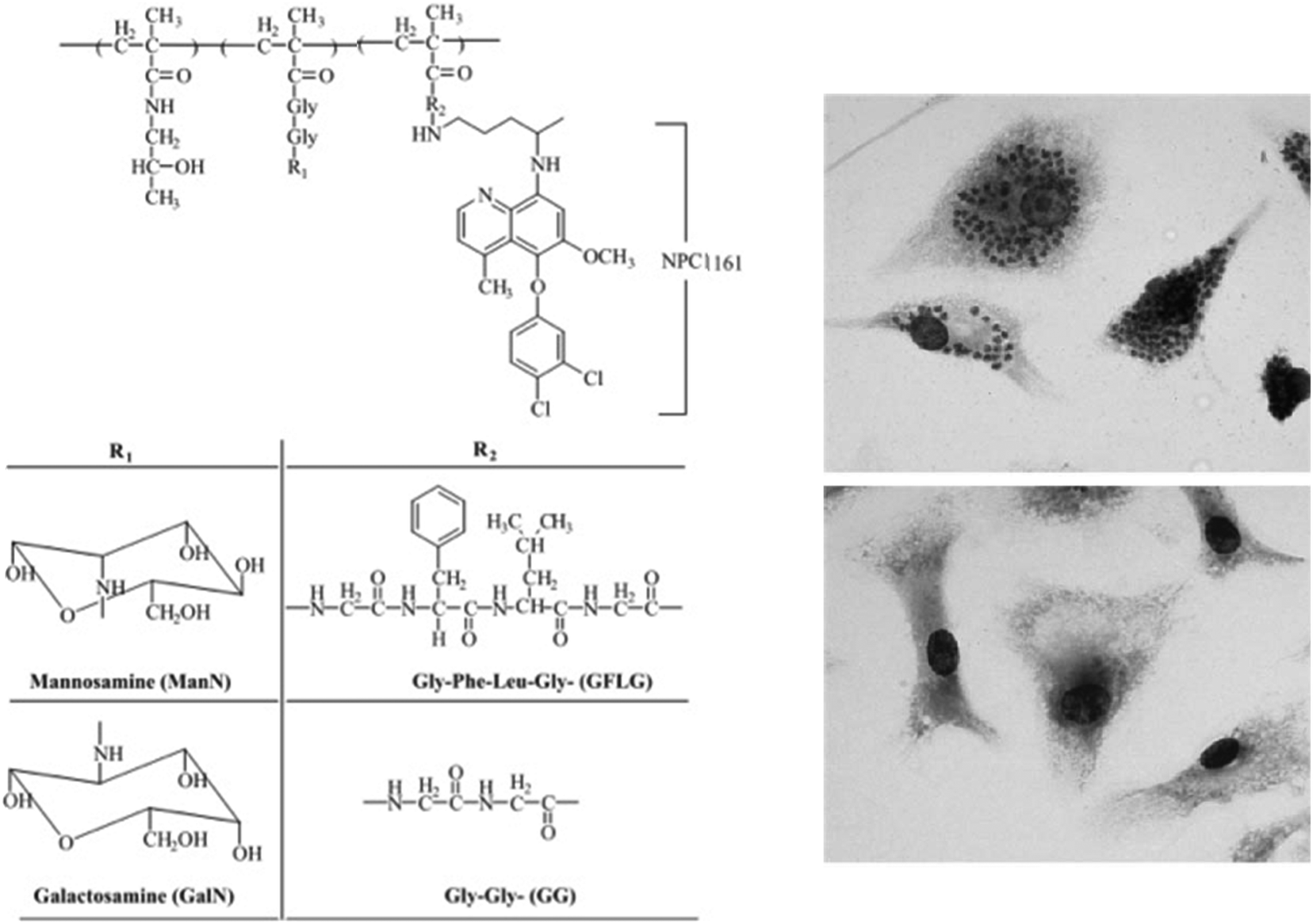 | ||
| Fig. 7 Left panel: Structure of HPMA copolymer–NPC1161 conjugates. Right panels: In vitro microscopic images of mouse peritoneal macrophages infected with L. donovani, prior to treatment (top) or after treatment (bottom) with polymer–drug conjugate. Reprinted with permission from ref. 39. Copyright 2004, Elsevier. | ||
Recently, Barros et al. prepared carbohydrate (mannan, MN) functionalized PLGA nanosphere in the treatment of murine VL.40 The authors demonstrated that MN-functionalized PLGA nanospheres were successfully internalized by murine macrophages due to the high affinity of the modified nanocarriers towards mannose receptor on macrophages. In addition, by nanoprecipitation technique, amphotericin B (AmB) was incorporated (encapsulation efficiency ∼57%) into MN-functionalized PLGA nanocarriers. In vivo experiments demonstrated that MN-functionalized PLGA nanocarrier containing AmB reduced in 99.1 ± 1.3% and 99.5 ± 1.1% the parasitic load in the spleen and liver, respectively; in comparison with as compared with the vehicle control group. Moreover, administration of MN-PLGA containing AmB in mice increased the production of important cytokines involved in the infection of VL, such as INF-γ, and nitric oxide (NO), which plays a key role in the organism defense against parasite infection. Therefore, AmB-containing MN-PLGA nanocarriers demonstrated potential application in the treatment of VL infection.
The behavior of dendrimers as potential antichagasic and antileishmanial prodrugs was analyzed in 2011 by Giarolla et al. by a molecular modeling study.41 The analyzed models contained myo-inositol (dendrimer core), L-malic acid (spacer), and active agents such as 3-hydroxyflavone, quercetin, and hydroxymethylnitrofurazone (NFOH). The authors modelled dendritic molecules with four, five, and six branched modules. The prodrug containing quercetin showed to be the most promising candidate due to the ester linkage next to the myoinositol, which should be hydrolyzed during dendrimer disassembly. In 2013 Daftarian et al. used a Pan-DR-binding epitope (PADRE)-derivatized-dendrimer (PDD), complexed with L-AmB in a L. major mouse model.42 They compared the therapeutic efficacy of low-dose PDD/L-AmB (6.25 mg per kg per day) with a full dose of L-AmB (37.5 mg per kg per day). For this aim, metacyclic promastigotes were injected intradermally on mice. They concluded that PDD reduced the effective dose and toxicity of L-AmB and resulted in a stronger parasite specific T-cell response (Fig. 8).
 | ||
| Fig. 8 Therapy with PDD/LAmB at low dose was as effective as that of LAmB at full dose. The skin lesions of the 2 groups were of comparable size despite different LAmB dosing. PDD conjugated with LAmB enhanced the efficacy of LAmB treatment by at least 6-fold. Reprinted with permission from ref. 42. Copyright 2013, Oxford University Press. | ||
The main goal of vaccination is the induction of a protective immune response against a specific pathogen. The physicochemical properties of polymer nanoparticles when used as adjuvants in a vaccine could mean a more effective immune response against Leishmania. There have been several efforts in the last 5 years concerning immunization against leishmaniasis through nanotechnology, mainly through the use of polymers. In 2010 Tafaghodi et al. took PLGA nanospheres as an antigen delivery system and Quillaja saponins (QS) as immunoadjuvant to enhance the immune response against autoclaved L. major (ALM).43 BALB/c mice were immunized three times in 3-week intervals using the following formulations: ALM plus QS loaded NS [(ALM + QS)PLGA], ALM encapsulated with PLGA NS [(ALM)PLGA], (ALM)PLGA plus QS, ALM plus QS, ALM alone, or phosphate buffer saline (PBS). The footpad's swelling size at the site of injection was measured in order to analyze the intensity of infection. Mice immunized with (ALM)PLGA showed a smaller footpad swelling and the strongest protection. On the other hand, (ALM + QS)PLGA group showed the least protection and highest swelling, while the (ALM)PLGA + QS, ALM + QS and ALM showed an intermediate protection with no significant difference. With these results the authors concluded that PLGA NS could increase the protective immune responses as a vaccine delivery system and that QS adjuvant could have a reverse effect on protective immune responses.
4 Metallic nanoparticles and carbon-based materials
Nanoparticles (NPs) are based on small well-defined aggregates of the noble metals in the zero valent state. The size and the shape of the nanoparticles can be controlled by the reducing agent, the capping agent, and the reaction conditions used in the preparation.44 Inherent properties of the noble metals, enhanced due to the greater surface area of the nanoparticulate form, could be of interest for the treatment of leishmaniasis. As an example, the production of reactive oxygen species (ROS) by silver NP is an antibacterial well-known effect and Leishmania parasites seem to be very sensitive to it as well. In 2011 Allahverdiyev et al. demonstrated an antileishmanial effect using Ag-NPs at concentrations ranging from 25 to 200 μg mL−1.45 They investigated the effects of Ag-NPs on biological parameters of L. tropica promastigotes such as morphology, metabolic activity, proliferation, infectivity, and survival in host cells in vitro. When exposed to Ag-NPs in the dark, L. tropica promastigotes lost their shape and their internal organelles were no longer distinguishable. Similarly, when exposed to Ag-NPs in UV light, the promastigotes membranes were disrupted and had an atypical appearance. Furthermore, Ag-NPs showed a significant antileishmanial effect by inhibiting the proliferation and metabolic activity of promastigotes by 1.5- to 3-fold, respectively in the dark, and 2- to 6.5-fold, respectively, under UV light. Ag-NPs also inhibited amastigotes survival in host cells, which was more significant when UV light was present.In 2012 Soflaei et al. analyzed the effect of antimony sulfide (Sb2S5) NPs upon L. infantum in vitro, which were synthesized with a biological method from Serratia marcescens that had been isolated from the Caspian Sea in northern Iran.46 This parasite is known to be a VL etiological agent. The correlation between cytotoxicity, concentration, and incubation time was assessed on the promastigote and amastigote stages of this parasite in the spleen macrophages of infected and uninfected BALB/c mice. Sb2S5 NPs showed a positive and dose-dependent effectiveness on proliferation of promastigote form. The 50% inhibitory concentration (IC50) of antimony sulfide NPs on promastigotes was calculated to be 50 μg mL−1. The authors also concluded that this drug could induce apoptosis in promastigotes, which makes these particles useful for elimination of the parasite (Fig. 9).
 | ||
| Fig. 9 Viability of mouse macrophages contained amastigotes of Leishmania infantum in 5 dilutions of antimony sulfide NPs during 24, 48, and 72 hours of incubation. Reprinted with permission from ref. 46. Copyright 2012, Creative Commons Attribution License. | ||
Metal oxide NPs, especially titanium dioxide (TiO2), silver oxide (Ag2O), zinc oxide (ZnO), and magnesium oxide (MgO) NPs, have been extensively explored by demonstrating significant antibacterial activity. Jebali et al. studied the antileishmanial effects of some NPs, including Ag-NPs, gold NPs (Au-NPs), TiO2-NPs, ZnO-NPs, and MgO-NPs on L. major parasites under UV, IR, and dark conditions (Fig. 10).47 They showed that the highest antileishmanial activity was observed for Ag-NPs, followed by Au-NPs, TiO2-NPs, ZnO-NPs, and MgO-NPs. Both UV and IR light increased antileishmanial properties of all the NPs. However, they observed that these NPs had cytotoxicity on macrophages. The authors concluded that the use of NPs for treatment of CL may have both positive and negative consequences. Similarly, Mohebali et al. also demonstrated in their work that Ag-NPs were effective for control of secondary infection of localized CL.48
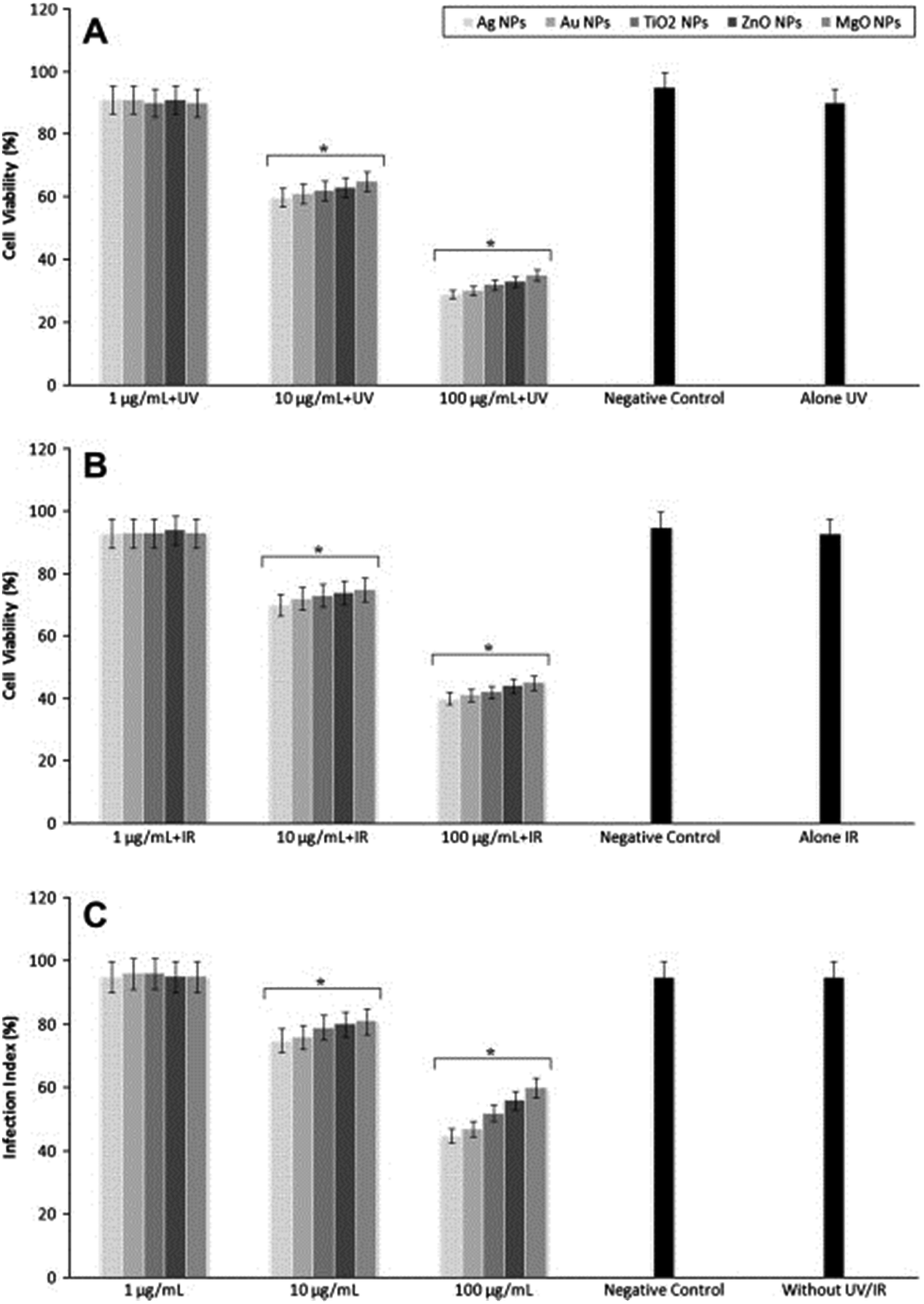 | ||
| Fig. 10 Proliferation of parasites after exposure with different nanoparticles under (A) UV light, (B) IR light, and (C) at dark conditions. Reprinted with permission from ref. 47. Copyright 2013, Elsevier. | ||
Prajapati et al. devised a novel way by using multi-walled carbon nanotube to deliver an antileishmanial drug to overcome drug-induced toxicity. AmB was linked to functionalized carbon nanotubes (f-CNTs) to yield AmB-f-CNTs. The drug carrier enhanced the drug's efficacy for inhibiting the growth of L. donovani, a parasite that causes VL. AmB-f-CNT was observed to be 14 folds more effective than AmB for inhibiting the growth of amastigotes. Furthermore, no toxicity to kidney and liver in mice was observed. Interestingly, a higher suppression percentage of parasites in the spleen with AmB-f-CNT (89.8%) was achieved compared to AmB (68.9%).49
5 Exogenous NO donors in the treatment of cutaneous leishmaniasis
The endogenous free radical nitric oxide (NO) is an important cellular signaling molecule that plays a key role in the defense against pathogens.50,51 NO was elected the ‘Molecule of the Year’ in 1992 due to its important physiological and pathophysiological actions.52In vivo, this ephemeral gas is synthesized by one of three NO synthase (NOS) isoforms: endothelial (eNOS), neuronal (nNOS), and inducible (iNOS). The isoforms differ in respect to regulation, amplitude, and duration of NO production, as well as cellular and tissue distribution.53 Both eNOs and nNOS are calcium-dependent isoforms and produce low concentrations of NO (pico-nano molar range) for short periods of time. At low concentrations, NO acts as intracellular signaling molecule.54 The third isoform, iNOS, which is mainly expressed in activated macrophages, is considered a component of the innate immune system and produces high concentrations of NO (micro-mili molar range).51,55 NO produced by mammalians iNOS has important cytostatic and cytotoxic effects against invading pathogens and parasites, including Leishmania protozoa.56,57 Indeed, iNOS deficient mice are unable to control the infection caused by Leishmania.58 In particular, activation of infected macrophages by interferon (IFN)-γ in CL increases the expression of iNOS killing parasite via NO pathway.59 Although the leishmanicidal mechanisms of NO are not fully elucidated, it is assumed that NO may have apoptosis-like death effectors, while the toxicity of exogenous NO donors can be achieved either by the release of free NO or by the S-nitrosation of important parasite/host cell proteins.60–62 Either endogenous or exogenous NO can inhibit intracellular and extracellular Leishmania parasite.63,64As Leishmania protozoa are able to compromise host macrophages decreasing iNOS activity, the administration of exogenous NO donors represents an interesting strategy to combat CL.65 Hence, application of exogenous NO/NO donors is aimed to supply the lack of endogenous NO production by infected macrophages. In this context, de Souza and collaborators incubated L. major and L. amazonensis promastigotes with the NO donor S-nitrosoglutathione (GSNO).64Fig. 11 shows the concentration dependent toxicity of GSNO on L. major (Fig. 11A) and L. amazonensis (Fig. 11B) promastigotes, after 24 h of incubation. In vitro 50% inhibitory concentrations (IC50) of 68.8 ± 22.86 and 68.9 ± 7.9 μmol L−1 were found for L. major and L. amanzonesis, respectively, upon GSNO treatment.64 These results demonstrate the leishmanicidal activity of administration of high concentrations of NO donor in protozoa cultures, highlighting their potent therapeutic effects to treat leishmaniasis.
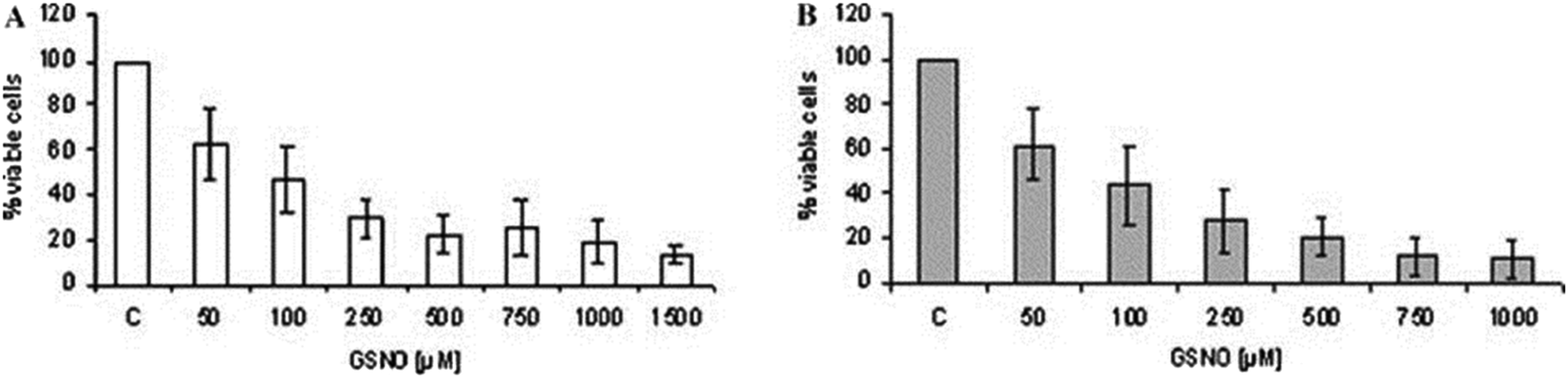 | ||
| Fig. 11 Percentage of cell viability of L. major (A) or L. amazonensis (B) promastigotes upon incubation with different concentrations of S-nitrosoglutathione (NO donor) after 24 h. Reprinted with permission from ref. 64. Copyright 2006, Elsevier. | ||
Ideally, exogenous NO donors should release sustained and high concentrations of NO for longer periods of time to kill Leishmania protozoa, leaving host tissues intact. In this context, several NO-releasing vehicles have been developed to delivery controlled amounts of NO in a safe manner. These approaches range from direct application of gaseous NO66 to NO-releasing nanomaterials.67 It should be noted that in CL parasites are located in the dermis and in deep subcutaneous tissues,68 which makes it difficult for the leishmanicidal agent to penetrate. The great advantage of using topical NO donors to combat CL is based on the fact that NO intrinsically has the property to rapidly diffuse though the dermis and lipid membranes, due to its lipophilicity and small size.69
Topical application of a cream containing the NO donor S-nitroso-N-acetylpenicillamine (SNAP) (final concentration 200 μmol L−1) in human CL was first reported by Lopez-Jaramillo et al. Administration of SNAP cream on patient lesions, caused by L. braziliensis, was performed 4 times a day. After one month treatment, the lesions were healed, since topical application of S-nitrosothiols, such as SNAP, promotes and accelerates wound contraction, in addition to their known cytotoxic effects.70,71 In a similar approach, a NO generating topical formulation comprised by acidified nitrite and ascorbic acid was applied on BALB/c mice lesions caused by L. tropica.68 This NO-generating formulation caused the cure of only 12% of the patients and healing of 28% of the ulcer lesions.68 The importance of these studies is that both works described the first approaches to topically administrate NO donors/generators to combat CL with no serious adverse effects.
However, due to the uncontrolled NO release profile from the formulations, the topical NO donors/generators need to be frequently reapplied, which impairs the adherence to the treatment. The lack of appropriate vehicles to stabilize NO donors has been limited the clinical uses of NO. Thus, the difficulties of promoting therapeutic amounts of NO, released in a sustained and controlled manner, have motived the development of new platforms to be used in the treatment of CL. Nanotechnological approaches represent a very promising strategy to increase the utility of NO in biomedical applications, such as in the treatment of CL. The combination of nanoparticles and NO donors are known to promote a controlled, sustained, and site direct NO release, at required concentrations for desired therapeutic applications.50,67
In this context, Lopez-Jaramillo et al. reported in 2010 the controlled and sustained NO delivery from a topical nanofiber NO releasing patch in the treatment of CL caused by L. (V) panamensis. A multilayer transdermal patch, produced by an electrospinning technique, produced a continuous and topical flux of NO release (3.5 μmol NO per cm2 per day for 20 days). In this nanomaterial, acidified nitrite and ascorbic acid were encapsulated in the polymer nanofibers and led to a controlled release of NO. The authors observed a 40% cure at 90 days follow-up in patients treated with NO-releasing nanofibers.72 Similarly, poly(lactide-co-glycolide) NPs containing the NO donor sodium nitroprusside (SNP) along with DOX were prepared as potential leishmanicidal agent against VL. Encapsulation SNP in the polymeric NP led to a sustained NO release for over 72 h, however the synergetic leishmanicidal activity still has to be demonstrated.73 Nanomaterials have the ability to guarantee a controlled NO release at high concentrations, directly to the target site. Due to the thermal and photochemical instability of NO donors, such as S-nitrosothiols,74 their incorporation in nanomaterials has the ability to greatly reduce the rates of NO release.67,71
Although the leishmanicidal actions of traditional NO donors have been confirmed in in vitro and in vivo studies64,75 and despite that NO-releasing nanomaterials have been successfully used in several biomedical applications,76 the combination of NO donors/generators with nanomaterials for combat leishmaniasis has not been deeper explored. Nanomaterials allied to NO is a convenient approach in the treatment of CL, as already greatly explored in other biomedical applications, such as in the promotion of wound healing, inhibition of platelet aggregation or antibacterial effects in cutaneous infections.71,77 In this scenario, several strategies have aimed to open new perspectives for the treatment of CL with controllable and sustainable release of therapeutic amounts of NO direct to the target tissues, with minimum side effects.
6 Nanoimmunization
Nanoparticles have not only been used as drug vehicles but also as promising vaccine carriers. Vaccines are composed of appropriate antigens responsible for induction of a protective immune response against a specific pathogen, boosted by adjuvants. Despite many antigens are immunogenic to the host, the lack of universal protection may result to the inability of the vaccine to elicit desirable immune response.78 Nanoparticles can help to deliver antigens to antigen-presenting cells (APCs) that play a crucial role in activating the host immune system.79The use of nanotechnology in delivering antigens and adjuvants have different aims: (i) to enhance their uptake by APCs,80 (ii) to generate Th1 type immune response,81 and (iii) to induce a stronger immunological effect due to a simultaneous delivery to the same APC, compared to the free antigen and adjuvant.82
In the last years, the research in Leishmania vaccines is trying to bridge the gap between humoral and cellular immune responses. Dendritic cells (DCs) are a class of specialized APCs that coordinate both innate and acquired immunity. They sense the presence of microorganisms and recognize conserved pathogen-associated molecular patterns. Mature DCs migrate from the infection site to the closer draining lymph node for antigen presentation to naïve T-cells.83
The major challenge in Leishmania immunotherapy lies in inducing a sustained life-long immunity against the parasite. Efficient leishmanicidal activity is obtained when naïve CD8 + T-cells located at the lymph nodes are primed with the CD4 + T-cell help. CD4 cells play a key role in the expansion of CD8 cells, providing the properties of memory cells. However, vaccines against Leishmania that reach clinical trials showed low efficacy due to scarce CD8 + T-cell response emerging from cross-presentation.84
The physicochemical properties of polymer nanoparticles when used as adjuvants in a vaccine could mean a more effective immune response against Leishmania. There have been several efforts in the last 5 years concerning immunization against leishmaniasis through nanotechnology, mainly through the use of polymers. In 2010 Tafaghodi et al. took PLGA nanospheres as an antigen delivery system and Quillaja saponins (QS) as immunoadjuvant to enhance the immune response against autoclaved L. major (ALM).43 BALB/c mice were immunized three times in 3-week intervals using the particulate antigen alone or in combination with the immunoadjuvant. Immunized mice were challenged with L. major at the footpad three weeks after the last booster and the lesion size was measured in order to analyze the intensity of infection. Mice immunized with particulate ALM antigen showed the smaller footpad swelling and the strongest protection in relation to the other formulations. The authors concluded that nanoparticles could be used as a vaccine delivery system.
Santos et al. tested two immunization strategies comparing (a) unique priming using a PGLA nanoparticle loaded with plasmid DNA encoding KMP-11 and (b) the above particulate vaccine in combination with a 21-day delayed booster with the same carrier loaded with recombinant protein KMP-11.85 Both strategies showed detectable cellular immune responses with pro-inflammatory and anti-inflammatory cytokines. After animal challenge with L. braziliensis, although lesion development was similar with both vaccination strategies, the parasite burden at the lesion size was significantly reduced with the approach that combines initial priming and a booster. Following the mice inoculation with L. braziliensis, the group immunized with recombinant PLGA nanoparticles, showed upregulation of IFN-γ and TNF-α, which are proteins that orchestrate an immune defense against pathogens, findings that could explain the greater parasite killing at the site (Fig. 12).
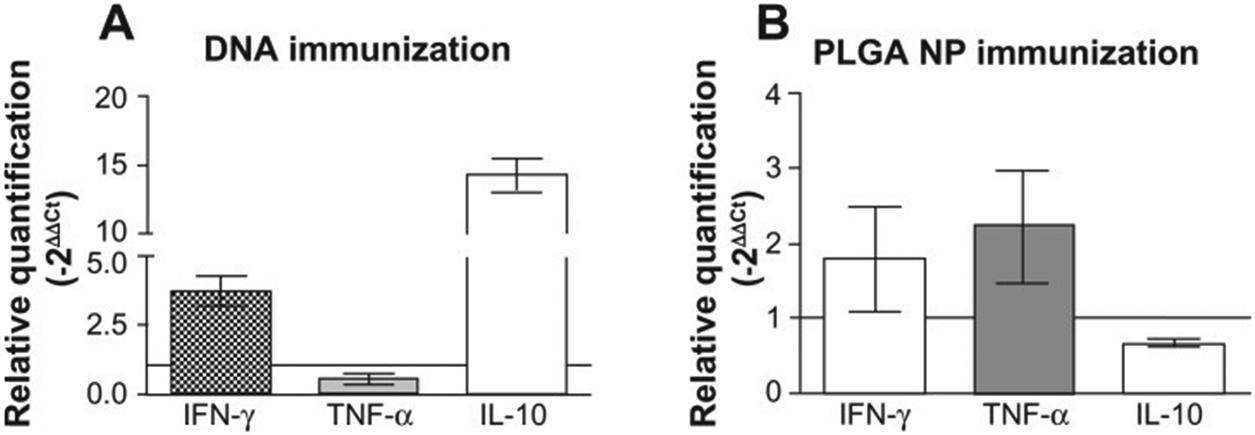 | ||
| Fig. 12 Cytokine expression at the ear dermis following a live challenge with parasites. (A) Mice immunized with plasmid DNA encoding L. infantum chagasi KMP-11. (B) Mice immunized with PLGA NP. Reprinted with permission from ref. 85. Copyright 2012, Dove Press. | ||
In 2011 Doroud et al. evaluated the suitability of cationic solid lipid nanoparticles (cSLN) as an adjuvant or delivery system for cisteine proteinases (CPs) of L. major as target antigens in C57BL/6 mice.86 They also determined the role of the C-terminal extension (CTE) in CPs in the protective response. The results indicated that encapsulation of CPs in cSLNs enhances the extent of protection, showing a significant reduction in the parasite burden of the draining lymph nodes. In addition, the CTE domain proved not to have a crucial production of protective responses against L. major infection.
Electroporation is an example of a physical vaccine DNA delivery method that it is traditionally used for gene delivery. It is believed to be a gold standard and it is defined as the application of controlled electric fields to facilitate cell permeabilization, leading to the enhancement of gene uptake into cells after injection of naked DNA. In 2013 Saljoughian et al. compared the potential of either a physical method (electroporation) and a chemical delivery system (cationic solid-lipid nanoparticles cSLN) to deliver a DNA vaccine harbouring the L. donovani A2 antigen along with L. infantum cisteine proteinases A and B without its unusual C-terminal extension (A2-CPA-CPB-CTE).87 Deliver by either electroporation or cSLN formulation showed protection of BALB/c mice against L. infantum through a strong Th1 immune response. The cSLNs as a nanoscale vehicle proved to be an interesting alternative as a delivery system.
Based on the previous successful results, Shahbazi et al. extended the research to a clinical trial using outbred dogs, since they are primary reservoirs of the parasite.88 Results showed that the administration of pcDNA-A2-CPA-CPB-CTE GFP vaccine as a prime-boost by either electroporation or cSLN formulation protects the dogs against L. infantum infection. But even more important than that, was the fact that vaccinated dogs were associated with significantly (p < 0.05) higher levels of IgG2, IFN-γ, and TNF-α and with low levels of IgG1 and IL-10 as compared to the control group, which correlates with a good Th1 immune response against the parasite. Protection was also correlated with a low parasite burden in bone marrow.
In the same way 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) nanoliposomes were used as an antigen delivery system and immunoadjuvant for soluble Leishmania antigens (SLA) by Firouzmand et al. in 2013. They demonstrated that SLA incorporated within the nanoliposomes are appropriate delivery systems to induce a Th1 type of immune response and protection against L. major infection in BALB/c mice.89 Groups of mice immunized with liposomal SLA showed very low number of parasites in the footpad and spleen in comparison with the control group, as well as the best Th1 response (high level of IFN-γ and low level of IL-4).
7 Advantages and limitations of nanoparticulate systems in the treatment of leishmaniasis
Traditional nanocarriers like liposomes and polymeric nanoparticles are readily internalized by macrophages in the liver and spleen, therefore they are the preferred nanocarriers employed in the treatment of macrophage parasitic diseases.15 Liposomes are definitely the most used nanocarrier in the treatment of leishmaniasis. The positive aspects of liposomes are the ability to load either hydrophilic or hydrophobic drugs, the possibility to surface modifications, and the fate of liposomes is the macrophage where the parasite exists. In particular, surface modifications of liposomes are known to significantly improve drug targeting, since macrophages express different receptors. Functionalization of liposomes with sugar can improve macrophage targeting since macrophage contains receptors that recognize sugar molecules. Similarly, positively charged liposomes are readily internalized by macrophages due to their interaction with negatively charged macrophage membrane. However, liposomes display some limitations such as instability that could lead to toxicity due to the leakage of the drug from the nanocarrier into the blood stream.17 Nanoemulsions are promising drug delivery systems due to their simple preparation and scale-up, ability to solubilize hydrophobic drugs and physicochemical stability.14The second most popular nanocarrier used in the treatment of leishmaniasis is polymeric nanoparticles, which can in part overcome some limitations of liposomes.90 The advantages of polymeric nanoparticles are the low toxicity, the possibility to design biodegradable systems, the cost-effectives, small size, possibility to surface functionalization and co-administration with others drugs. Among the polymers, biodegradable and biocompatible ones such as PLGA are preferred used. Polysaccharides such as chitosan are also employed. By changing the polymer, the physicochemical characteristic of the nanoparticles, such as zeta potential, can be modulated.91 As liposomes, polymeric nanoparticles are internalized by macrophages, possess high surface area that can be used for functionalization improving targeting and biocompatibility, and stability in biological medium.16 Polymeric nanoparticles are more stable compared to liposomes, thus extravasation of the drug before the nanocarrier reach the target site (macrophage) can be avoided.
Metallic nanoparticles and carbon-based nanomaterials have been emerging as versatile nanocarriers in the treatment of leishmaniasis. From the revised literature, further studies are necessary to explore the advantages of these nanocarriers in this arena. In a similar manner, dendrimers are nanocarriers with great potential to carry and delivery antileishmanial drugs due to their ability to load with amounts of the drug on their branched surface, improving the drug bioavailability.91
Although the recent advantages on the development of efficient antileishmanial releasing nanocarriers, there are some challenges to be overcome. For example, the development of efficient oral nanoformulations for the treatment of leishmaniasis, with low costs. In comparison with conventional pharmaceutical drugs, the nanostructure formulations have higher costs. However, it should be noted that traditional drugs often require substitutions for treatment due to lack of efficiency and undesired side effects, which can in turn result in further economic investments.16
8 Conclusions
The crux of discovering a new chemical entity for any disease or a disorder is to make sure it is safe, efficacious, effective, and relatively inexpensive. Small molecules are known to be effective, but their ability to reach its target is limited by many intrinsic physico-chemical traits. The efficacy of these small molecules could be further enhanced by various advanced techniques, e.g., by encapsulation or conjugation into/with nanocarriers like liposomes, polymeric nanoparticles, dendrimers, carbon-based materials, among others.31 Similarly, the systemic toxicity of the small molecule drug can be reduced using this techniques and by achieving enhanced bioavailability which results in increased patient compliance. Conversely, the early diagnosis of the disease is equally critical and a very challenging aspect of treatment. In this direction, particularly for infectious diseases like leishmaniasis, the U.S. Army Medical Material Development Activity (USAMMDA) has conducted market research to survey available or emerging technologies for a diagnostic device to detect CL. Ideally the diagnosis of such diseases must be able to be used in point-of-care settings and for the early treatment.Treating leishmaniasis with first-line drugs such as AmB, paromomycin, and miltefosine face many drawbacks since such treatments are toxic and expensive. Furthermore many of the Leishmania parasites are now resistant to these drugs. Nano-based delivery systems deliberate many clinical advantages, e.g., liposomal formulation of AmB is unquestionably effective, but could also be expensive. As we described in this review, in recent years there have been several reported studies as far as the treatment against leishmaniasis through nanotechnology is concerned. Some of them have shown encouraging results that have allowed us to think that nanotechnology might play a prudent role in the future, due to its many advantages especially in comparison to the current antileishmanial treatment. Liposomes, NCs, polymers, and NPs have been the object of much study, because nanotechnology may not only be a valuable tool in antileishmanial treatment but in prevention (vaccines) as well. Interestingly, few nanotechnology based formulations are being introduced in clinic for the treatment of leishmaniasis. Recently, nano-liposomal AmB gel was approved in Iran for the treatment of CL, fungal, and topical diseases.
As we already showed, the validity of in vitro and animal model projects using nanotechnology as a treatment against leishmaniasis is extremely important. However, none of the recently published studies mentioned further in vivo and clinical tests, which could bring new challenges due to the large difference between the two scenarios. Until today AmBisome® is the only nanotechnology based antileishmanial drug, which has use in the clinical practice, but, despite its proven efficacy and low toxicity among other features that make it an excellent alternative, its use remains restricted in some areas due to its high cost. Further efforts should be made in order to have more drugs based on this promising technology in our future antileishmanial drugs’ arsenal.
Acknowledgements
A. B. S. acknowledges support from FAPESP (Proc. 2012/17053-7), the Brazilian Network on Nanotoxicology (MCTI/CNPq). R. M. R. acknowledges PI12/00104 of the Instituto de Salud Carlos III and CYTED 214RT0482 (Ministerio de Economía y Competitividad (MINECO)). M. C. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG)/German Research Foundation via SFB1112 (Project A04), the Bundesministerium für Bildung und Forschung (BMBF) through the NanoMatFutur award (13N12561), the Helmholtz Virtual Institute, Multifunctional Biomaterials for Medicine, and the Freie Universität Berlin Focus Area Nanoscale. A. B. S. and M. C. acknowledge the CONNECT Program of the Alexander von Humboldt Foundation. The authors acknowledge Dr P. Winchester for proof reading and Dr W. Fischer for graphical support.Notes and references
- G. J. Grimaldi and R. Tesh, Clin. Microbiol. Rev., 1993, 6, 230–250 Search PubMed.
- M. den Boer, D. Argaw, J. Jannin and J. Alvar, Clin. Microbiol. Infect., 2011, 17, 1471–1477 CrossRef CAS PubMed.
- M. A. Danesh-Bahreini, J. Shokri, A. Samiei, E. Kamali-Sarvestani, M. Barzegar-Jalali and S. Mohammadi-Samani, Int. J. Nanomed., 2011, 6, 835–842 CAS.
- D. Liu and J. E. Uzonna, Front. Cell. Infect. Microbiol., 2012, 2, 83 Search PubMed.
- F. L. Ribeiro-Gomes and D. Sacks, Front. Cell. Infect. Microbiol., 2012, 2, 59 Search PubMed.
- J. A. Guerra, S. R. Prestes, H. Silveira, L. I. Coelho, P. Gama, A. Moura, V. Amato, M. Barbosa and L. C. Ferreira, PLoS Neglected Trop. Dis., 2011, 5, e980 Search PubMed.
- R. Balana-Fouce, R. M. Reguera, J. C. Cubria and D. Ordonez, Gen. Pharmacol., 1998, 30, 435–443 CrossRef CAS PubMed.
- S. Croft and P. Olliaro, Clin. Microbiol. Infect., 2011, 10, 1478–1483 CrossRef PubMed.
- A. Musa, E. Khalil, A. Hailu, J. Olobo, M. Balasegaram, R. Omollo, T. Edwards, J. Rashid, J. Mbui, B. Musa, A. A. Abuzaid, O. Ahmed, A. Fadlalla, A. El-Hassan, M. Mueller, G. Mucee, S. Njoroge, V. Manduku, G. Mutuma, L. Apadet, H. Lodenyo, D. Mutea, G. Kirigi, S. Yifru, G. Mengistu, Z. Hurissa, W. Hailu, T. Weldegebreal, H. Tafes, Y. Mekonnen, E. Makonnen, S. Ndegwa, P. Sagaki, R. Kimutai, J. Kesusu, R. Owiti, S. Ellis and M. Wasunna, PLoS Neglected Trop. Dis., 2012, 6, e1674 CAS.
- A. H. Mohamed-Ahmed, S. Brocchini and S. L. Croft, Curr. Opin. Infect. Dis., 2012, 25, 695–702 CrossRef CAS PubMed.
- S. K. Bhattacharya, P. K. Sinha, S. Sundar, C. P. Thakur, T. K. Jha, K. Pandey, V. R. Das, N. Kumar, C. Lal, N. Verma, V. P. Singh, A. Ranjan, R. B. Verma, G. Anders, H. Sindermann and N. K. Ganguly, J. Infect. Dis., 2007, 196, 591–598 CrossRef CAS PubMed.
- J. J. Berman, Expert Opin. Drug Metab. Toxicol., 2008, 9, 1209–1216 CrossRef PubMed.
- S. Sundar, T. K. Jha, C. P. Thakur, P. K. Sinha and S. K. Bhattacharya, N. Engl. J. Med., 2007, 356, 2571–2581 CrossRef CAS PubMed.
- P. Prahu, V. Patravale and M. Joshi, Curr. Nanosci., 2012, 8, 491–503 CrossRef.
- K. Jain and N. K. Jain, Drug Discovery Today, 2013, 18, 23–24 CrossRef PubMed.
- N. M. Khalil, A. C. Mattos, T. C. M. Carraro, D. B. Ludwing and R. M. Mairnades, Curr. Pharm. Des., 2013, 19, 7316–7329 CrossRef CAS PubMed.
- T. T. H. Pharm, P. M. Loiseau and G. Barrat, Int. J. Pharm., 2013, 454, 539–552 CrossRef PubMed.
- S. Costa Lima, R. Silvestre, D. Barros, J. Cunha, M. Baltazar, R. Dinis-Oliveira and A. Cordeiro-da-Silva, Nanomedicine, 2013, 10, 1021–1030 CrossRef PubMed.
- E. L. Romero and M. J. Morilla, Expert Opin. Drug Delivery, 2008, 5, 805–823 CrossRef CAS PubMed.
- S. G. Suresh P Vyas, Int. J. Nanomed., 2006, 4, 417–432 CrossRef.
- R. L. Taylor, D. M. Williams, P. C. Craven, J. R. Graybill, D. J. Drutz and W. E. Magee, Am. Rev. Respir. Dis., 1982, 125, 610–611 CrossRef CAS PubMed.
- G. Lopez-Berestein, R. Mehta, R. Hopfer, K. Mills, L. Kasi, K. Mehta, V. Fainstein, M. Luna, E. Hersh and R. Juliano, J. Infect. Dis., 1983, 147, 939–945 CrossRef CAS PubMed.
- C. Tremblay, M. Barza, C. Fiore and F. Szoka, Antimicrob. Agents Chemother., 1984, 26, 170–173 CrossRef CAS PubMed.
- G. Lopez-Berestein, V. Fainstein, R. Hopfer, K. Mehta, M. P. Sullivan, M. Keating, M. G. Rosenblum, R. Mehta, M. Luna, E. M. Hersh, J. Reuben, R. L. Juliano and G. P. Bodey, J. Infect. Dis., 1985, 151, 704–710 CrossRef CAS PubMed.
- F. C. Szoka Jr., D. Milholland and M. Barza, Antimicrob. Agents Chemother., 1987, 31, 421–429 CrossRef CAS.
- M. Balasegaram, K. Ritmeijer, M. Lima, S. Burza, G. Ortiz Genovese, B. Milani, S. Gaspani, J. Potet and F. Chappuis, Expert Opin. Emerging Drugs, 2012, 17, 493–510 CrossRef CAS PubMed.
- M. El-Tonsy, Parasitologists United Journal, 2010, 3, 19–26 Search PubMed.
- J. Roychoudhury, R. Sinha and N. Ali, PLoS One, 2011, 6, e17376 CAS.
- A. P. Perez, A. Casasco, P. Schilrreff, M. V. D. Tesoriero, L. Duempelmann, M. J. Altube, L. Higa, M. J. Morilla, P. Petray and E. L. Romero, Int. J. Nanomed., 2014, 9, 3335–3345 Search PubMed.
- S. Kansal, R. Tandon, P. Dwivedi, P. Misra, P. Verma, A. Dube and P. Mishra, J. Antimicrob. Chemother., 2012, 67, 2650–2660 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- M. Molina, M. Asadian-Birjand, J. Balach, J. Bergueiro and M. Calderón, Chem. Soc. Rev., 2015, 44, 6161–6186 RSC.
- J. Khandare, A. Mohr, M. Calderon, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268–4277 CrossRef CAS PubMed.
- M. Calderón, M. Quadir, M. Strumia and R. Haag, Biochimie, 2010, 92, 1242–1251 CrossRef PubMed.
- S. Mondal, P. Roy, S. Das, A. Halder, A. Mukherjee and T. Bera, PLoS One, 2103, 8, e81492 Search PubMed.
- P. Roy, S. Das, T. Bera, S. Mondol and A. Mukherjee, Int. J. Nanomed., 2010, 5, 1113–1121 CAS.
- S. Nicoletti, K. Seifert and I. Gilbert, Bioorg. Med. Chem., 2012, 18, 2559–2565 CrossRef PubMed.
- G. Koczan, A. C. Ghose, A. Mookerjee and F. Hudecz, Bioconjugate Chem., 2002, 13, 518–524 CrossRef CAS PubMed.
- A. Nan, S. L. Croft, V. Yardley and H. Ghandehari, J. Controlled Release, 2004, 94, 115–127 CrossRef CAS PubMed.
- D. Barros, S. A. C. Lima and A. Cordeiro-da-Silva, Nanomedicine, 2015, 10, 387–403 CrossRef CAS PubMed.
- J. Giarolla, K. Pasqualoto, D. Rando, M. Zaim and E. Ferreira, J. Mol. Model., 2012, 18, 2257–2269 CrossRef CAS PubMed.
- P. M. Daftarian, G. W. Stone, L. Kovalski, M. Kumar, A. Vosoughi, M. Urbieta, P. Blackwelder, E. Dikici, P. Serafini, S. Duffort, R. Boodoo, A. Rodríguez-Cortés, V. Lemmon, S. Deo, J. Alberola, V. L. Perez, S. Daunert and A. L. Ager, J. Infect. Dis., 2013, 208, 1914–1922 CrossRef CAS PubMed.
- M. Tafaghodi, M. Eskandari, M. Kharazizadeh, A. Khamesipour and M. R. Jaafari, Trop. Biomed., 2010, 27, 639–650 CAS.
- Y. Tauran, A. Brioude, A. Coleman, M. Rhimi and B. Kim, World J. Biol. Chem., 2013, 26, 35–63 Search PubMed.
- A. E. Allahverdiyev A, M. Bagirova, C. Ustundag, C. Kaya, F. Kaya and M. Rafailovich, Int. J. Nanomed., 2011, 6, 2705–2714 CrossRef PubMed.
- S. Soflaei, A. Dalimi, F. Ghaffarifar, M. Shakibaie, A. R. Shahverdi and M. Shafiepour, J. Parasitol. Res., 2012, 2012, 756568 Search PubMed.
- A. Jebalia and B. Kazemi, Toxicol. In Vitro, 2013, 27, 1896–1904 CrossRef PubMed.
- M. Mohebali, M. Rezayat and K. Gilani, Daru J. Fac. Pharm., 2009, 17, 285–289 CAS.
- V. Prajapati, K. Awasthi, S. Gautam, T. Yadav, M. Rai, O. Srivastava and S. Sundar, J. Antimicrob. Chemother., 2011, 66, 874–879 CrossRef CAS PubMed.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- D. Schairer, J. Chouake, J. Nosanchuk and A. Friedman, Virulence, 2012, 3, 271–279 CrossRef PubMed.
- E. Culotta and D. Koshland, Science, 1992, 258, 1862–1865 CAS.
- W. K. Alderton, C. E. Cooper and R. G. Knowles, Biochem. J., 2001, 357, 593–615 CrossRef CAS PubMed.
- D. Wink and J. Mitchell, Free Radicals Biol. Med., 1998, 25, 434–456 CrossRef CAS PubMed.
- F. Aktan, Life Sci., 2004, 75, 639–653 CrossRef CAS PubMed.
- P. Tripathi, Indian J. Biochem. Biophys., 2007, 44, 310–319 CAS.
- S. Patel, S. Kumar, A. Jyoti, B. S. Srinag, R. S. Keshari, R. Saluja, A. Verma, K. Mitra, M. K. Barthwal, H. Krishnamurthy, V. K. Bajpai and M. Dikshit, Nitric Oxide, 2010, 22, 226–234 CrossRef CAS PubMed.
- A. Diefenbach, H. Schindler, M. Röllinghoff, W. Yokoyama and C. Bogdan, Science, 1999, 284, 951–955 CrossRef CAS PubMed.
- S. Elcicek, M. Bagirova and A. Allahverdiyev, Exp. Parasitol., 2013, 237–242 CrossRef CAS PubMed.
- P. Holzmuller, D. Sereno, M. Cavaleyra, I. Mangot, S. Daulouede, P. Vincendeau and J. Lemesre, Infect. Immun., 2002, 70, 3727–3735 CrossRef CAS PubMed.
- M. A. Dea-Ayuela, L. Ordonez-Gutierrez and F. Bolas-Fernandez, Int. J. Med. Microbiol., 2009, 299, 221–232 CrossRef CAS PubMed.
- D. S. Lima-Junior, D. L. Costa, V. Carregaro, L. D. Cunha, A. L. Silva, T. W. Mineo, F. R. Gutierrez, M. Bellio, K. R. Bortoluci, R. A. Flavell, M. T. Bozza, J. S. Silva and D. S. Zamboni, Nat. Med., 2013, 19, 909–915 CrossRef CAS PubMed.
- G. L. Colassanti, M. Mattu, T. Persichini, L. Salvati, G. Venturini and P. Ascenzi, Int. J. Mol. Med., 2002, 9, 131–134 Search PubMed.
- G. F. de Souza, J. K. Yokoyama-Yasunaka, A. B. Seabra, D. C. Miguel, M. G. de Oliveira and S. R. B. Uliana, Nitric Oxide, 2006, 15, 209–216 CrossRef PubMed.
- N. L. Diaz, M. Fernandez, E. Figueira, R. Ramirez, I. B. Monsalve and F. J. Tapia, Clin. Exp. Dermatol., 2003, 28, 288–293 CrossRef CAS PubMed.
- A. Ghaffari, C. Miller, B. McMullin and A. Ghahary, Nitric Oxide, 2006, 14, 21–29 CrossRef CAS PubMed.
- A. Seabra and N. Durán, J. Mater. Chem., 2010, 20, 1624–1637 RSC.
- R. N. Davidson, V. Yardley, S. L. Croft, P. Konecny and N. Benjamin, Trans. R. Soc. Trop. Med. Hyg., 2000, 94, 319–322 CrossRef CAS PubMed.
- A. Seabra, E. Pankotai, M. Fecher, A. Somlai, L. Kiss, L. Biro, C. Szabo, M. Kollai, M. de Oliveira and Z. Lacza, Br. J. Dermatol., 2007, 156, 814–818 CrossRef CAS PubMed.
- J. Georgii, T. Amadeu, A. Seabra, M. de Oliveira and A. Costa, J. Tissue Eng. Regener. Med., 2011, 5, 612–619 CrossRef CAS PubMed.
- A. Seabra and N. Duran, Curr. Nanosci., 2012, 8, 520–525 CrossRef CAS.
- P. Lopez-Jaramillo, M. Y. Rincon, R. G. Garcia, S. Y. Silva, E. Smith, P. Kampeerapappun, C. Garcia, D. J. Smith, M. Lopez and I. D. Velez, Am. J. Trop. Med. Hyg., 2010, 83, 97–101 CrossRef CAS PubMed.
- S. Panday, R. Verma and A. Misra, J. Biomed. Nanotechnol., 2011, 7, 213–215 CrossRef.
- S. Shishido, A. Seabra, W. Loh and M. de Oliveria, Biomaterials, 2003, 24, 3453–3553 CrossRef.
- S. G. Costa ISF, M. G. de Oliveira and I. A. Abrahamsohn, J. Antimicrob. Chemother., 2013, 68, 2561–2568 CrossRef PubMed.
- A. Kutner and A. Friedman, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 502–514 CAS.
- P. Prabhu, V. Patravale and M. Joshi, Curr. Nanosci., 2012, 8, 491–503 CrossRef CAS.
- C. B. Palatnik-de-Sousa, Vaccine, 2008, 26, 1709–1724 CrossRef CAS PubMed.
- D. Doroud and S. Rafati, Expert Rev. Vaccines, 2012, 11, 69–86 CrossRef CAS PubMed.
- K. Sehgal, K. M. Dhodapkar and M. V. Dhodapkar, Immunol. Lett., 2014, 162, 59–67 CrossRef CAS PubMed.
- K. D. Newman, J. Samuel and G. Kwon, J. Controlled Release, 1998, 54, 49–59 CrossRef CAS PubMed.
- M. Diwan, M. Tafaghodi and J. Samuel, J. Controlled Release, 2002, 85, 247–262 CrossRef CAS PubMed.
- R. Medzhitov, Nature, 2007, 449, 819–826 CrossRef CAS PubMed.
- E. M. Janssen, E. E. Lemmens, T. Wolfe, U. Christen, M. G. v. Herrath and S. P. Schoenberger, Nature, 2003, 421, 852–856 CrossRef CAS PubMed.
- D. Santos, M. Carneiro, T. de Moura, K. Fukutani, J. Clarencio, M. Soto, S. Espuelas, C. Brodskyn, A. Barral, M. Barral-Netto and C. de Oliveira, Int. J. Nanomed., 2012, 7, 2115–2127 CAS.
- D. Doroud, F. Zahedifard, A. Vatanara, A. R. Najafabadi and S. Rafati, Parasite Immunol., 2011, 33, 335–348 CrossRef CAS PubMed.
- N. Saljoughian, F. Zahedifard, D. Doroud, F. Doustdari, M. Vasei, B. Papadopoulou and S. Rafati, Parasite Immunol., 2013, 35, 397–408 CrossRef CAS PubMed.
- M. Shahbazi, F. Zahedifard, N. Saljoughian, D. Doroud, S. Jamshidi, N. Mahdavi, S. Shirian, Y. Daneshbod, S. Hamid Zarkesh-Esfahani, B. Papadopoulou and S. Rafati, Vet. Parasitol., 2015, 212, 130–139 CrossRef CAS PubMed.
- H. Firouzmand, A. Badiee, A. Khamesipour, V. Heravi Shargh, S. H. Alavizadeh, A. Abbasi and M. R. Jaafari, Acta Trop., 2013, 128, 528–535 CrossRef CAS PubMed.
- R. Tyagi, S. Lala, A. K. Verma, A. K. Nandy, S. B. Mahato, A. Maitra and M. K. Basu, J. Drug Targeting, 2005, 13, 161–171 CrossRef CAS PubMed.
- R. Sharma, U. Agrawal, N. Mody and S. P. Vyas, Biotechnol. Adv., 2015, 33, 64–79 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |