
Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters
Vikas S.
Padalkar
* and
Shu
Seki
 *
*
Department of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: vikaspadalkar@gmail.com; seki@moleng.kyoto-u.ac.jp
First published on 27th October 2015
Abstract
Solid state emitters based on excited state intramolecular proton transfer (ESIPT) have been attracting considerable interest since the past few years in the field of optoelectronic devices because of their desirable unique photophysical properties. The photophysical properties of the solid state ESIPT fluorophores determine their possible applicability in functional materials. Less fluorescence quantum efficiencies and short fluorescence lifetime in the solid state are the shortcomings of the existing ESIPT solid state emitters. Designing of ESIPT chromophores with high fluorescence quantum efficiencies and a long fluorescence lifetime in the solid state is a challenging issue because of the unclear mechanism of the solid state emitters in the excited state. Reported design strategies, detailed photophysical properties, and their applications will help in assisting researchers to overcome existing challenges in designing novel solid state ESIPT fluorophores for promising applications. This review highlights recently developed solid state ESIPT emitters with focus on molecular design strategies and their photophysical properties, reported in the last five years.
 Vikas S. Padalkar | Vikas Padalkar completed his graduation (2005) and post-graduation (2007) from the University of Pune, and received his PhD degree in Chemistry from the University of Mumbai (2011). Prior to obtaining his doctorate degree he has worked with Merck Pharmaceuticals, Mumbai. He has worked as a post-doctoral research fellow at the Institute of Chemical Technology, Mumbai (2011–March 2014) on project “Stand-off detection of explosives based on immunochemical techniques”. He joined Osaka University in April, 2014 (Professor Seki Laboratory) as a JSPS post-doctoral research fellow, and currently he is continuing his JSPS-research with Professor Shu Seki in Kyoto University, Japan. His research interests include development of fluorescent organic materials for functional applications. |
 Shu Seki | Shu Seki graduated from the University of Tokyo in 1993, and received his PhD degree in 2001 from Osaka University. He has worked as researcher at the Argonne National Laboratory, USA (1993), and the Delft University of Technology, Netherlands (2001). He joined Osaka University, Japan as a faculty member in 1995 and served for 18 years as assistant (1995–2001), associate (2002–2009) and full professor (2009–2015). Since 2015 he has been appointed as full professor in Department of Molecular Engineering, Graduate School of Engineering, Kyoto University, Japan. His research is primarily focused on the physical chemistry of condensed matter, functional organic materials, nanomaterials and radiation chemistry. He has published more than 430 SCI papers and reviews in rated journals and is an author for 20 book chapters. |
1. Introduction
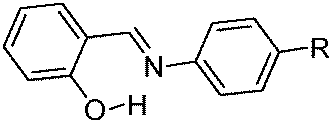
Excited state intramolecular proton transfer (ESIPT) [all abbreviations are summarized in the abbreviation section before references section] is a photochemical process that produces a tautomer with a different electronic structure from the original excited form.1 It is a four-level photo-cycle (E → E* → K* → K) scheme implemented by the enol (E) → keto (K) photo-tautomerisation process1–3Fig. 1. The ESIPT phenomenon was reported first for salicylic acid by Waller in 1950s.4 After the discovery of ESIPT, many researchers studied the fundamental photophysics, proton transfer dynamics, pico-second kinetics, vibronically resolved spectroscopy, kinetics and thermodynamics at the singlet state, femtosecond and time-resolved ESIPT fluorophores.3,5–9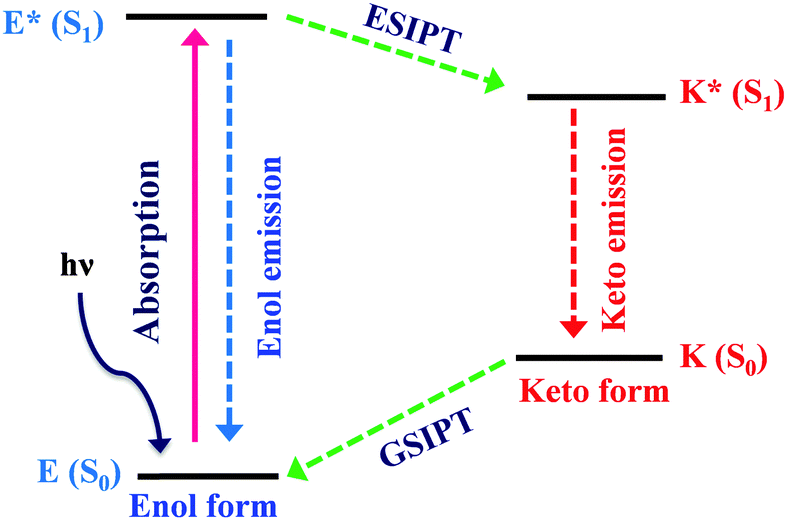 | ||
| Fig. 1 Four level ESIPT process. | ||
Recently, Park et al.,3 Wang et al.,9 Zhao et al.,2 and Chuo et al.,8,10 critically reviewed the photophysical properties, synthetic strategies and potential applications of the ESIPT fluorophores.
The remarkable properties of the ESIPT fluorophores are a large Stokes shift (∼200 nm),3 dual emission,3 an ultrafast process,11,12 and spectral sensitivity to the surrounding medium.13 The pre-requisite for ESIPT is the presence of an intramolecular hydrogen bond (H-bond) between the proton donor (–OH and –NH2) and the proton acceptor (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– and –CO) groups in close proximity to each other in a molecule.2,3,9,10,14–16 ESIPT is extremely fast (kESIPT > 1012 s−1) and is able to proceed even in rigid glass at very low temperature.11 The rate of ESIPT processes depends on donor/acceptor groups and the surrounding medium.17 Broad, tunable and dual emissions are attractive properties of ESIPT chromophores that cover the whole visible domain which can result in the production of white light.18,19 Most of the ESIPT chromophores show dual emission, short wavelength emission due to the excited state enol form (E*) (normal emission) and the longer one due to the excited state keto form (K*) (ESIPT emission) through a four-level photocycle.3,8,9 In addition, this process generates different transient species.3,8,9 Due to transient changes involved in the ESIPT process, ESIPT emission gets easily affected by its micro-environment, leading to a change in the fluorescence properties.20 The spectral properties of the ESIPT fluorophores depend on the hydrogen bonding, rotamerisation process, acidity/basicity of the surrounding medium, and substituents present on the donor and acceptor units.2,3,8,10 The fluorescence properties of the ESIPT fluorophores are tunable by changing these parameters.2,3,8
N– and –CO) groups in close proximity to each other in a molecule.2,3,9,10,14–16 ESIPT is extremely fast (kESIPT > 1012 s−1) and is able to proceed even in rigid glass at very low temperature.11 The rate of ESIPT processes depends on donor/acceptor groups and the surrounding medium.17 Broad, tunable and dual emissions are attractive properties of ESIPT chromophores that cover the whole visible domain which can result in the production of white light.18,19 Most of the ESIPT chromophores show dual emission, short wavelength emission due to the excited state enol form (E*) (normal emission) and the longer one due to the excited state keto form (K*) (ESIPT emission) through a four-level photocycle.3,8,9 In addition, this process generates different transient species.3,8,9 Due to transient changes involved in the ESIPT process, ESIPT emission gets easily affected by its micro-environment, leading to a change in the fluorescence properties.20 The spectral properties of the ESIPT fluorophores depend on the hydrogen bonding, rotamerisation process, acidity/basicity of the surrounding medium, and substituents present on the donor and acceptor units.2,3,8,10 The fluorescence properties of the ESIPT fluorophores are tunable by changing these parameters.2,3,8
To date, most studied ESIPT fluorophores have been derivatives of 2-(2′-hydroxyphenyl)benzimidazole (HBI),21–26 2-(2′-hydroxyphenyl)benzoxazole (HBO)27–30 and 2-(2′-hydroxyphenyl)benzothiazole (HBT).20,28,31–36 Along with HBIs, HBOs, and HBTs, derivatives of quinoline,37–39 benzophenones,40 flavones,41–43 anthraquinones,44,45 benzotriazoles,46 thiodiazoles,47 salicylidene aniline,48 quinoxalines,49 benzazoles,12 chromones,50 coumarins,51,52 and naphthalimides34,53 have been reported for the ESIPT study. Besides, a novel class of imidazo[1,2-a]pyridines showing ESIPT was recently reported by Araki et al.54–57 Because of the unique photophysical properties, ESIPT fluorophores have been applied in various fields, such as molecular logic gates,58 newer arenas of biotechnology,9,59 fluorescence imaging,60,61 radiation scintillators,41,62 UV-absorbers,63 and for fundamental photophysical dynamics studies.64 Reported ESIPT molecules are highly emissive in solution but weakly emissive in the solid state.2,3,9 Recently, solid state emitters with high fluorescence quantum efficiencies have attracted wide attention in the field of optoelectronic devices and laser applications.65–69 ESIPT chromophores which are highly emissive in rigid media but non-emissive (or weakly emissive) in solution are more appropriate fluorophores for optoelectronic devices.66,69–71
To date, many articles describing synthesis methods, extensive photophysics and applications of ESIPT in solution have been reported, however ESIPT inspired solid state emitters have not been explored to a large extent. In this review article, the solid state ESIPT emitters and their photophysical properties, applications, and designed strategies have been critically summarized. First, the fundamentals of solid state emitters have been briefly summarized by considering their molecular design strategies. Secondly, a short summary has been made of those solid state ESIPT emitters which were reported before 2011 and which are highlighted by Park et al.3 Thirdly, the innovative concepts of solid state emitters reported in the last five years have been discussed.3 In the last part, the conclusion and prospects for developing novel solid state emitters for high-tech applications have been summarized.
2. Scope and limitations
In this article, a critical review of the recent literature relevant to solid state emitters based on the ESIPT mechanism has been presented. The strategies for molecular design, absorption, emission properties, quantum efficiencies, fluorescence lifetime, and applicability of the ESIPT solid state emitters have been summarized and discussed. The structure-photophysics relation and application of the ESIPT fluorophores have been highlighted with the help of representative examples.More than 4018 research articles related to ESIPT have been published by researchers since 1950s. Synthesis methods and photophysical properties of the ESIPT chromophores in solution have been well summarized in the literature in the form of review articles.2,3,8,9,64 In 2011, Park et al. summarized some strategies to achieve solid state emission for ESIPT chromophores by discussing representative examples.3 Afterward many new strategies and solid state ESIPT chromophores have been developed but not yet summarized to assist researchers in overcoming existing shortcomings in the practical implementation of ESIPT systems into actual optoelectronic devices. In order to have recent updates on strategies for designing novel ESIPT solid state emitters and considering our research background in the field of ESIPT,24,27,28,31,32,37,72–75 here we have critically reviewed the papers which were published in these 5 years. In this review, we have mainly focused on solid state emitters and their properties in the solid-state. References such as patents, abstracts, and summaries of the conference presentations are not included in this review.
3. Solid state ESIPT emitters
Most of the reported organic chromophores are highly emissive in solution with high fluorescence quantum efficiencies, but weakly emissive (or non-fluorescent) in a rigid medium due to the aggregation-caused quenching mechanism (ACQ).76 To achieve solid state emission, it is necessary to suppress the other radiationless deactivation pathways which occur at the excited state. Different strategies3,70,77–80 were exploited to obtain solid state emissions for various types of ESIPT chromophores such as (i) controlling the mode of molecular packing, (ii) aggregation-induced emission (AIE), (iii) aggregation-induced enhanced emission (AIEE), (iv) restriction of intramolecular rotation (RIR) etc. In addition to the above mentioned strategies for solid state emission, various new strategies have been reported by researchers recently. It is well known that ESIPT chromophores emit with multiple bands with a high Stokes shift which covers a broad range of spectrum.2,3,8 In most of the ESIPT chromophores, fluorescence quantum efficiencies of the large Stokes shifted ESIPT emission (K*) are generally low due to non-radiative deactivation pathways occurring at the K* state.2,3,8 ESIPT chromophores which emit at the red region with very high fluorescence quantum efficiencies in the solid state are attracting considerable attention in functional materials. Approaches such as dendrimer encapsulation, aggregation-induced emission (AIE), aggregation-induced enhanced emission (AIEE), the restriction of intramolecular rotation (RIR), polymer doping, and the restriction of twisted intramolecular charge transfer (RTICT) mechanisms have been considered responsible for solid state emission and the enhancement of quantum efficiencies. Various strategies of ESIPT emission in solvent versus solid state (crystalline state/polymorphic form/supported rigid medium) emission have been summarized below.(1) Hydrogen bonding: intramolecular H-bonding is a key factor for an efficient ESIPT process because the proton transfer process takes place through a characteristic four-level photocycle (5- or 6-membered cyclic transition state).3,8,9 In polar solvents, in addition to intramolecular H-bonding, intermolecular H-bonding between the acidic proton of chromophores (hydroxy/amino groups of ESIPT chromophores) and solvents molecules is possible.20 Polar solvents (alcohols) acting only as hydrogen bond acceptors can hamper the ESIPT process by the formation of intermolecular hydrogen bonding with solvent molecules, which results in the suppression of keto (K*) emission.81 In the solid state, hampering of the ESIPT process through intermolecular H-bonding with solvents molecules is omitted which causes the enhancement of K* emission in the solid state.
(2) Restriction of twisted intramolecular charge transfer: a twisted intramolecular charge transfer (TICT) process in the excited keto state (K*) after ESIPT is one of the pathways for fluorescence quenching of ESIPT fluorescence in solution.82,83 TICT is significantly influenced by the geometrical properties of the intramolecular H-bond associated with the basic atom (–N) of the ring structure.84 TICT and strength of the intramolecular H-bond are co-operative in the Keto (K*) state of ESIPT molecules, which determines the emission properties.3 TICT deactivation is prominent in solution, which causes the suppression of keto (K*) emission. However, in rigid media keto (K*) emission is significant because of the restriction of the TICT (rigidochromism).3,85
(3) Restriction of the intramolecular rotation: restriction of the intramolecular rotation (RIR) is also a key factor in the enhancement of fluorescence quantum efficiencies in solution and rigid media of ESIPT fluorophores.3,26,86,87 RIR effectively suppresses the non-radiative pathways via vibronic and rotational relaxation. RIR is more efficient in the solid state in comparison to solvent due to physical constraints in the solid state (restriction of free rotations).3,26,86,87 The RIR process helps in maintaining suitable geometry for intramolecular H-bonds as well as in suppressing the non-radiative pathways by means of intermolecular vibronic relaxation (except few cases).3,26,86,87
(4) Restriction of cis–trans isomerization: cis–trans isomerization of the ESIPT keto form deactivates the excited state which causes fluorescence quenching in solution.84,88–90 However, in rigid media cis–trans transformation of the excited state keto form is restricted through tight molecular packing.84,88–90 This conformational restriction significantly blocks the non-radiative relaxation pathways and fascinates the radiative decay which results in the enhancement of fluorescence in the solid state.84,88–90
(5) Aggregation effect: the limited vibrational/rotational relaxations are considered to be the origin of fluorescence emission in the solid sate over solution. The type of aggregation and characteristics of molecular stacking determine the fluorescence quantum efficiencies of ESIPT chromophores.3 Generally, J-aggregation (slip-stacking/head-to-tail interaction) enhances the fluorescence emission over H-aggregation (face-to-face interaction) for ESIPTs.91,92 The stacking of ESIPT fluorophores depends on the nature of group present on the ESIPT molecules. In common, steric/bulky group's causes slip-stacking i.e. J-aggregation.
(6) Torsion/planarity of enol conformers: the stability of enol conformers depends on torsion between the proton donor ring and the acceptor ring.93–95 In the solid state torsion/twisting is less in comparison to solution because of physical constraints. In most of the cases torsion is coplanar in the solid state, which is the origin for the enhancement of fluorescence in the crystalline state. However, in solution efficient radiationless decay processes occur due to twisting motion.93–95 The torsion is dependent on molecular motion/intermolecular interaction with neighbouring molecules/packing-induced conformational fixation and/or alteration at a single molecular level. The mode of molecular packing determines the ESIPT emission.
Based on these approaches, recently developed ESIPT fluorophores, photophysical properties and their applications have been summarized in detail in the next section.
4. Recent progress in the solid state ESIPT emitters
In this section, recently developed strategies to suppress the deactivation pathways occurring at the excited state are summarized. Various fluorophores and approaches to improve fluorescence quantum efficiencies in the solid state, which have been used by researchers, are highlighted below and details of photophysical properties and applications are discussed in the next section.(a) White light generation from single molecules through the RIR mechanism18,33,66,90,96,97
(b) Aggregation-induced emission92,98–100
(c) Aggregation-induced enhanced emission84,85
(d) Polymer based ESIPT chromophores101
(e) ESIPT chromophores with polymorphic properties55,94,95,99,102–104
(f) ESIPT-coupled aggregation-induced enhanced emission105
These various approaches have been broadly classified into three main sections for simplicity
(i) Solid state ESIPT emitters through single molecules
(ii) Solid state ESIPT emitters based on the polymorphic form (molecular packing)
(iii) Solid state ESIPT emitters through AIE/AIEE processes
The reported solid-state ESIPT fluorophores have been mainly used as organic light emitting diodes (OLEDs)/white light organic limiting diodes (WOLEDs). The various approaches for the generation of white-light emission using ESIPT chromophores have been highlighted below and detailed discussion is covered in the next section.
(a) White-light-emission (WLE) through a single molecule dyad, consisting of two non interacting chromophores with ESIPT units:87 the two interactive chromophores having blue and orange-emitting ESIPT emission have been used for white-light generation. In this approach, the concept of the frustrated energy transfer strategy between two units was used for the generation of WLE. The generated white-light emission is the sum of emissions from the individual components. The WLE by this approach is independent of concentration.
(b) Mixture of proton-transfer and nonproton-transfer chromophores (ESIPT and non-ESIPT):19 ESIPT and non-ESIPT chromophores having similar UV-absorption properties but different luminescence properties have been used. Mixture of ESIPT chromophores with yellow emission and non-ESIPT chromophores with blue emission generates white light by simultaneous irradiation of UV light. The white luminescence composes of two independent emissions without involving energy transfer processes. The use of compounds with similar molecular structures is more advantageous, because these compounds are expected to have similar physical properties.
(c) Mixture of three chromophores (two ESIPT and one non-ESIPT):17 white light generation by mixing three chromophores having yellow, red and blue emission. In this approach, proton transfer molecules with yellow and red emission and non-proton transfer molecules with blue emission have been used.
(d) Based on solvatochromism properties (solvent polarity and ability for forming H-bonds):66 the emission properties of the ESIPT chromophores are sensitive towards microenvironments.106 This desired characteristic of ESIPT chromophores helps in tuning the fluorescence emission. Intra-and intermolecular H-bonding in the ESIPT process generates different rotameric isomers/transition species (cis-enol, cis-keto/trans-keto/deprotonated species etc.) in non-polar, polar and polar-protic media, which emit at different wavelengths resulting in white light emission.
(e) Mixture of nanodots and ESIPT chromophores:107 a mixture of quantum dots (blue emission) and keto emission of ESIPT chromophores generates white-light emission. In this approach, emission tunability could be achieved simply by changing the relative amount of the two species.
(f) Tuning of acidic and basic centers of ESIPT chromophores:108 the emission properties of the ESIPT chromophores are dependent on the electron nature and the position of donor/acceptor groups. White-light emission could be generated by tuning the position and strength of donors/acceptors. These groups affect the acidity and basicity of donor and acceptor groups responsible for ESIPT due to the mesomeric effect.
(g) Altering the surrounding medium (e.g. polymer matrices or rigid media):57,109 homogenous dispersion of two or more ESIPT chromophores (different Stokes shift) with different emission wavelengths in a polymer matrix generates white light emission. Poly(N-vinylcarbazole) (PVK) as a blue-light emitting and hole-transporting polymer host material has been used for white light emitting electroluminescence. This approach is mainly based on the principle of limited energy transfer between different ESIPT chromophores.
(h) Based on the halochromism properties of ESIPT chromophores:101 ESIPT emission is highly sensitive towards acidic and basic media. In an acidic medium, protonation of imine nitrogen causes a blue shift and in a basic medium deprotonation of acidic hydrogen generates phenoxide ions which causes a red shift. This helps in tuning the emission properties which generate white light emission by controlling the acidity and basicity of the medium.
(i) Controlling the molecular packing of ESIPT chromophores (e.g. polymorphs):55,56 the emission properties of the ESIPT chromophores are tunable by controlling the mode of molecular packing. The difference in the modes of molecular packing affects the electron state of the proton transfer species and results in different luminescence color.
(j) AIE and AIEE processes generate white light emission of ESIPT chromophores:105 in the aggregate state, a blue or a red shift of enol (E*) and keto (K*) emission or the appearance of new emission bands results in white-light emission.
(k) Single ESIPT system18: Fine tuning the energetics of ESIPT (systematic balance between the destruction of aromaticity and the compensation of π-conjugation).
(l) Panchromatic emission97via modulating the substitution patterns of ESIPT chromophores.
5. Solid state ESIPT emitters through single molecules
Generation of white light through a mixture of two or more organic chromophores is very simple and more convenient over white light from single/individual molecules. However it suffers from several limitations.110,111 Long term color stability, phase separation and differential aging are some of the limitations of white light generated from a mixture of chromophores. White light generation from single molecules112,113 would be more ideal, and it gives perfect color stability and easy reproducibility in the fabrication of white light organic light emitting diodes (WOLEDs). ESIPT is well known for dual emission or broad emission; these unique properties can be utilized for the generation of white light by tuning the microenvironment.66,106 ESIPT chromophores generally absorb in the ultra-violet region and emit in the visible region. Enol (E*) and keto (K*) emission cover the white light spectrum. Park et al. reviewed some ESIPT chromophores which generate white light and are emissive in the solid state.3 In this section we have summarized solid state ESIPT emitters reported in the last five years.In early 2011, Wang and coworkers reported full color emitting organic materials based on 2-(2′-hydroxyphenyl)benzothiazole (BTZ) with superior ESIPT characteristics.33 Different BTZ chromophores were synthesized by modifying the 4- and 5-positions of the phenyl ring attached to the thiazole ring with different substituted amino groups. Studied BTZ chromophores are summarized below (Scheme 1).
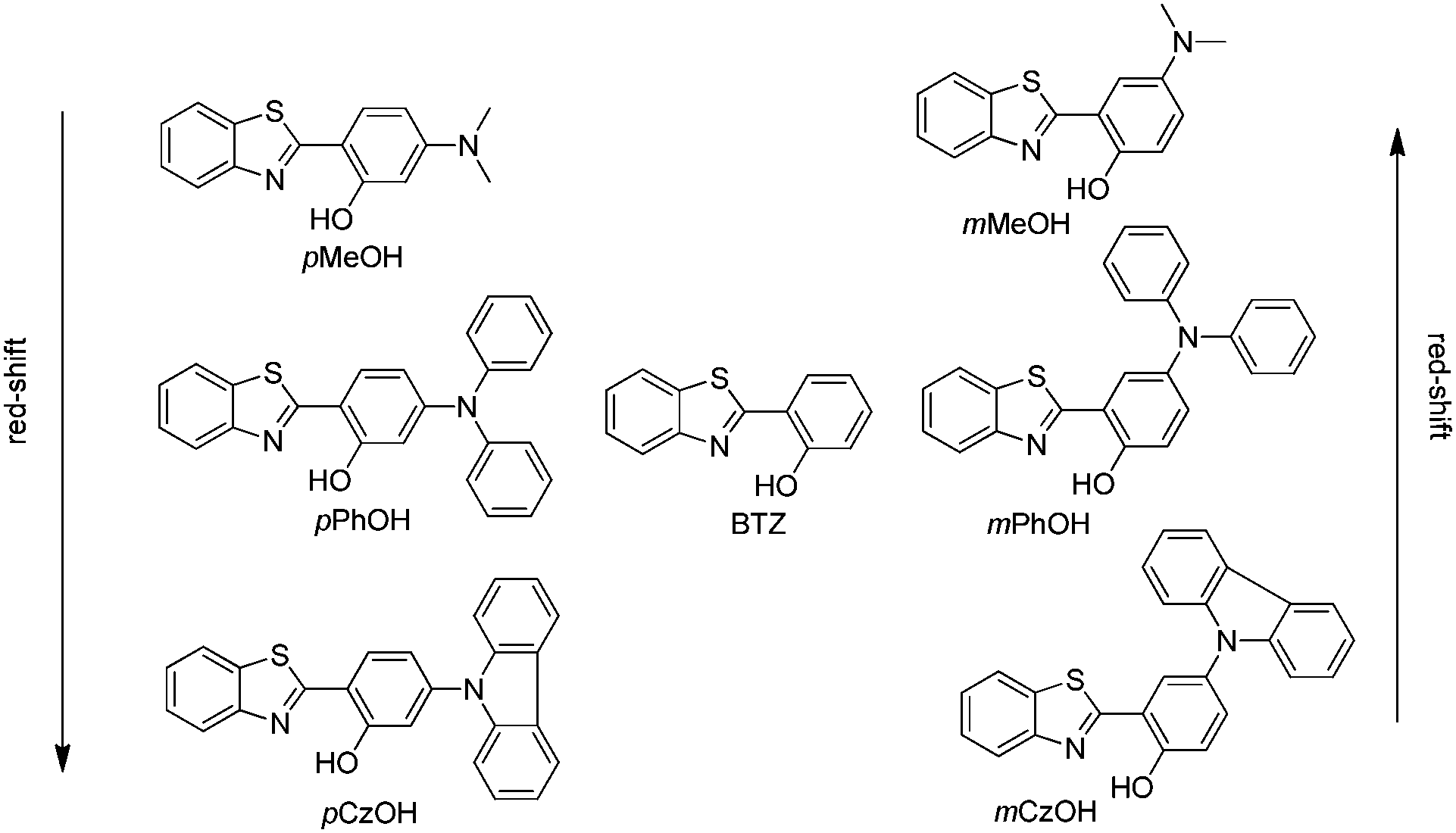 | ||
| Scheme 1 Structure of BTZs. | ||
In BTZ chromophores, the benzothiazole unit acts as an electron acceptor and the substituted amino group as an electron donor. The emission spectra and fluorescence quantum efficiencies of m-OH and p-OH BTZ chromophores were reported in the solid state. In the solid state, the emission spectra of m-substituted groups showed a red shift in comparison to the p-substituted amino group [m-MeOH (λem = 630 nm, ΦFL = 5.65%), m-PhOH (λem = 600 nm, ΦFL = 37.54%), m-CzOH (λem = 532 nm, ΦFL = 66.86%), p-MeOH (λem = 443 nm, ΦFL = 29.07%), p-PhOH (λem = 461 nm, ΦFL = 30.16%), p-CzOH (λem = 502 nm, ΦFL = 91.68%)]. In the studied BTZ chromophores, the emission properties are dependent on the position and nature of the electron donor, the m-OH type of BTZ showed blue shifted emission, while the p-OH type of BTZ showed red shifted emission with increasing electron-donating ability of the substituted amino group. The solid state emission spectra cover the entire visible light region and are highly emissive in the solid state Fig. 2.
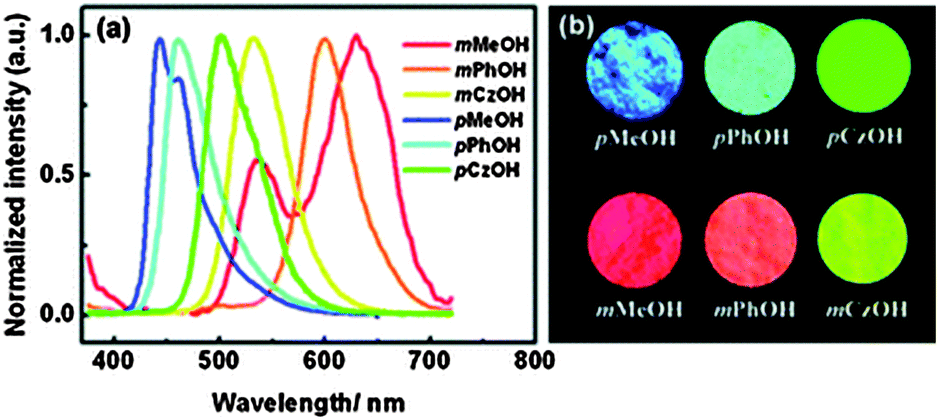 | ||
| Fig. 2 (a) Solid state emission spectra of BTZ derivatives, (b) photoluminescence image of BTZ derivatives. Reproduced with permission.33 Copyright 2011 The Royal Society of Chemistry. | ||
The single-crystal X-ray analysis showed that the HBT unit of m-OH and p-OH types of chromophores exhibits planar nature Fig. 3 (suitable for ESIPT) with a small dihedral angle between HBT and amino substituted groups. The absence of intermolecular interaction helps to suppress the non-radiative pathways resulting in solid state emission Fig. 3. The high fluorescence quantum efficiencies were considered owing to the favorable geometry for ESIPT and strong intra-molecular hydrogen bonding instead of intermolecular interactions. This is the first report which describes extremely high quantum efficiencies reaching up to ∼92% in their solid state for single ESIPT molecules.
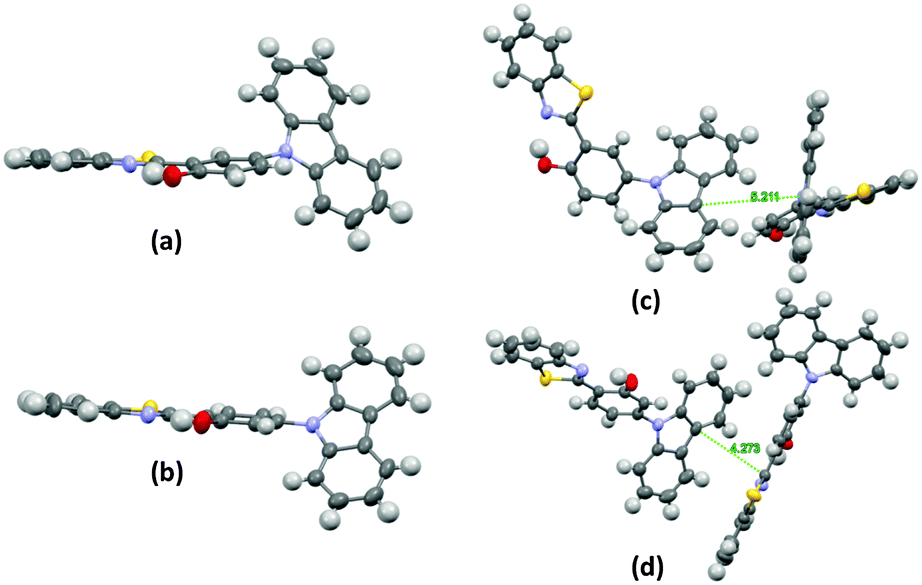 | ||
| Fig. 3 Single crystal of m-CzOH and p-CzOH (a) m-CzOH and (b) p-CzOH molecular structure with thermal ellipsoids drawn at the 50% probability level; (c) m-CzOH and (d) p-CzOH intermolecular relationship. Reproduced from CIF file.33 | ||
The electroluminescence (EL) devices were fabricated for each BTZ derivatives to evaluate EL characteristics. BTZ derivatives (m-OH and p-OH) were used as emitting materials for device fabrication. 4,4-Bis[N-(1-naphthyl)-N-phenyl-amino]biphenyl (NPB) was used as a hole-transporting material, and 1,3,5-tri(1-phenyl-1H-benzo[d]imidazol-2-yl)phenyl (TPBI) as both the electron-transporting and hole-blocking layer. m-CzOH showed good EL properties with device configuration [ITO/NPD (15)/m-CzOH (5 nm)/TPBI (75 nm)/LiF (0.5 nm)/Al (200 nm)] in comparison to other BTZ derivatives. The m-CzOH based device reached a maximum brightness of 7866 cd m−2 at 14.5 V and a maximum current efficiency of 2.80 cd A−1 at 6.5 V. Wang et al. concluded that device performance depends on the thickness of emitting layers.
Recently, Chou and colleagues have reported the hydroxy analogues of green fluorescent protein (GFP) chromophores90 (Scheme 2). GFP contains (Z)-4-(4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one (HBDI) chromophores in which emission properties are tunable by changing the substitution pattern at the C(1) position. Synthesis, photophysical and luminescent properties of the seven o-hydroxy analogues of GFP which are solid state emitters have been summarized by Chou et al.
 | ||
| Scheme 2 Hydroxy analogues of GFP. | ||
HBDI exhibits two possible conformers (o-HBDI-Z form and o-HBDI-E forms) due to exo-C(5)–C(4)–C(3) bonds. Compounds 1a–1g exist mainly in Z-conformers in nonpolar solvents and the solid state. This conformation leads to a seven membered-ring-hydrogen bond, from which the ESIPT takes place, instead of an eight-member-hydrogen bond transfer process via E-conformers. All studied compounds have the same core and differ in substitution at the C(9) position.
The photophysical properties of o-HBDI-Z are dependent on substitution patterns as well as solvents. Electron donor groups at the C(9) position of HBDI showed a significant red shift in absorption and emission spectra in comparison to electron acceptor groups at C(9) of HBDI in solution and the solid state.
In a nonpolar solvent (cyclohexane), compounds showed red shifted emission over the polar solvent (acetonitrile) which indicates that ESIPT emission is more favorable in a nonpolar solvent for studied compounds. In the solid state, due to the restriction of exo-C(5)–C(4)–C(3) bond rotation, intense tautomer emission (keto emission) with high quantum yields (ΦFL = 9–90%) was observed for 1a–1g. However, the restriction of exo-C(5)–C(4)–C(3) bond rotation is not significant for compounds containing electron donors at the C(9) position which causes lowering of quantum efficiencies (1f: ΦFL = 85%, τ = 7.4 ns; 1e: ΦFL = 84%, τ = 7.1 ns; 1d: ΦFL = 40%, τ = 4.3 ns; 1c: ΦFL = 59%, τ = 5.9 ns; 1b: ΦFL = 19%, τ = 3.7 ns; 1a: ΦFL = 9%, τ = 5.9 ns). The significant enhancement of fluorescence quantum efficiency (10 fold; ΦFL = 90%, τ = 10.2 ns) was observed for electron acceptor nitro groups in comparison to phenyl groups at the C(9) position. Tautomer emission (keto*) spectra of compounds in the solid state cover the visible region Fig. 4. Chromophores 1b, 1d and 1f are highly emissive in the solid state (Fig. 5) in comparison with other studied analogues.
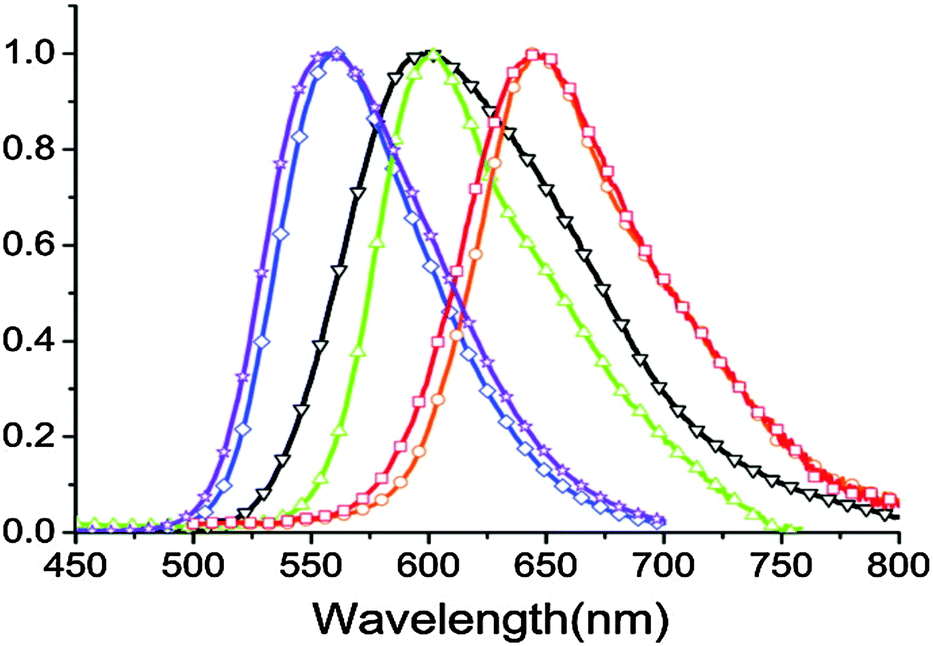 | ||
| Fig. 4 Solid state emission spectra of compounds 1a–1f (1a – red, 1b – orange, 1c – green, 1d – black, 1e – blue, 1f – purple). Reproduced with permission90 Copyright 2011 American Chemical Society. | ||
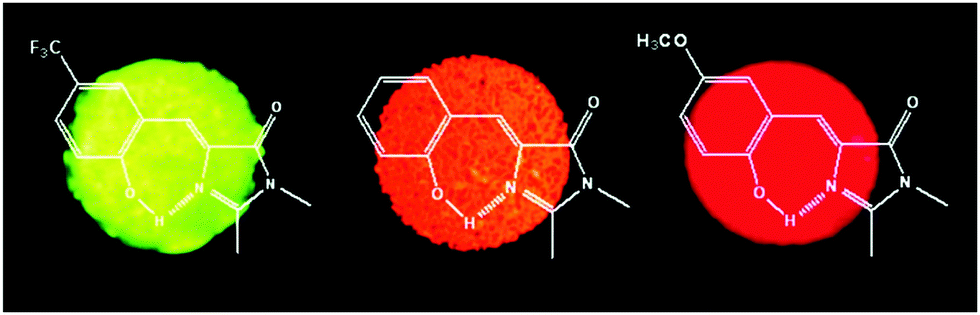 | ||
| Fig. 5 Solid-state fluorescence of compounds 1b, 1d and 1f. Reproduced with permission.90 Copyright 2011 American Chemical Society. | ||
The applicability of o-HBDI-Z as emitters in electroluminescence was tested by fabricating an OLED device. GFP analogue 1d was used as an emitting material for device fabrication. 4,4′-N,N-Dicarbonyl-1,1′-biphenyl (CBP) was used as a host, conducting polymer poly(ethylene dioxythiophene)/poly(styrene sulfonate) (PE-DOT:PSS) was used as the hole-injection layer, 4,4-bis[N-(1-naphthyl)-N-phenyl-amino]biphenyl (NPB), and 4,4′,4′′-tri(N-carbazolyl)triphenylamine (TCTA) were used as hole-transport layers, 1,3,5-tri(1-phenyl-1H-benzo[d]imidazol-2-yl)phenyl (TPBI) was used as both the electron-transporting and hole-blocking layer, LiF was used as an electron-injection layer and Al as a cathode. The device configuration was [ITO/PE-DOT:PSS (30 nm)/NPB (20 nm)/TCTA (5 nm)/CBP: 1d (5 wt%, 25 nm)/TPBI (50 nm)/LiF (0.5 nm)/Al (100 nm)]. The device showed a maximum brightness of 5790 cd m−2 at 17.5 V with CIE coordinates (0.55, 0.43) and a maximum external efficiency of 0.40% at 0.82 cd A−1 and a power efficiency of 0.24 lm W−1.
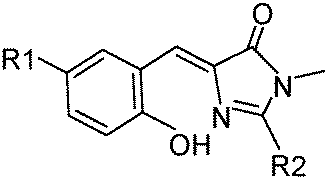
7-Hydroxy-1-indanone I based ESIPT molecules exhibiting good fluorescence quantum efficiencies in the crystalline form and in solution were reported by the Chou group18 (Scheme 3). Two ESIPT molecules II and III showing dual emission were synthesized by the chemical modification of 7-hydroxy-1-indanone at C(2)–C(3) positions.
 | ||
| Scheme 3 7-Hydroxy-1-indanone based ESIPT molecules. | ||
Chromophores I, II and III contain a hydroxyl group as a proton donor and a carbonyl as an acceptor with suitable geometry for the ESIPT process. Indene I showed long wavelength emission (λex = 315 nm, λem = 530 nm; Δλ = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1; ΦFL = 1.2%, τ = 1.0 ns) in cyclohexane, while it is non-fluorescent in the solid state. Dual emissions were reported for II (λem = 468 nm; ΦFL = 1.9%; τ = 1.2 ns and λem = 535 nm; ΦFL = 1.4%; τ = 1.2 ns) and III (λem = 450 nm; ΦFL = 0.6%; τ = 0.67 ns and λem = 621 nm; ΦFL = 2.0%; τ = 0.66 ns) in cyclohexane. However, only III showed dual emission in the solid state which is blue shifted emission (λem = 435 nm and 580 nm; overall ΦFL = 10%) in comparison to emission in cyclohexane. The blue shift was assigned to π–π stacking in between the tetracyclic planes resulting in H-aggregation Fig. 6.
000 cm−1; ΦFL = 1.2%, τ = 1.0 ns) in cyclohexane, while it is non-fluorescent in the solid state. Dual emissions were reported for II (λem = 468 nm; ΦFL = 1.9%; τ = 1.2 ns and λem = 535 nm; ΦFL = 1.4%; τ = 1.2 ns) and III (λem = 450 nm; ΦFL = 0.6%; τ = 0.67 ns and λem = 621 nm; ΦFL = 2.0%; τ = 0.66 ns) in cyclohexane. However, only III showed dual emission in the solid state which is blue shifted emission (λem = 435 nm and 580 nm; overall ΦFL = 10%) in comparison to emission in cyclohexane. The blue shift was assigned to π–π stacking in between the tetracyclic planes resulting in H-aggregation Fig. 6.
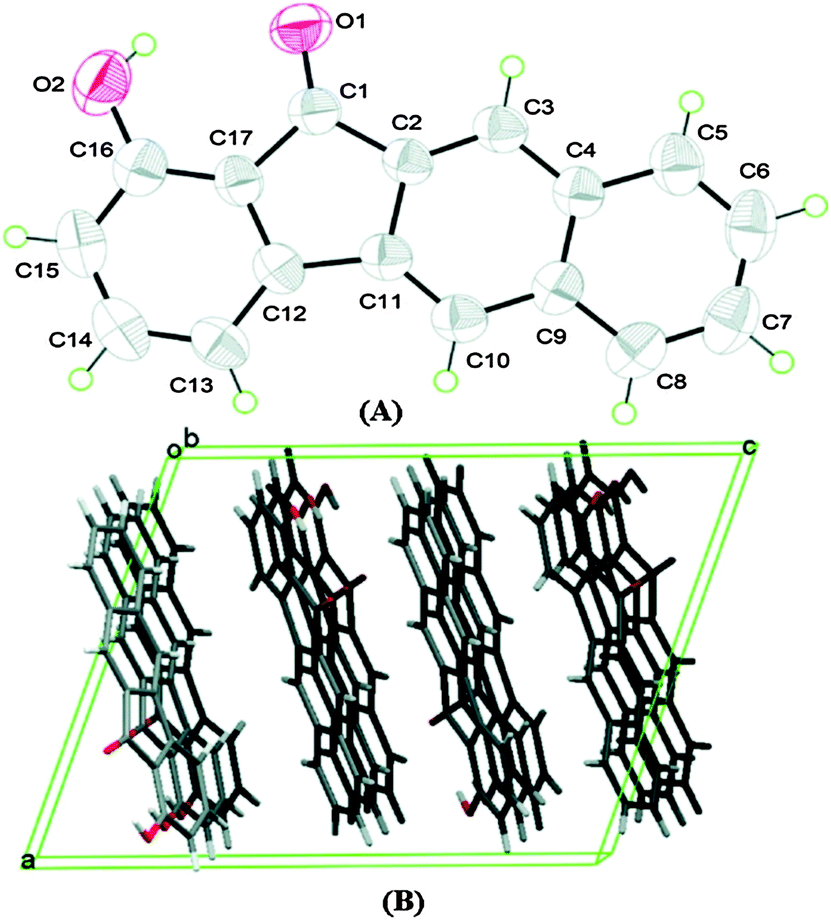 | ||
| Fig. 6 (A) Molecular structure of III with thermal ellipsoids drawn at the 50% probability level. (B) Packing view of III, viewed along the b axis. Reproduced with permission.18 Copyright 2011 American Chemical Society. | ||
In the solid state, dual emission of III covers the entire white light spectrum with CIE (x, y) = (0.30, 0.27). The electroluminescent properties of indene III were investigated using a multi-layer OLED device structure with PE-DOT:PSS as a hole-injection layer, a NPB hole transport layer, TCTA was used to decrease the barrier between NPB and emitting layer. CBP was used as host for the emitter III. TPBI was used as both the hole-blocking and electron-transporting layer. An EL device was fabricated with ITO/PE-DOT:PSS-(30 nm)/NPB (15 nm)/TCTS (5 nm)/5 wt% III doped CBP (25 nm)/TPBI (50 nm)/LiF (0.5 nm)/Al (100 nm) configuration. The maximum external quantum efficiency, current and power efficiency were 0.11%, 0.20 cd A−1 and 0.06 lm W−1 respectively with the CIE coordinates of (x, y) = (0.26, 0.35). The effect of voltage on EL spectra was also studied by authors. The EL spectra were constant when applied voltage was below 16 V (x, y) = (0.26, 0.35), however a slight shift in CIE coordinates was observed at 20 V (x, y) = (0.28, 0.40) Fig. 7. This color shift was due to energy transfer from the normal excited state to the tautomer excited state at higher voltage. The EL spectra of devices incorporating III as a dopant at different voltages are shown in Fig. 7.
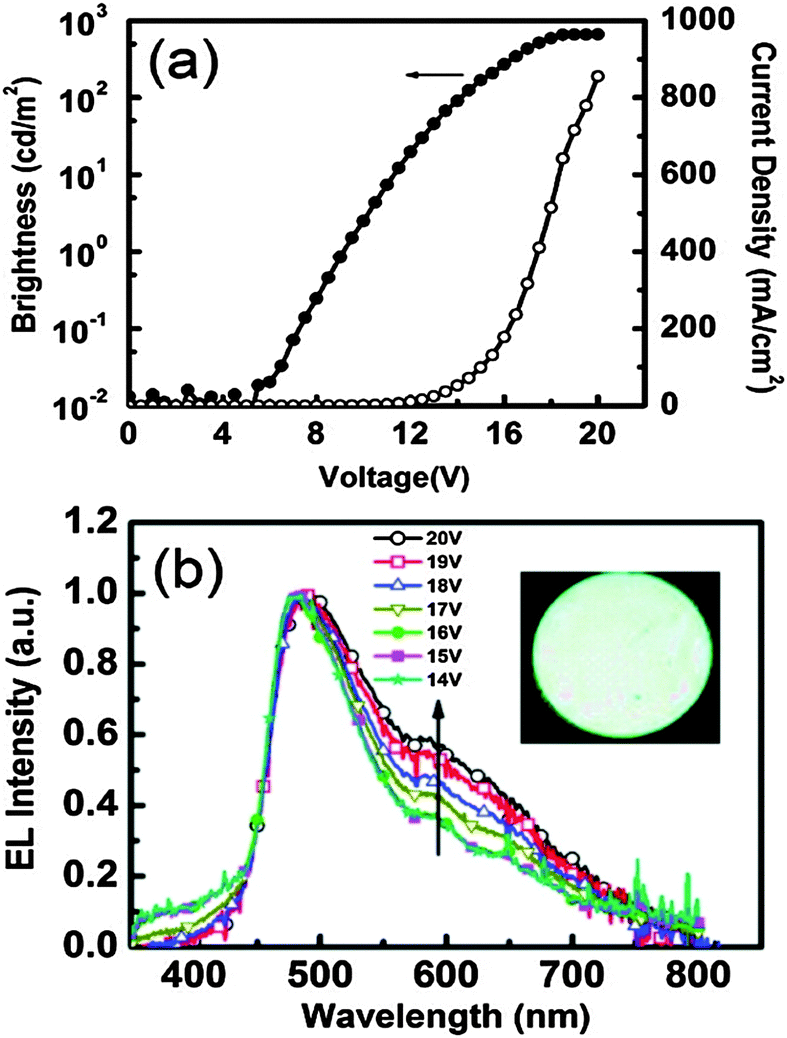 | ||
| Fig. 7 (a) Current density–voltage–luminance (J–V–L) characteristics. (b) EL spectra for devices incorporating III as a dopant at different voltages. Inset: Photo of the device in operation. Reproduced with permission.18 Copyright 2011 American Chemical Society. | ||
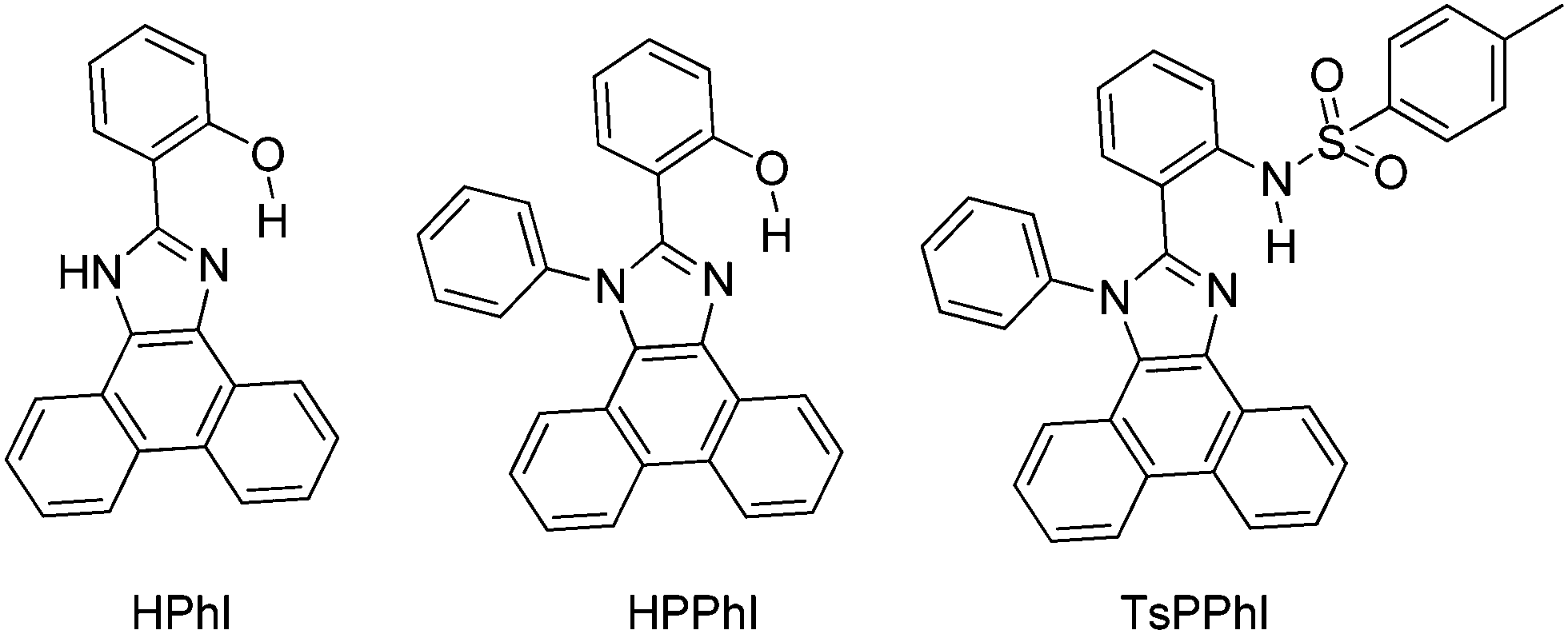
Synthesis and optical properties of aryl and di-aryl substituted phenanthroimidazoles (Scheme 4) were reported by Manoj Kumar Nayak in 2012.114 High fluorescence quantum efficiencies in the solid state (ΦFL = 39–68%) were reported for synthesized chromophores.
 | ||
| Scheme 4 Aryl and di-aryl substituted phenanthroimidazoles. | ||
In the design of the phenanthroimidazoles, twisted phenyl groups were purposefully incorporated on central imidazole nitrogen to explore the steric effect and the bond rotation effect on the ESIPT process and photophysical properties. Similar types of chromophores, which are solid state emitters, were reported by Park et al. in 2005 for optoelectronic applications.26
In the solid state (film) high fluorescence quantum efficiencies (ΦFL = 39–68%) were reported in comparison to quantum efficiencies (ΦFL = 0.7–49%) in solution. From theoretical calculation using DFT, it was noted that 2-(1H-phenanthro[9,10-d]imidazol-2-yl)phenol (HPhI) is planar, while 2-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol (HPPhI) and 4-methyl-N-(2-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)benzenesulfonamide (TsPPhI) are non-planar. The torsion angles between imidazole and the 2-phenyl ring are slightly twisted, whereas the phenyl ring attached to imidazole nitrogen is almost perpendicular to the chromophore plane. This twisting causes the prevention of direct π–π stacking of chromophores and maintaining a proper intermolecular distance for ESIPT, which effectively suppresses non-radiative decay. TsPPhI showed very high fluorescence quantum efficiency with bright emission in the blue-green region at room temperature. This could be due to slip-stacked packing present in molecules instead of complete π–π stacking. The free rotation occurring in solution for HPPhI and TsPPhI is restricted due to aggregation which results in the enhancement of fluorescence efficiencies. Park et al. have already concluded that the incorporation of a steric group on the ESIPT chromophore would improve the emission properties by suppressing other non-radiative pathways.26
Subsequently, Ziessel et al. have reported N-alkylated-2-(2-hydroxyphenyl)benzimidazole (HBI) and N-arylated-9,10-phenanthroimidazole (HPI) ESIPT chromophores (Scheme 5) and their borate complexes.21 Here we have considered only ESIPT chromophores instead of borate complexes. All studied compounds showed absorption in the ultra-violet region with a high absorption coefficient due to the π–π* transition of polyaromatics and emission in the near-visible or visible region. The fluorescence quantum efficiencies of the chromophores in the solid state were measured in KBr pellets. HPI3 showed high quantum efficiency in comparison to other HPI and HBI chromophores (Table 1).
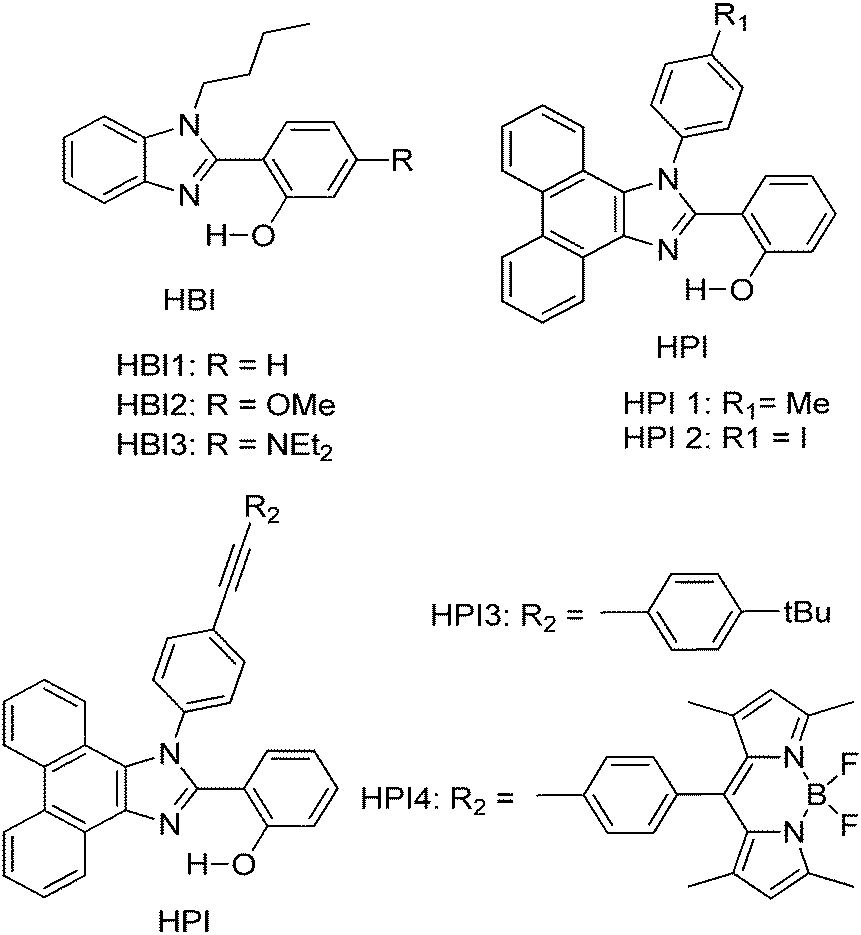 | ||
| Scheme 5 N-Alkylated-2-(2-hydroxyphenyl)benzimidazole (HBI) and N-arylated-9,10-phenanthroimidazoles (HPI) ESIPT chromophores. | ||
| Comps | λ abs (nm) | λ em (nm) | Δλ (cm−1) | Φ FL (%) | τ (ns) |
|---|---|---|---|---|---|
| Nr: not reported. | |||||
| HPI4 | 370 | 473 | 5900 | Nr | 0.3 |
| HPI3 | 374 | 474 | 5600 | 28 | 3.2 |
| HPI1 | 379 | 473 | 5200 | 12 | 1.6 |
| HBI3 | 371 | 447 | 4600 | 4 | 0.9 |
| HBI2 | 333 | 465 | 8500 | 3 | 2.9 |
| HBI1 | 372 | 415 | 2800 | 4 | 2.9 |
From crystallographic analysis of HPI1, it was found that the phenol group is planar with a HPI core, while an aryl (tolyl) group is almost perpendicular to the HPI core Fig. 8. This planar nature with a suitable bond distance and geometry enhances the ESIPT process. Aryl/alkyl groups restrict the intramolecular rotation which helps to achieve solid state emission and enhancement of quantum efficiencies.
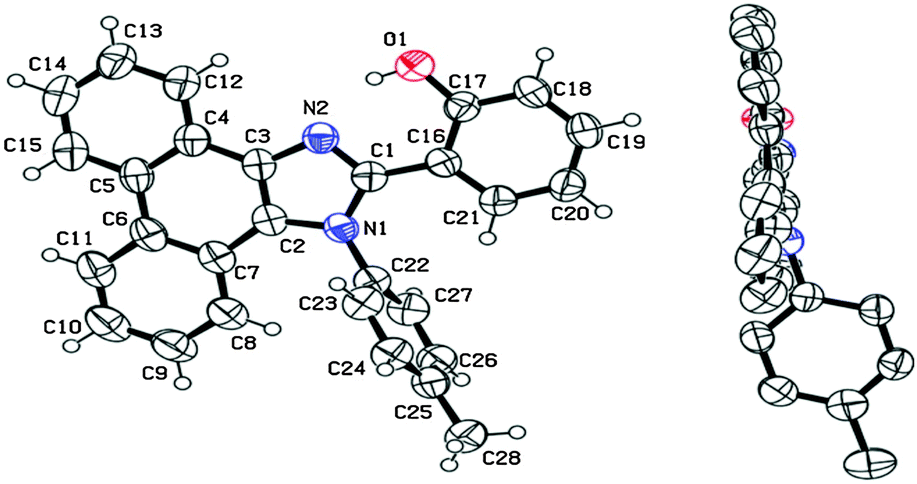 | ||
| Fig. 8 X-Ray single crystal data of HPI1. Reproduced with permission.21 Copyright 2013 American Chemical Society. | ||
Thanikachalam and coworkers have reported HPI (Scheme 6) based solid state chromophores115 similar to HPI derivatives reported by Ziessel et al.21 and Park et al.25
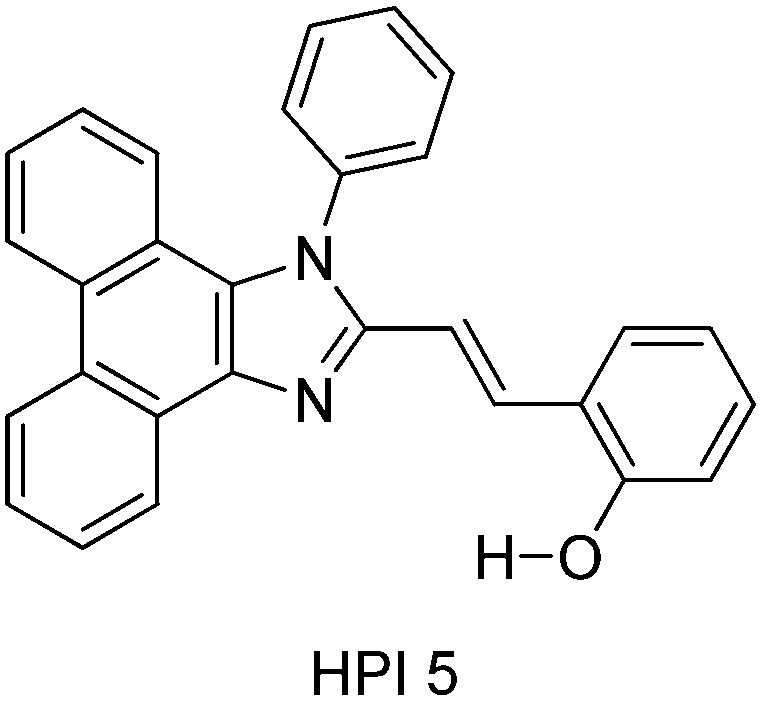 | ||
| Scheme 6 Structure of HPI5. | ||
In HPI5, ESIPT occurs through the eight membered cyclic transition state, instead of the common five and six membered proton transfer process. The chromophore showed 63% fluorescence quantum efficiency in the solid film, which is higher than the fluorescence quantum efficiency in the solvent (51%). The high fluorescence quantum efficiency in the solid state was assigned to π–π staking and the steric effect of phenyl groups in the solid state.
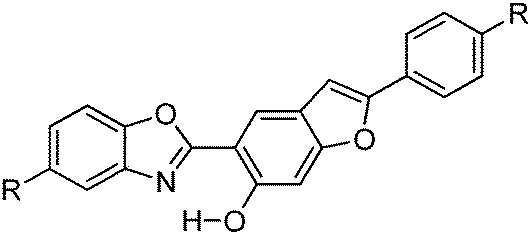
Recently, white light emitters were reported by the Ziessel group108 by tuning the 2-(2′-hydroxybenzofuran)benzoxazole (HBBO) ESIPT unit (Scheme 7). Different electron donor (–NBu2, –OMe and –tBu) and acceptor (–CF3, –CN, –CO2Me) groups were incorporated on the fused hydroxybenzofuran system B1–B7 to achieve white light emission.
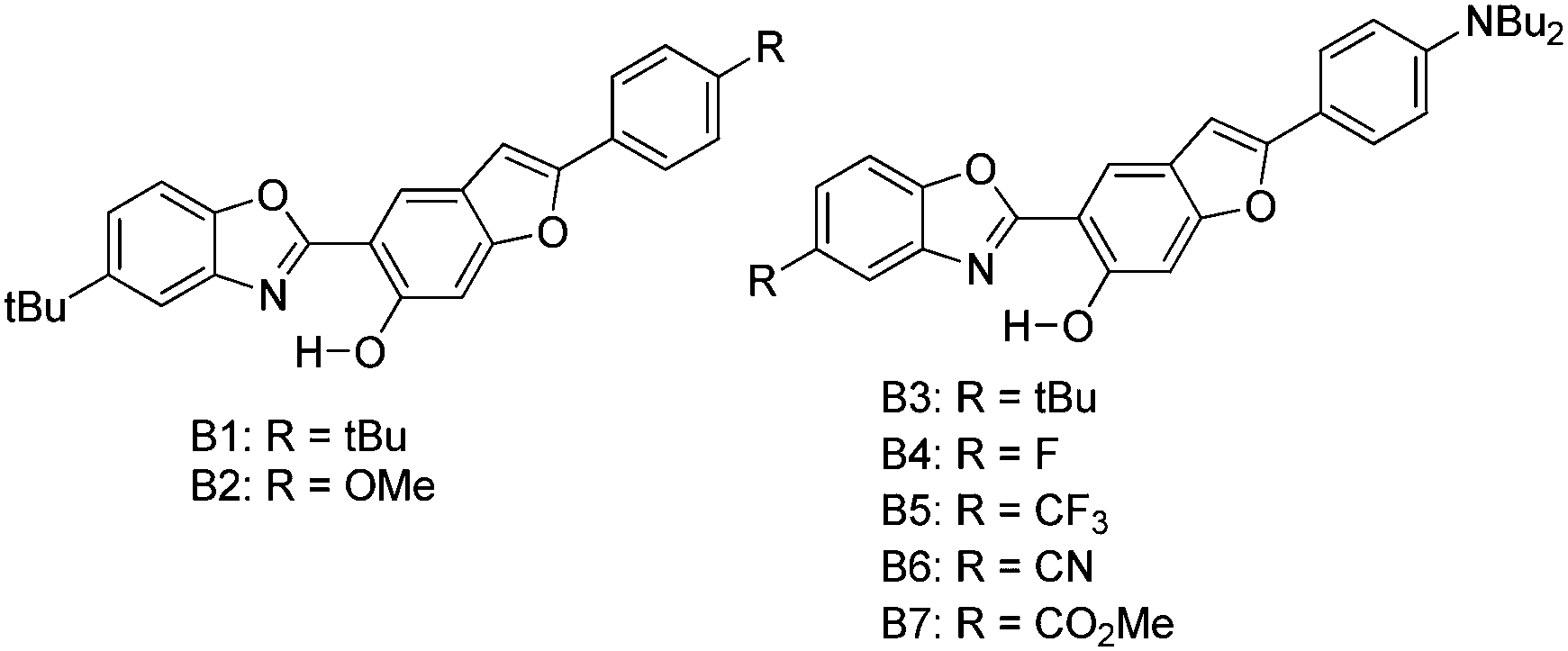 | ||
| Scheme 7 2-(2′-Hydroxybenzofuran)benzoxazole (HBBO) ESIPT derivatives. | ||
All chromophores showed similar absorption spectra in solution between 358 and 396 nm. The absorption spectra are weakly dependent on substitution patterns present on the aromatic system. Intense single or dual emission bands located between 407 and 588 nm with quantum efficiencies up to 25% were reported in different solvents. The fluorescence intensity of the compounds depends on electro-negativity of attracting groups present on the hydroxybenzofuran core. The photophysical properties have been studied in the solid state as dispersed in KBr matrix pellets and polymethyl methacrylate/polystyrene films. Dual emissions (E* and K*) were observed for B3–B7 chromophores in rigid media, which are identical to the emission spectra observed in solution for most of the chromophores Fig. 9.
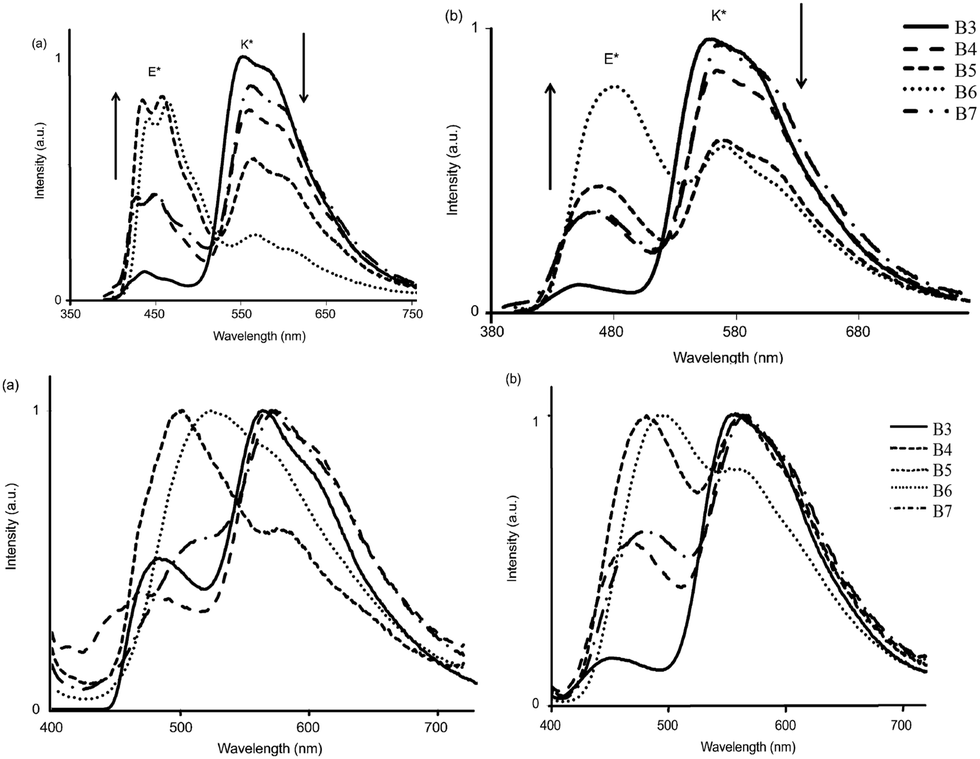 | ||
| Fig. 9 Emission spectra of B3–B7 [top] (a) in cyclohexane (b) toluene [bottom] (a) in KBr pellets (b) PMMA–PS film. Reproduced with permission.108 Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The fluorescence quantum efficiencies of the compounds in the solid state (ΦFL = 8–54%) are much higher than that of the solution (ΦFL = 1–25%) and dependent on substitution patterns. The electron donor and acceptor groups increase or decrease the acidity and basicity of phenolic hydrogen and imidazole nitrogen, which helps to enhance the ESIPT process. In the case of B5 and B6, the proton transfer in the excited state is more favorable among the series because of the appropriate substitution patterns present on benzazole and benzafuran rings. Ziessel and coworkers concluded that in the case of HBBO series chromophores, low fluorescence quantum efficiencies were observed in solution due to the enhanced rate of non-radiative activation, while in the solid state, the interaction with the environment suppresses the non-radiative process. As a consequence, an enhancement in fluorescence quantum efficiencies with white light emission in the solid state was observed.
Simultaneously, panchromatic luminescence based on ESIPT julolidine chromophores L1 and L3 have been (Scheme 8) reported by Ziessel et al.97 Chromophores exhibit panchromatic photoluminescence covering the whole visible region in organic solvents and solid state. The authors used julolidine as a strong donor to achieve red shifted emission, as well as to favor chelation with metals. Four methyl groups were introduced on the julolidine core to increase water solubility and to prevent concentration quenching in the solid state. ESIPT chromophores L1 and L3 were converted to boranil derivatives BL1 and BL3 by complexation with BF3 Et2O to obtain high quantum efficiencies by harvesting the ESIPT process.
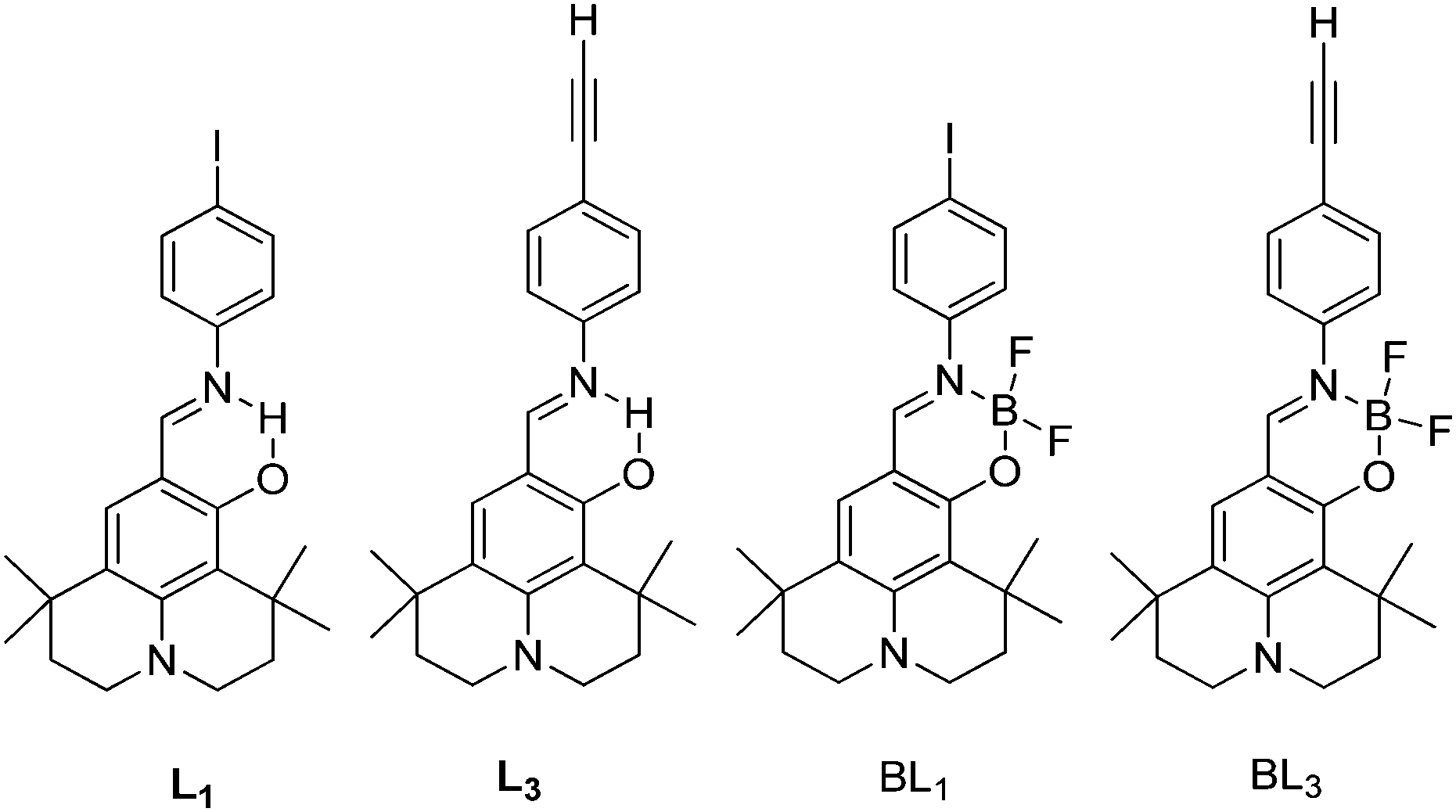 | ||
| Scheme 8 Julolidine based ESIPT chromophores and borate complexes. | ||
All compounds showed absorption at ∼450 nm with a very high extinction coefficient. The absorption properties are independent of solvent polarity. Chromophores L1 and L3 showed dual emission due to the ESIPT process, E* emission ∼470 nm and K* emission ∼590 nm. The emission pattern of boranil is completely different than ESIPT ligands. After complexation blue shifted emissions were observed with a significant enhancement of the fluorescence quantum efficiencies in solution (Table 2). The prototropic behavior of L1 and L3 was also studied in the acetonitrile solvent. Absorption spectra showed a red shift in acidic acetonitrile solution in comparison to pure acetonitrile solution. The disappearance of K* emission was observed for both ESIPT chromophores with the appearance of a new peak between E* and K* emission.
| Comps | Solvent | λ emE* (nm) | λ emK* (nm) | Φ FL (%) | τ (ns) |
|---|---|---|---|---|---|
| Nr: not reported. | |||||
| L1 | Toluene | 440 | 588 | 0.7 | 0.12 |
| DCM | 468 | 596 | 0.5 | 0.13 | |
| Methanol | 462 | 586 | 0.3 | 0.07 | |
| L3 | Toluene | 464 | 590 | 0.9 | 0.12 |
| DCM | 478 | 602 | 0.7 | 0.14 | |
| Methanol | 466 | 592 | 0.4 | 0.08 | |
| BL1 | Toluene | 467 | Nr | 52 | 1.1 |
| DCM | 473 | Nr | 69 | 1.6 | |
| Methanol | 471 | Nr | 69 | 1.8 | |
| BL3 | Toluene | 471 | Nr | 48 | 1.0 |
| DCM | 477 | Nr | 71 | 1.6 | |
| Methanol | 475 | Nr | 72 | 1.7 | |
The panchromatic behavior of compounds was also studied in a condensed medium and the results showed that in a glossy medium compounds showed white light emission Fig. 10. In the solid state, L1 gave more balanced white light emission having color coordinates (x, y) = (0.34, 0.36) in comparison to L3 due to iodine atoms present on the phenyl ring. In short, white light emitters with high fluorescence quantum yields were synthesized by the chemical modification of the p-position of the phenyl ring.
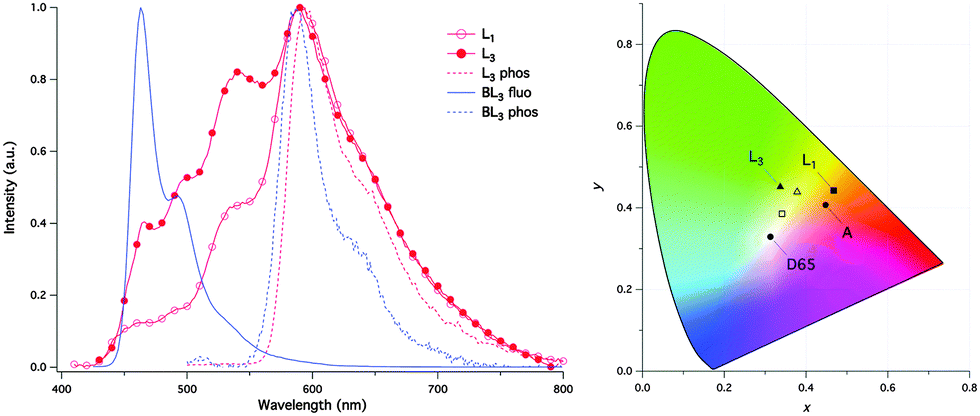 | ||
| Fig. 10 (left) Normalized luminescence spectra of Ln and BLn in CH3OH:CH2Cl2 1:1 glass; (right) CIE coordinates of L1 and L3 in CH3OH:CH2Cl2 1:1 glass at 77 K (filled symbol) and as powder film at rt (open symbol) and of the standard illuminates A (tungsten lamp) and D65 (noon daylight). Reproduced with permission.97 Copyright 2015 The Royal Society of Chemistry. | ||
Blue-white-yellow color-tunable ESIPT chromophores (Scheme 9) sensitive towards the solvent polarity and emissive in the solid state have been reported by Sakai et al.66 2,4-Dibenzothiazolylphenols (2,4-DBTP) and 2,6-dibenzothiazolylphenols (2,6-DBTP) chromophores were used for blue-white-yellow light generation. 2,4-DBTP (λex = 365 nm, λem = 570 nm, ΦFL = 68%) and 2,6-DBTP (λex = 365, λem = 576, ΦFL = 62%) showed bright yellow fluorescence in the solid state Fig. 11. Similar blue shifted emissions were observed for 2,4-DBTP (λex = 378 nm, λem = 554 nm, ΦFL = 9%) and 2,6-DBTP in chloroform with a significant decrease of fluorescence quantum efficiency. The emission bands at 570, 576 and 554 nm were assigned to K* emission in the solid state and in chloroform. The mixture of solvents (DMF and CHCl3) was used for tuning the fluorescence properties of DBTPs. At the ratio of DMF:CHCl3 as 1:1, 2,4-DBTP showed three emission bands covering the entire range of visible light (generation of white light) Fig. 11. In addition to K* and E* emission, a new emission peak was observed between E* and K* and assigned to A* emission (the formation of an anion) (Scheme 10).
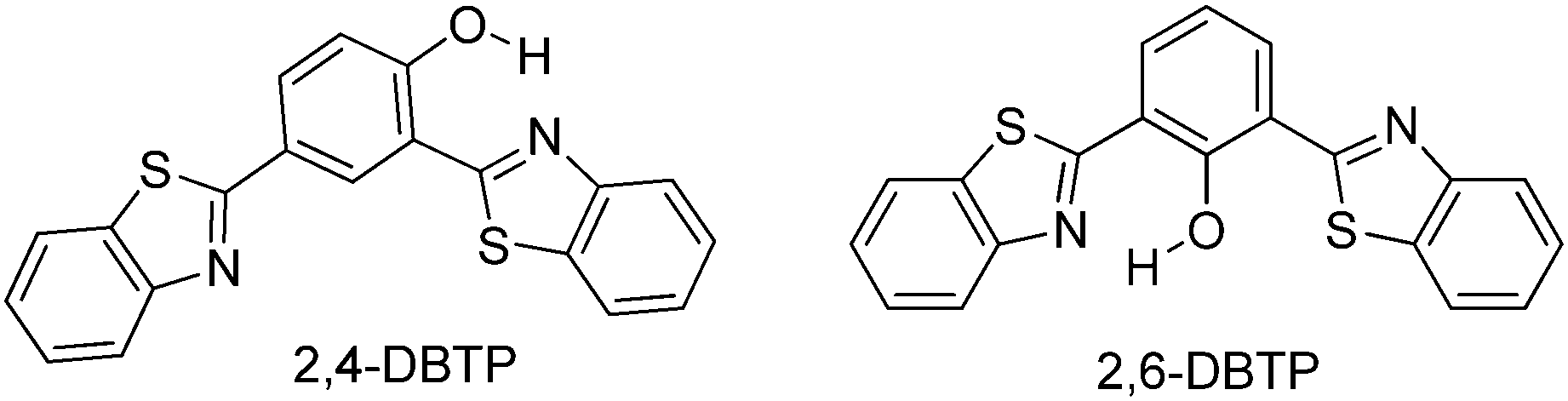 | ||
| Scheme 9 Dibenzothiazolylphenols (2,4-DBTP and 2,6-DBTP) chromophores. | ||
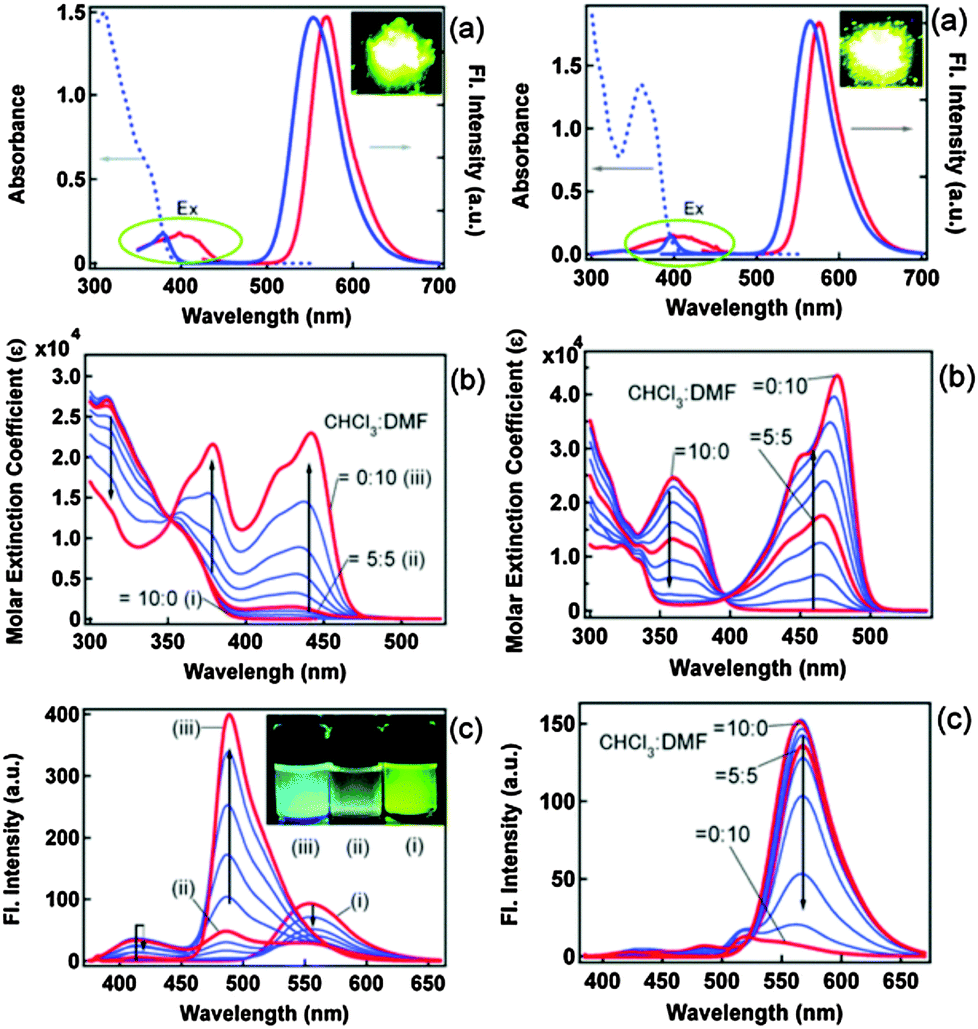 | ||
| Fig. 11 (a) Fluorescence and excitation spectra of 2,4-DBTP (left) 2,6-DBTP (right) in the solid state (red) and in chloroform (blue). (b) Absorption and (c) fluorescence spectral changes by changing the CHCl3/DMF (v/v) ratio. Reproduced with permission.66 Copyright 2013 The Royal Society of Chemistry. | ||
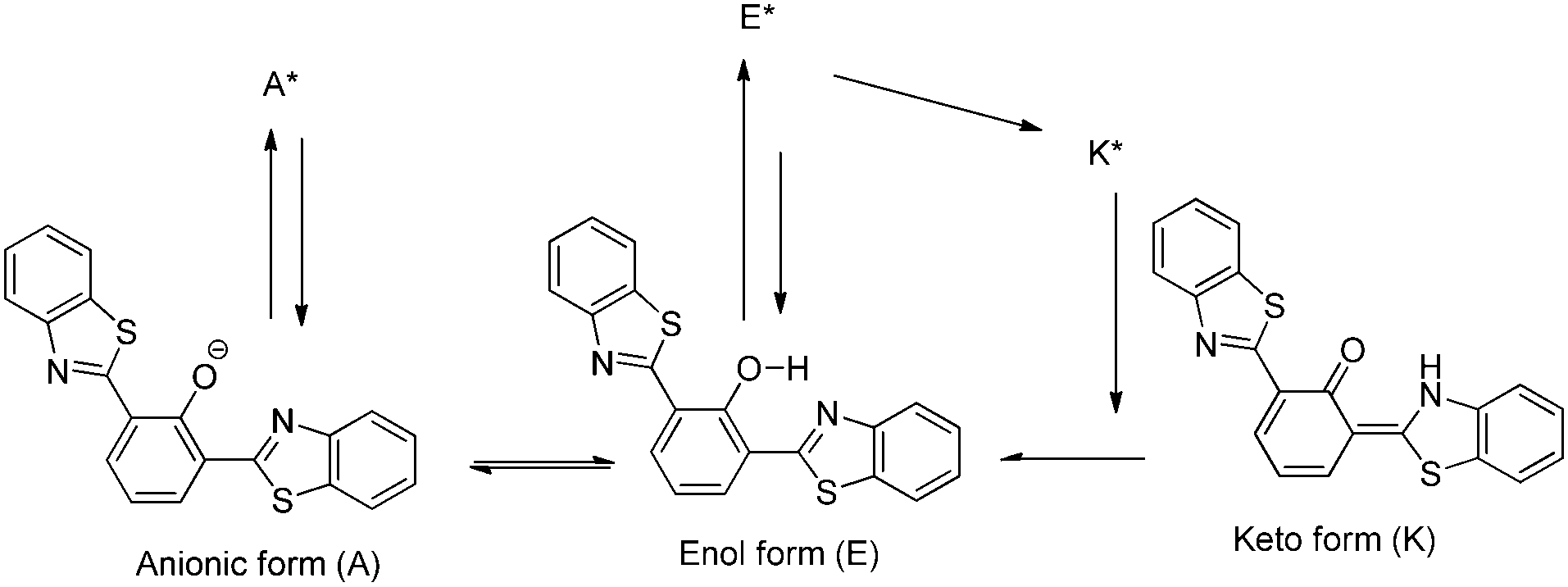 | ||
| Scheme 10 E*, K* and A* emission pathway. | ||
The absorption properties of 2,4-DBTP and 2,6-DBTP in the mixture of solvents are identical. However, 2,6-DBTP was not found to be suitable for white light emission because of overlapping of K* (λem 565 nm) and A* (λem 521 nm) emissions. The fluorescence quantum efficiency of 2,4-DBTP was identical (ΦFL = 68%) in the crystalline state and in the spin coated thin film, which indicates that 2,4-DBTP showed no interference with perylene matrices. The film of 2,4-DBTP and perylene-doped methylmethacrylate prepared in the chloroform solvent emits white light Fig. 12.
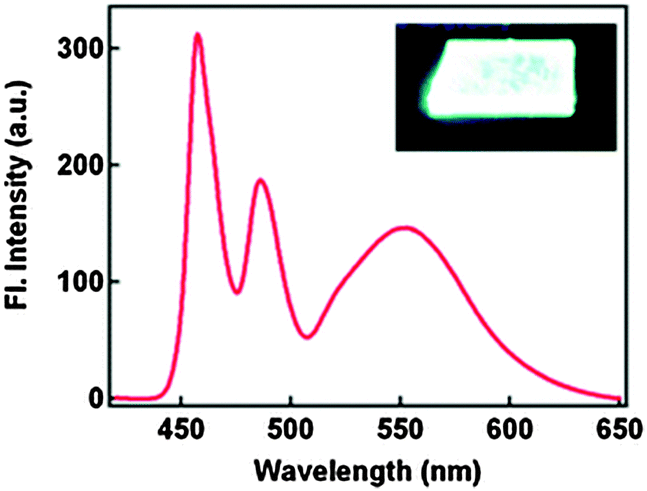 | ||
| Fig. 12 Fluorescence spectrum of the 2,4-DBTP and perylene co-doped PMMA film. Reproduced with permission.66 Copyright 2013 The Royal Society of Chemistry. | ||
Simultaneously, Sakai et al. also reported solid state red emissive ESIPT chromophores based on 2,6-dibenzothiazolylphenols (2,6-DBTP) (Scheme 11) by introducing an alkoxy group at the 4-position.103 Four different alkoxy groups namely –OMe, –OEt, –OPr and –OBt were synthesized and their optical properties were studied.
 | ||
| Scheme 11 2-Dibenzothiazolylphenols. | ||
2,6-DBTPs were fluorescent in solution as well as in the solid state. The red shifted emission was observed for alkoxy substituted 2,6-DBTPs (R = alkoxy: λem = ∼619 nm, R = H: λem = 565). In chloroform 2,6-DBTPs showed identical fluorescence properties for all alkoxy derivatives. However, the fluorescence properties of the alkoxy derivatives were different in the solid state. In the solid state, –OMe and –OEt showed red shifted emission, while –OPr and –OBt showed blue shifted emission in comparison to emission in chloroform Fig. 13. This indicates that the fluorescence properties of DBTPs in the solid-state significantly depend on the length of alkyl groups.
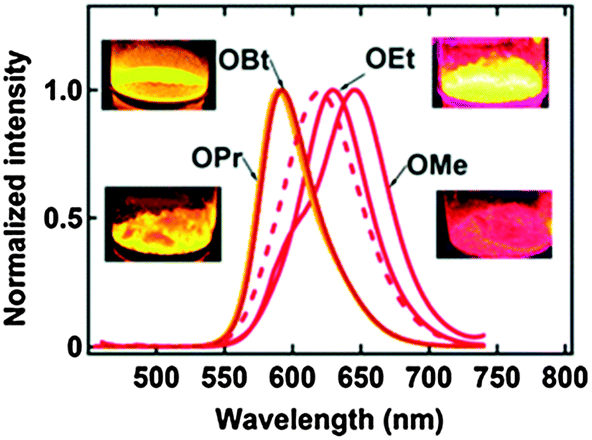 | ||
| Fig. 13 Fluorescence spectra of crystalline powder samples (λex = 400 nm), the dotted line is the spectrum in chloroform (same for all alkoxy), all inset photographs under irradiation with a 365 nm UV lamp. Reproduced with permission.103 Copyright 2014 The Royal Society of Chemistry. | ||
The difference in solid state fluorescence properties was assigned to the mode of molecular packing (J- and H-aggregation) described by X-ray single crystal data of –OMe and –OPr. In –OMe the H-bonded benzothiazole moieties involved in the ESIPT have slipped configuration (J-aggregation) Fig. 14, while in the –OPr crystal, its configuration is eclipsed (H-aggregation) Fig. 14. The three planar π-units of –OMe and –OPr are almost in plane. In the case of –OMe the torsion angle between phenol and H bonded BT (θ = 3.7°) is little larger than –OPr (θ = 0.2°) which causes weaker hydrogen bonding in –OMe than –OPr. In ESIPT chromophores, it is well known that when the H-bond strength is strong, the fluorescence quantum yields will be high.116 This supports the quantum yield trends of alkoxy derivatives (–OMe: λem = 647 nm, ΦFL = 17%, –OEt: λem = 633 nm, ΦFL = 32%, –OPr: λem = 589 nm, ΦFL = 38%, –OBt: λem = 592 nm, ΦFL = 44% in spin coated polymer matrices). The high fluorescence quantum efficiencies and solid state fluorescence properties were supported by TD-DFT computations and the difference in solid state fluorescence was well explained by Davydov exciton coupling theory. In summary, alkoxy derivatives showed solid state emission with very high fluorescence quantum efficiencies even with the absence of a bulky group to prevent intermolecular interactions.
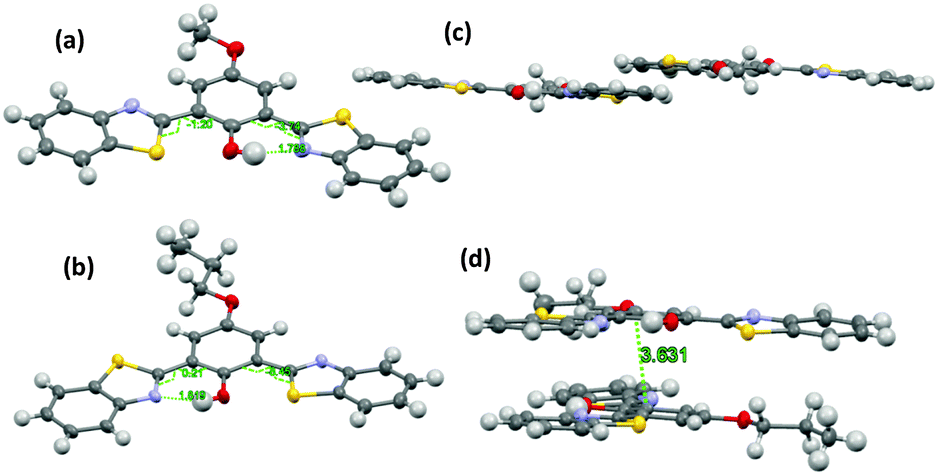 | ||
| Fig. 14 Molecular structure of (a) –OMe and (b) –OPr with 50% probability; (c) packing of –OMe (d) packing of –OPr. Reproduced from CIF file.103 | ||
Among the studied alkoxy derivatives, the –OEt derivative is the best red non-doped red emitter in terms of color with moderate fluorescence quantum efficiency (λem = 633 nm, ΦFL = 32%). The fluorescence properties of the –OEt derivative are comparable with best fluorescent non-doped red emitters which are reported in the literature. They all are large molecules and contain bulky groups in order to avoid twisting around the single bonds. Reported non-doped red emitters are dithienylbenzothiazole117 (λem = 624 nm, ΦFL = 62%), perylene tetracarboxylic acid bisimide118 (λem = 635 nm, ΦFL = 59%), spirobifluorene119 (λem = 646 nm, ΦFL = 46%), 1,4-bis(alkenyl)-2,5-dipiperidinobenzene120 (λem = 641 nm, ΦFL = 34%) and mesitylboryl-substituted bithiophene121 (λem = 657 nm, ΦFL = 30%).
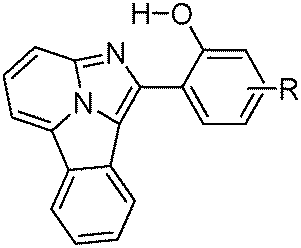
Fused π-conjugated benzo[a]imidazo[5,1,2-cd]indolizine ESIPT fluorophores (Scheme 12) have been reported by Gryko et al.122 Benzo[a]imidazo[5,1,2-cd]indolizines derivatives were prepared from 2-(2′-hydroxy-phenyl)-imidazo[1,2-a]pyridines by cyclo-addition followed by dehydration reaction.
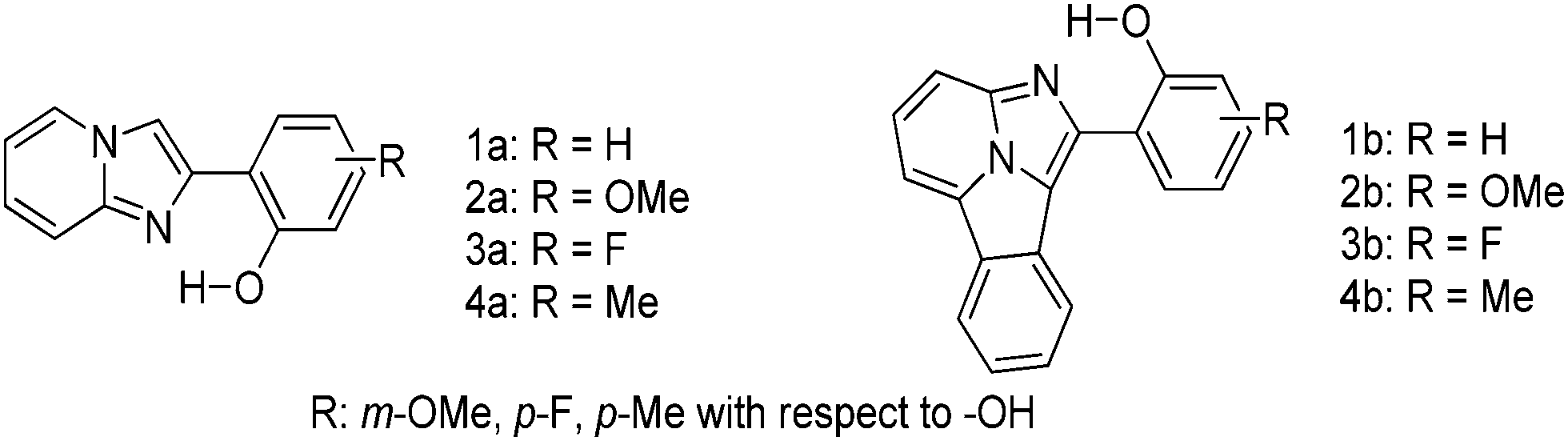 | ||
| Scheme 12 π-Conjugated benzo[a]imidazo[5,1,2-cd]indolizine and 2-(2′-hydroxy-phenyl)-imidazo[1,2-a]pyridines ESIPT fluorophores. | ||
The effect of π-conjugation on the ESIPT process was well studied by comparing photophysical properties of benzo[a]imidazo[5,1,2-cd]indolizines with structural analogues 2-(2′-hydroxy-phenyl)-imidazo[1,2-a]pyridines reported by Araki et al.94,95
In the solid state π-conjugated benzo[a]imidazo[5,1,2-cd]indolizines showed red shifted absorption and emission in comparison to corresponding imidazo[1,2-a]pyridines (Table 3). However, lowering of fluorescence quantum yields (ΦFL = 18–26%) was observed for a π-conjugated system. The absorption maxima of π-conjugated compounds 1b–4b were bathochromically shifted by ∼100 nm in comparison to corresponding imidazo[1,2-a]pyridines 1a–4a. This red shift can be assigned to extended π-conjugations.
| Comps | λ abs (nm) | λ em (nm) | Φ FL (%) | Comps | λ abs (nm) | λ em (nm) | Φ FL (%) |
|---|---|---|---|---|---|---|---|
| 1a | 331 | 494 | 39 | 1b | 410, 435 | 612 | 26 |
| 2a | 336 | 520 | 41 | 2b | 416, 443 | 568 | 24 |
| 3a | 332 | 522 | 37 | 3b | 410, 435 | 626 | 18 |
| 4a | 332 | 529 | 31 | 4b | 414, 439 | 626 | 21 |
Except for compound 2b, all compounds 1b and 3b–4b showed single broad emission which covers entire visible light. The blue shifted emission for 2b may be due to electron donor methoxy groups present on the phenyl ring Fig. 15.
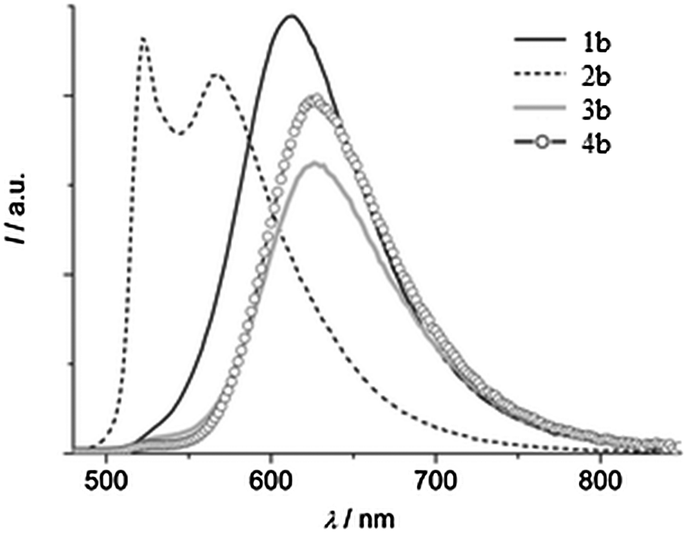 | ||
| Fig. 15 Emission spectra of compounds 1b–4b in the solid state (λex = 440 nm). Reproduced with permission.122 Copyright 2014 The Royal Society of Chemistry and the Centre National de la Recherche Scientifique. | ||
Absorption and emission spectra of both series depend on solvent polarity. In a polar aprotic solvent, the emission spectra consist of two bands which are characteristics of ESIPT, while in a polar protic (methanol) solvent single hyposochromic emission, which corresponds to normal emission, was observed for all the compounds. In a nonpolar solvent, π-conjugated benzo[a]imidazo[5,1,2-cd]indolizines showed high fluorescence quantum efficiencies in comparison to imidazo[1,2-a]pyridines. Based on the results of Araki and coworkers for imidazo[1,2-a]pyridine derivatives, Gryko et al. concluded that π-conjugated imidazo pyridine maintained planar conformation in rigid media, because of the restricted rotation of 2-hydroxyphenyl substituents. The lowering of quantum efficiencies of 1b–4b in solid in comparison to imidazo[1,2–a]pyridines can be assigned to mode of molecular packing present in solid state.
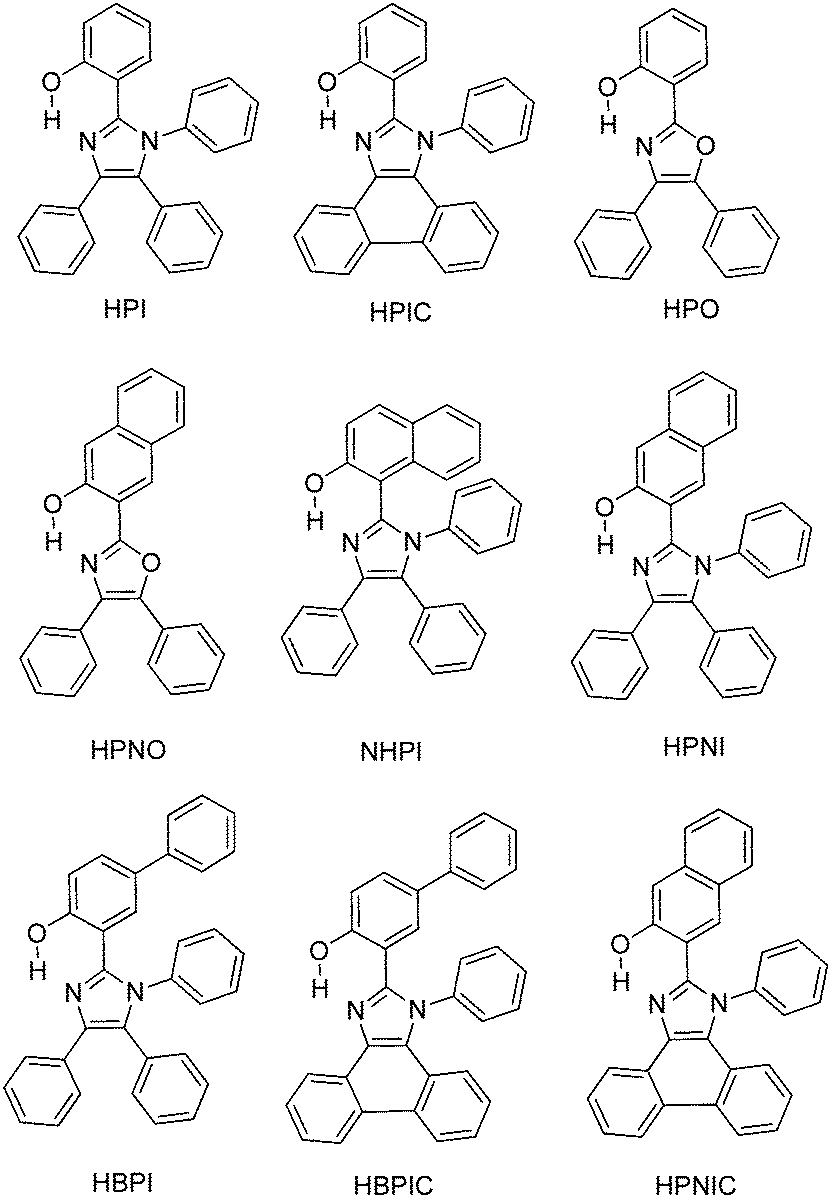
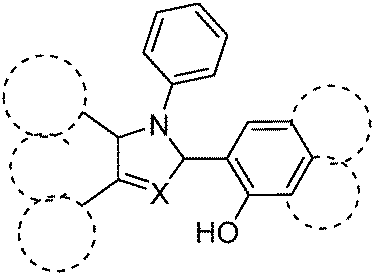
Color tuning of imidazole based ESIPT chromophores by chemical alteration was reported by Park et al.25 Nine different chromophores (Scheme 13) were synthesized and their photophysical properties were studied in solvents and in solid thin films.
 | ||
| Scheme 13 Structures of N-aryl-HBI and HBO. | ||
The concept of a nodal plane model,44,123 the extension of effective conjugation25,124 and the modification of heterocyclic rings17 were used for the development of color tunable chromophores. All these chromophores showed emission in the visible region in chloroform and in the solid state Fig. 16.
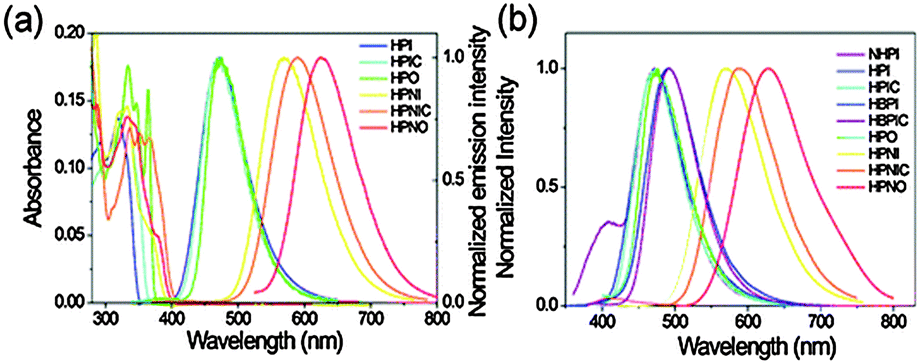 | ||
| Fig. 16 UV-Vis absorption and normalized photoluminescence spectra of synthesized molecules (a) chloroform (b) solid film in PMMA. Reproduced with permission.25 Copyright 2012 PCCP Owner Societies. | ||
In the nodal plane (Nagaoka et al.) model,123 the stabilization effect of the aromatic ring at the 2 position of the imidazole ring can be predicated by considering the nodal pattern of wave function and delocalization of electrons in the excited state. In the excited state the extended conjugation along with a nodal plane stabilizes the excited state keto and helps in the lowering of K* energy. In NHPI (1-(1,4,5-triphenyl-1H-imidazo-2-yl)phenol), HPNI (3-(1,4,5-triphenyl-1H-imidazol-2-yl)naphthalen-2-ol), HPNIC (3-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-naphthalen-2-ol) and HPNO (3-(4,5-diphenyloxazol-2-yl)-naphthalen-2-ol) contain an extended conjugation at the 2-position of imidazole (Table 4).
| Comps | λ abs (nm) | λ em (nm) | Φ FL (%) | Φ FL (%) | τ (ns) |
|---|---|---|---|---|---|
| a Φ FL: fluorescence quantum efficiency in chloroform. b Φ FL: fluorescence quantum efficiency in the polymer matrix (PMMA). | |||||
| NHPI | 343 | 475 | 2.6 | 16 | 0.2 |
| HPNI | 333 | 570 | 5.3 | 22 | 3.2 |
| HPNIC | 365 | 590 | 3.4 | 15 | 2.0 |
| HPNO | 333 | 630 | 0.6 | 4 | 0.8 |
| NHPI | 343 | 475 | 2.6 | 15 | 0.2 |
| HPIC | 364 | 470 | 17 | 34 | 1.3 |
| HBPIC | 368 | 495 | 29 | 56 | 2.7 |
| HPO | 333 | 475 | 13 | 19 | 1.4 |
| HPI | 318 | 470 | 15 | 35 | 1.7 |
HPNI, HPNIC, and HPNO showed long red shifted ESIPT emission due to the stabilization of K* through the effective delocalization of electrons from the aromatic naphthalene ring (Table 4). However, NHPI showed blue shifted K* emission because the naphthalene ring lies out of the nodal plane. Park et al. used the strategy for the extension of conjugation length to tune the fluorescence properties. In the case of HPIC (2-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol), HBPIC (3-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-[1,1′-biphenyl]-4-ol), and HPNIC, the extension of conjugation as well as planarity were achieved to obtain a bathochromic shift (Table 4). In addition to above two strategies, electron donors and acceptors also affect the energies of the excited species. In HPO (2-(4,5-diphenyloxazol-2-yl)phenol) and HPNO (3-(4,5-diphenyloxazol-2-yl)naphthalen-2-ol) electron acceptor imidazole units were replaced by strong electron acceptor oxazole units to change the color properties.
These three strategies help in tuning the fluorescence properties of compounds upon altering the energy gaps between the HOMO and the LUMO of enol and keto forms. To support these observations, theoretical calculations were reported by Park et al., HOMO and LUMO energies and band gaps were evaluated computationally, by a cyclic voltammetry technique and optical spectroscopy. These results conclude that the strength of the electron acceptor and the donor, and the length of conjugation change the energies of the HOMO and the LUMO which results in a change in the color properties.
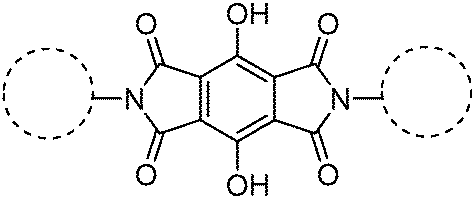
Recently, polyimide (P2H-DC) and imide (P2H-Ch) based ESIPT fluorophores (Scheme 14) exhibiting red fluorescence in solution and in solid thin films were reported by Ando et al.101 Polyimide P2H-DC and imide P2H-Ch contain hydroxy groups susceptible to the ESIPT process, while polyimide PM-DC and imide PM-Ch are non-ESIPT chromophores. Photophysical properties of the ESIPT and non-ESIPT chromophores were studied purposefully, to study the importance of –OH groups on color properties of imides. Initially, fluorescence properties of imides PM-Ch and P2H-Ch in chloroform were studied and results clearly indicate that hydroxy derivative P2H-Ch showed dual emission (E* and K*), while PM-Ch showed single normal emission. Photophysical properties of the polyimide were studied on solid thin films. The absorption properties of P2H-DC (λabs = 407 nm) and PM-DC (λabs = 317 nm) were similar to monomers P2H-Ch (λabs = 421 nm) and PM-Ch (λabs = 320 nm). The colorless PM-DC film showed weak absorption at 317 nm, while the P2H-DC film showed an absorption peak at 407 nm along with a shoulder peak at 418 nm. The emission properties of P2H-DC (λem = 642 nm, ΦFL = 10%) and PM-DC (λem = 439 nm) were completely different, but coincided well with emission bands of P2H-Ch (keto emission band in chloroform) and PM-Ch. This clearly indicates that the red emission band of P2H-DC in the solid state corresponds to ESIPT emission with a considerable enhancement of fluorescence quantum efficiency.
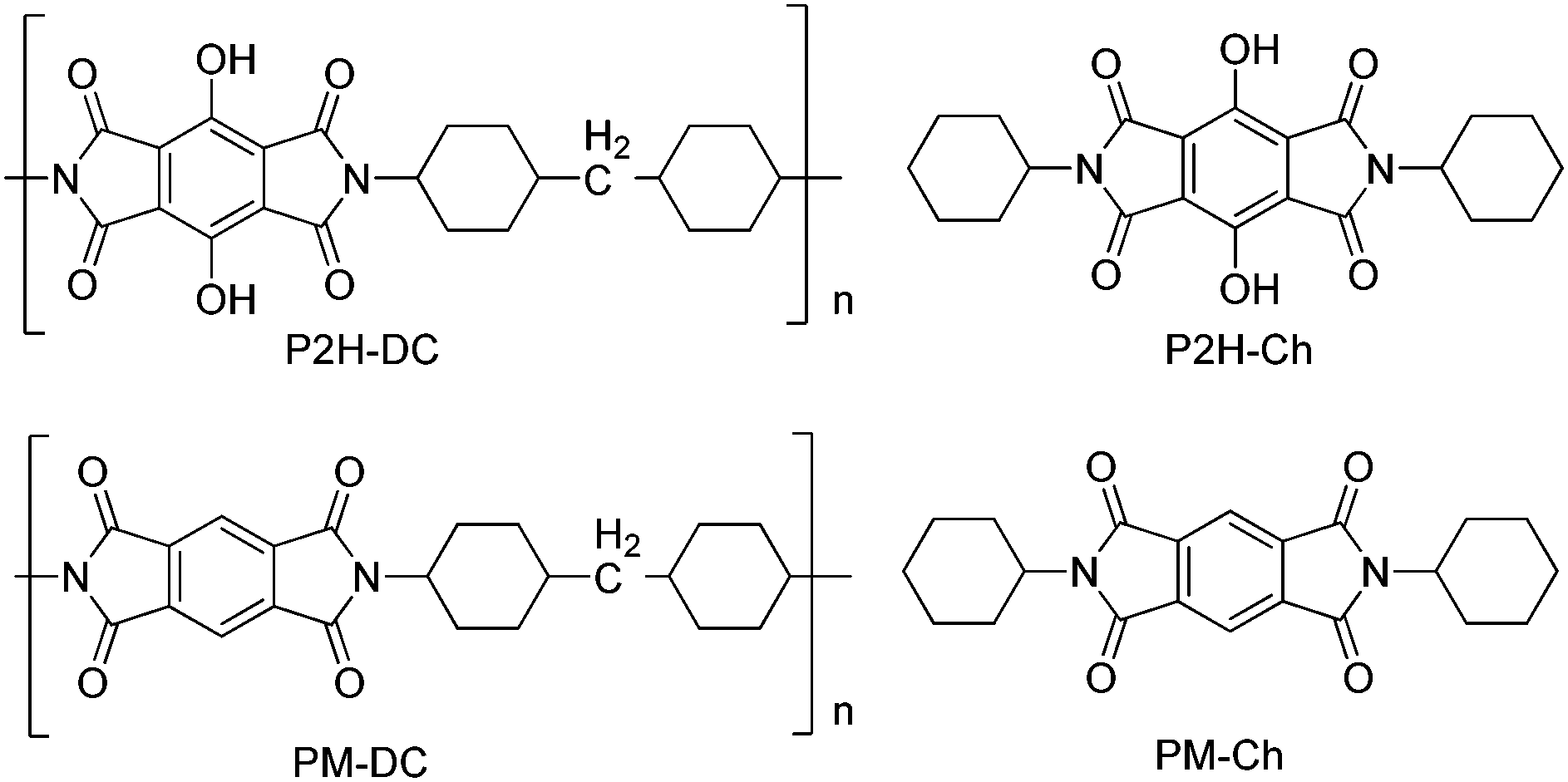 | ||
| Scheme 14 Structures of ESIPT–polyimide and imide and non-ESIPT analogues. | ||
The effect of pH on absorption spectra of polyimide P2H-DC was studied by authors for the pH range of 11–14. In basic media (pH = 11) a new absorption peak appeared at 529 nm with a gradual decrease of intensity of the 407 nm peak. At pH 14 a new absorption band was observed at 630 nm with a change of film color from yellow-red-deep blue. The appearance of new peaks was due to the formation of mono and di-phenoxide ions in basic media even in the solid state.
In summary, the polyimide was concluded to show red emission in solid state due to ESIPT process.
Recently, highly emissive ESIPT solid state emitters based on fluorene–HBT motifs have been reported by Padalkar et al.125 (Scheme 15). Padalkar and co-workers synthesized different fluorescent compounds containing two HBT units attached to the 7,7′-position and different alkyl groups at the 9,9-position on the fluorene unit. Compounds absorb (∼325–344) in the UV region and emit (∼405–552 nm) in the visible region. The absorption spectra of the compounds in the solid state and in solution are almost identical. However, the emission spectra of compounds are different in different media. Compounds showed single emission in the solid state, chloroform, toluene and DMF and multiple emissions in methanol and acetonitrile. The single emission in the solid state is assigned to keto emission, and multiple emissions in methanol and acetonitrile are assigned to cis-enol, cis-keto, protonated cis-enol and phenoxide ions. The fluorescence quantum efficiencies of the compounds in the solid state (ΦFL = 54–68%) are much higher than that of the solution of different polarities (ΦFL = 1–11%). The fluorescence lifetime in the solid state is between 3.48–5.21 ns, while it is significantly less in chloroform (0.52–0.75 ns). The high fluorescence quantum efficiencies in the solid state were assigned to slip-stacking and strong intramolecular hydrogen bonding which was confirmed by single X-ray crystal data as well as DFT computations by Padalkar et al.
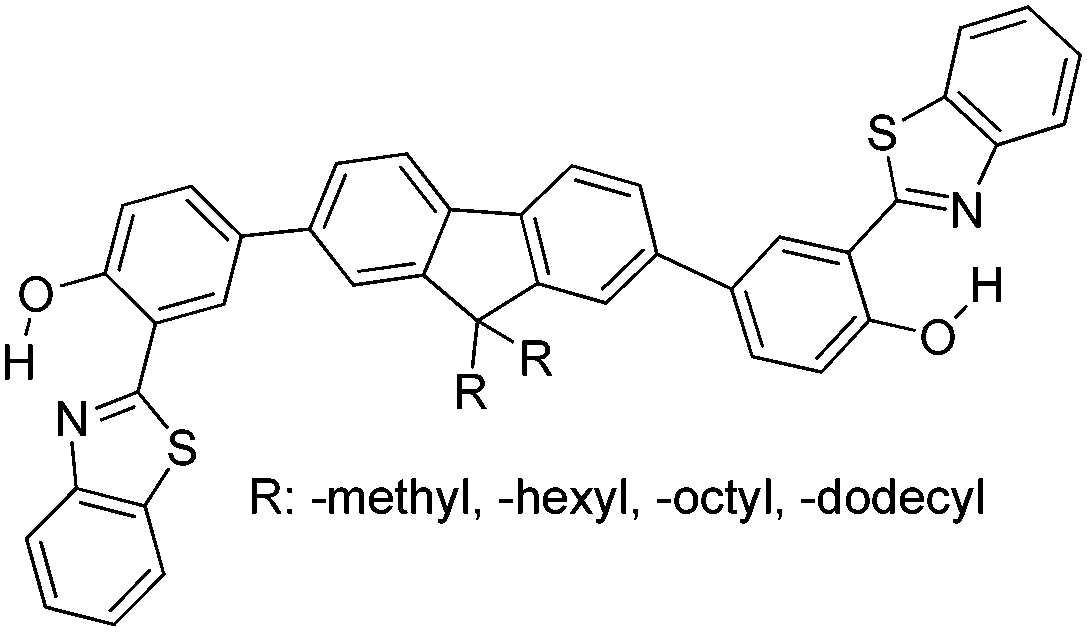 | ||
| Scheme 15 Structures of fluorene–HBT ESIPT molecules. | ||
A short summary of solid state ESIPT emitters as a single molecule is summarized in Table 5.
| ESIPTs structure | Spectral and photophysical data | Ref. |
|---|---|---|
| Φ FL: fluorescence quantum yields, λLem: luminescence emission, τ: fluorescence lifetime, RIR: restriction of intramolecular rotation, CIE: CIE 1931 chromaticity diagram, AIE: aggregation induced emission. | ||
|
White light emission; ΦFL = 5.65–91.68%; λem = 443–630 nm; λLem = 460–648 nm; planar structure; intramolecular H-bonding interaction; Fl depends on nature and position of amino substituted groups. Highest quantum efficiency reported till date for single small ESIPT molecule. | 33 |
|
White light emission; ΦFL = 9.0–90%; λem = 560–649 nm, τ = 4.3–10.2 ns; Z-conformation; seven membered ESIPT; RIR effect; Fl properties depends on the electron donating/withdrawing/resonance properties of the substituents. Analogues of GFP chromophore. Radiationless quenching process can be reduced by substitution of bulky groups at C4 position. | 90 |
|
White light emission; ΦFL = ∼10%; λem = 435–580 nm, nearly planar structure; π–π stacking; H-aggregation; 5-fold enhancement of quantum efficiency in solid state; CIE (0.30, 0.27); π–π stacking can be minimized by introducing a bulky substituents on tetracyclic moiety. | 18 |
|
Φ FL = 39–68%; λem = 375–480 nm; τ = 0.2–3.2 ns; slip-stacking; offset face-to-face stacking; hydroxy phenyl ring is co-planar with imidazole, N-aryl ring perpendicular to imidazole ring, N-aryl rings suppress the non-radiative pathways or π–π stacking | 21 |
| 114 | ||
| 115 | ||
|
White light emission; ΦFL = 8–54%; λem = 451–580 nm; CIE (0.34, 0.39); Fl depends on substitution patterns and position; electron-donating groups on benzofuranol side increases the pKa of compounds and acceptor on the benzoxazole decreases the basicity of cyclic amines. These combined effects provide broad dual-emission band. | 108 |
|
White light emission; panchromatic luminescence; dual emission; intramolecular H−interaction; λem = 465–590 nm; τ = 11.6–54.6 ns; CIE (0.34, 0.38); methyl groups helps for RIR; Fl dependent on groups attached at p-position with respect to imine N; prototropic behaviour. | 97 |
|
White light emission; blue-white-yellow emission; ΦFL = 68%; λem = 570 nm (2,4-DBTP); ΦFL = 62%; λem = 576 nm (2,6-DBTP); generation of white light due to K*, E* and A* emission. | 66 |
|
Non-doped red emitters; ΦFL = 17–44%; λem = 589–647 nm; CIE (0.57–0.65, 0.34–0.41); J- and H-aggregation; solid state emission depends on nature of alkoxy group; quantum efficiency is proportional to strength of intramolecular H-bond. Best non-doped red emitters reported till date (small molecule) | 103 |
|
Φ FL = 18–27%; λem = 612–626 nm; 100 nm red shift for benzo[a]imidazo[5,1,2-cd]indolizines relative to corresponding imidazo[1,2-a]pyridines; extending the length of conjugation causes red shift, but quenching in quantum efficiency (∼half). | 122 |
|
Polymorph dependence emission; white emission; ΦFL = 4–56%; λem = 450–630 nm; τ = 0.2–2.77 ns; color tuning: nodal plane approach, extension of conjugation, modification of heterocyclic ring and strength of electron donor and acceptor; intense Fl due to twisted phenyl ring. | 25 |
|
Red fluorescence; ΦFL = 10%; λem = 642 nm; pH dependent Fl properties; thermally stable ∼350 °C; halochromism. | 101 |
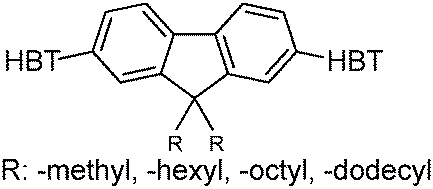
|
Yellow emitters, ΦFL = ∼68%; λem = 544–552 nm; τ = 3.48–5.21 ns; slip-stacking, AIE, thermally stable ∼440 °C; 10−60 fold enhancement of quantum efficiency in solid state in comparison to solution. | 125 |
Recently, a few more small molecules which are emissive in the solid state have been reported, but their detailed photophysical properties have not been explained by respective authors. The reported chromophores, which are derivatives of bis-imidazole,126 coumarin,127 benzothiazole-benzamide,128 benzothiazole hydroxy naphthalene-1-carbaldehyde,129 bis-benzothiazole130 and N-salicylidine anilines131 are summarized in Table 6.
| ESIPTs structure | Spectral and photophysical data | Ref. |
|---|---|---|
|
Two-photon absorption; yellow-green emission; ΦFL = 7–16%; λem = 500–506 nm (solvents); 2λOPA = 726 and 772 nm (dichloromethane); intramolecular H-interaction. | 126 |
|
Dual emission; S1 and S3 emission; ΦFL = 4%; λem = 363 and 514 nm; τ = 8.7 and 2.4 ns (toluene); green-blue solid state emission; λem = ∼520 nm. | 127 |
|
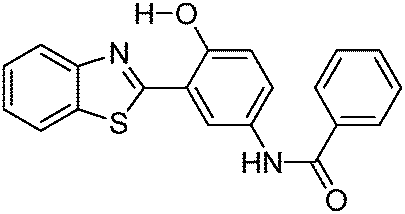
Solid state emission; λem = ∼550 nm. | 128 |
|
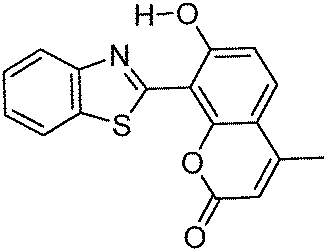
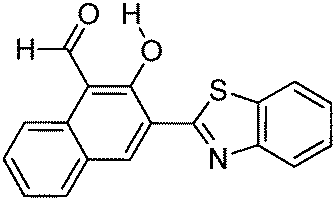
Multiple emission; λem = ∼500, 550 and 650 nm; absorption and emission properties are solvent dependent; ΦFL = 5–11% (solvents). –OH and –CHO responsible for ESIPT. | 129 |
|
Solid state emission; λem = ∼440, ∼580, and ∼595 nm. | 130 |
|
Thermochromic; photochromic; λem = ∼500; planar conformation; CH–π interaction; cis–trans isomerization; π–π stacking; close packed configuration. | 131 |
6. Solid state ESIPT emitters based on polymorphic form (molecular packing)
Imidazo[1,2]pyridine is an important class of heterocyclic compounds widely used in the medicinal field.132,133 Recently 2-(2′-hydroxyphenyl)imidazo[1,2]pyridine (Scheme 16) has attracted huge attention due to its unique photophysical properties.54–57 These types of chromophores undergo excited state intramolecular proton transfer upon photoexcitation, which helps to enhance the fluorescence properties of the chromophores. The fluorescence properties of 2-(2′-hydroxyphenyl)imidazo[1,2]pyridine contain an ESIPT unit which was first studied by Acuna and Douhal in 1995,7,134,135 and recently Gryko and Cyranski groups reported additional substituted ESIPT imidazo[1,2]pyridines.122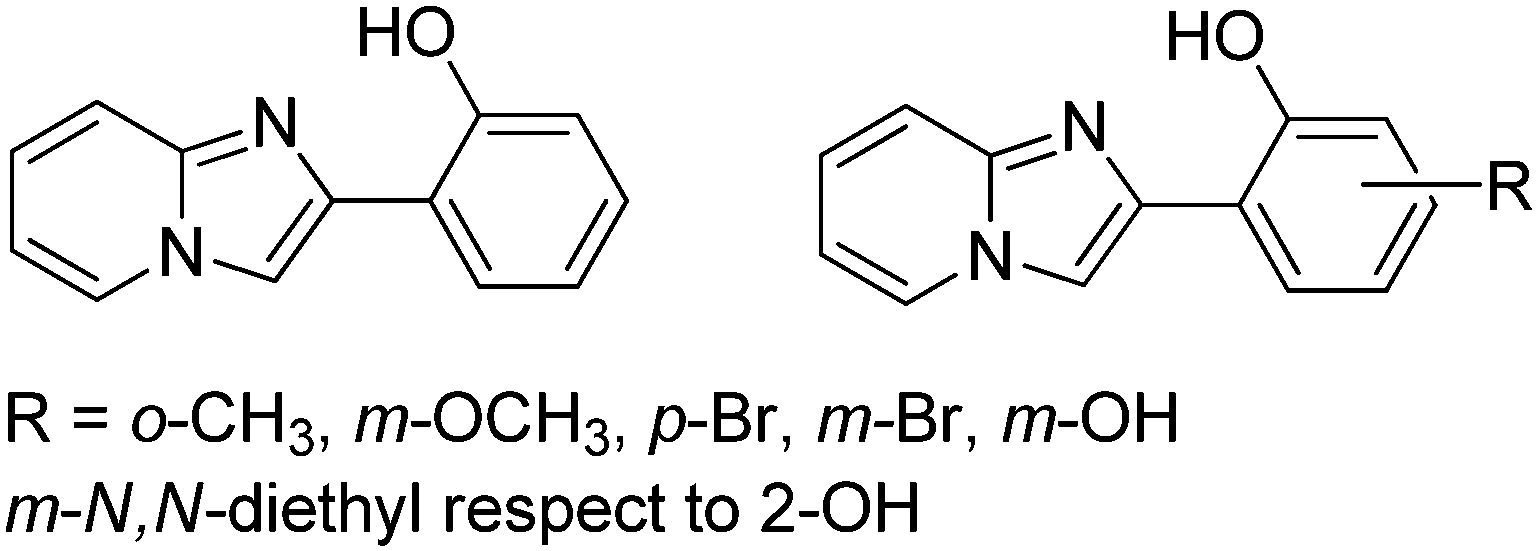 | ||
| Scheme 16 2-(2′-Hydroxyphenyl)imidazo[1,2]pyridines. | ||
Extensive research on photophysical properties of 2-(2′-hydroxyphenyl)imidazo[1,2]pyridines in the solution, crystalline state, in polymorph state, and in polymer matrices was performed by Araki and coworkers since 2008.54–57 The various parameters such as polymer matrices, crystal structures, types of substituents, and molecular packing were studied to understand the ESIPT process of 2-(2′-hydroxyphenyl)imidazo[1,2]pyridines experimentally as well as theoretically.54–57,94,95 In 2008, Araki and coworker reported the effective mechanism for switching of organic solid state luminescence for 2-(2′-hydroxyphenyl)imidazo[1,2]pyridine.56 Two crystal polymorphs of different colors (blue-green (BG) and yellow (Y)) due to the ESIPT phenomenon were studied in detail.
The emission maxima of compound 1 was reported in protic ethanol and compared with the emission spectrum of compound 2. Compound 1 showed weak fluorescence in the blue region (E* emission; λex = 332 nm, λem = 377 nm, ΦFL = 8%, τ = 2.63 ns in THF) and large Stoke's shifted emission (1a* emission;=602 nm, ΦFL = 2%, τ = 0.5 ns in THF) in the orange region, while only blue emission (E* emission; λex = 332 nm, λem = 378 nm, ΦFL = 11%, τ = 2.56 ns in THF) was observed for compound 2. This result clearly indicates that dual emissions in compound 1 were due to intramolecular hydrogen bonding. Blue emission is assigned to the locally excited state and orange emission is the ESIPT emission from 1a* (Scheme 17).
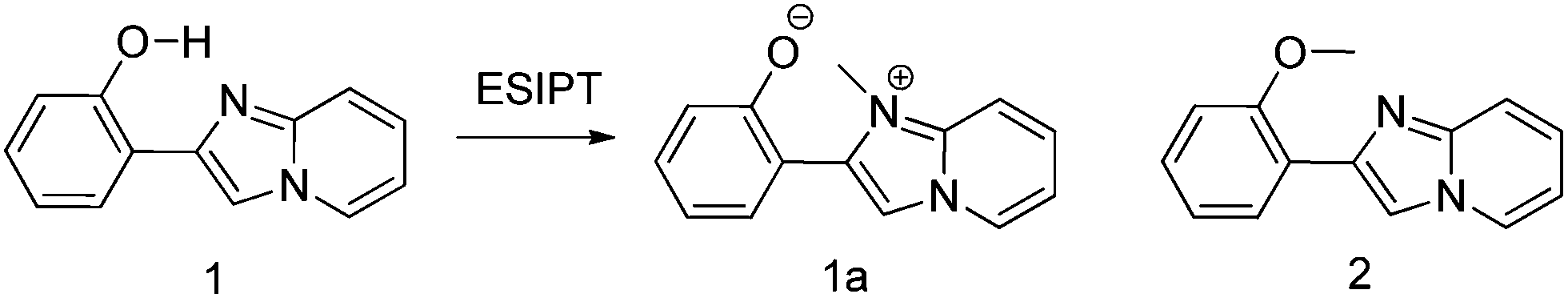 | ||
| Scheme 17 Structure of compounds 1, 1a and 2. | ||
In the frozen state compound 1 showed blue shifted ESIPT emission (1a* emission;=521 nm, τ = 5.2 ns in THF at 77 K). Similar to the frozen state, identical absorption and emission patterns were observed for compound 1 in amorphous (λex = 337 nm, λem = 527 nm, ΦFL = 39%, τ = 6.39) and polymorphic forms (polymorph-BG: λex = 339 nm, λem = 496 nm, ΦFL = 50%, τ = 5.91; polymorph-Y: λex = 337 nm, λem = 529 nm, ΦFL = 37%, τ = 5.84) with little change in ESIPT emission. However a significant enhancement in quantum efficiencies was observed in rigid media in comparison to dilute solution. The enhancement of quantum efficiencies and solid state emission was explained on the basis of molecular packing in amorphous and polymorphic forms. The dihedral angle between phenyl and imidazopyrine rings is close to zero for BG and Y polymorphs (blue-green polymorph θ = 5.8°) (yellow polymorph θ = 1.3°), this indicates that both polymorphs are planar in nature. In polymorph-Y compound 1 is packed with co-planar conformation, while in BG-polymorph the conformation is a twisted one. This packing-induced small deviation of the molecular conformation may affect the ESIPT process. Molecular packing is a key factor for switching the color properties of the ESIPT imidazo[1,2]pyridines. In polymorph-Y and -BG the molecular packing of compound 1 is thermally controlled reproducible packing, which was confirmed from DSC and TGA data. Based on these results Araki et al. concluded that the luminescence properties of ESIPT imidazo[1,2]pyridine 1 are switchable in the solid state with high quantum efficiencies.
After the discovery of polymorph dependent ESIPT luminescence for imidazo[1,2]pyridines, white light luminescence properties of the imidazo[1,2]pyridines were reported by Araki and coworkers.19 Mixtures of ESIPT and non-ESIPT chromophores were used for the generation of white light.19 Prior to this report based on ESIPT, Park and coworker87 have reported white light generation using two ESIPT chromophores having different Stokes shift and Chou et al.17 also reported white light generation in solution using ESIPT and normal luminophores.
In the design of white-light emitters, hydroxy and methoxy derivatives of 2-phenylimidazo[1,2-a]pyridine (PIP) chromophores (Scheme 18) having similar absorption properties and different emission properties were used.19 Authors claimed that the use of compounds of similar molecular structures is more advantageous, because these compounds may have similar physical properties. Before these ESIPT based white light emitter strategies, the generation of white light has been reported by mixing blue- and yellow-emitting species having proper combination of donor and acceptors.16,19 In this case, no donor–acceptor relationship exists for white light generation. 2-Hydroxy derivatives H1 (λex = 335, λem = 529 nm, ΦFL = 37%, (x, y) = (0.36, 0.55)) and H2 (λex = 330, λem = 536 nm, ΦFL = 31%, (x, y) = (0.41, 0.50)) exhibiting ESIPT and 2-methoxy derivatives M1 (λex = 312, λem = 382 nm, ΦFL = 24%, (x, y) = (0.23, 0.22)) and M2 (λex = 348, λem = 422 nm, ΦFL = 15%, (x, y) = (0.17, 0.09)) having no intramolecular hydrogen bond were used for white light generation in solution and solid film. All compounds showed identical absorption spectra in the UV region, while emission patterns were different for ESIPT and non-ESIPT chromophores. Hydroxy derivatives H1 and H2 emit with large Stokes shift (ESIPT process) and methoxy derivatives M1 and M2 emit with normal Stokes shift (normal emission).
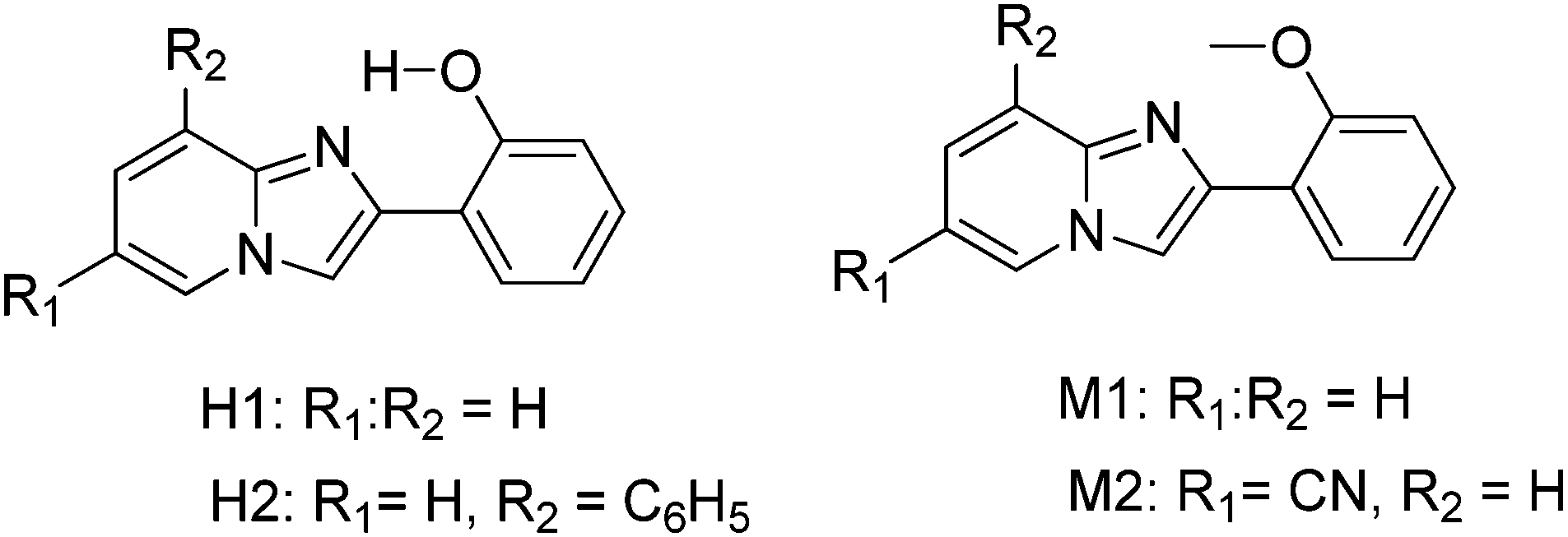 | ||
| Scheme 18 Hydroxy and methoxy derivatives of 2-phenylimidazo[1,2-a]pyridines. | ||
The absorption band should not trail into the visible region to achieve pure white light emission in the solid state. The same excitation wavelength was used to achieve yellow and blue luminescence in the present study. A mixture of various ratios of hydroxy: methoxy was tested to achieve white-light luminescence in the solid state. At a ratio of 1:100 by weight (H1:M1), bright whitish luminescence (prominent λem = 386 nm, and broad λem = 470–650 nm) with CIE coordinates (x, y) = (0.32, 0.40) was observed. The mixture of H2:M2 (3:10 by weight) displayed a bright white luminescence at CIE (x, y) = (0.30, 0.31), which is almost pure white light Fig. 17. The combination of ESIPT-H2 and non ESIPT-M2 in the polymer matrix [poly(methyl methacrylate) (PMAA)] generates a white light emissive film Fig. 17. Araki et al. have concluded that whitish luminescence is a summation of the luminescence of ESIPT and non-ESIPT chromophores and both types of chromophores emit independently. The white light generated from the mixture of H2 and M2 on vapor deposited films (ΦFL = 18%, (x, y) = (0.30, 0.31)) as well as in the PMAA matrix (ΦFL = 22%, (x, y) = (0.36, 0.33)) showed similar luminescent properties at an optimized H2:M2 ratio and remained unchanged for a long time.
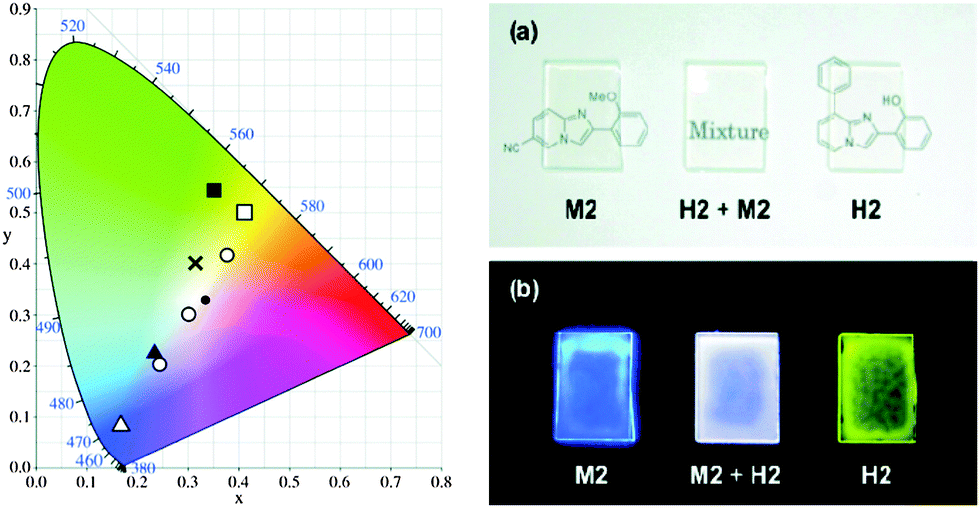 | ||
| Fig. 17 (left) A CIE chromaticity diagram of H1 (filled square), H2 (open square), M1 (filled triangle), M2 (open triangle), a mixture of H1 + M1 (1:100; cross), and H2 + M2 (1:10, 3:10, and 10:10; open circle; from left to right). A black dot indicates the ideal white (x, y = 0.33, 0.33). (right) Images of PMMA films containing M2, H2 + M2 (11:7), and H2 (from left to right) under (a) room light and (b) a UV lamp (365 nm). Reproduced with permission.19 Copyright 2011 American Chemical Society. | ||
Simultaneously, the effect of substitution on ESIPT emission of imidazo[1,2-a]pyridine in rigid media was studied by Araki et al.54 Various electron donor and acceptor groups on phenyl and imidazopyridine (Scheme 19) cores were studied in solution as well as in the polymer matrix (PMAA).
 | ||
| Scheme 19 Substituted imidazo[1,2-a]pyridines. | ||
All compounds showed absorption in the UV region (λabs = 333–341 nm) in THF and (λabs = 331–351 nm) in a PMAA film. This result concludes that substitution has little influence on the absorption spectra. Except a few compounds (2, 10, 13–14), all the compounds showed dual emission (normal and ESIPT emission) in THF with a significant change in the emission pattern. The emission pattern depends on the nature as well as the position of substituents present in the molecules. The absorption and emission properties of the compounds were also studied in frozen THF. In frozen media the absorption spectra were similar to THF solution.
However blue shifted emissions (50–90 nm) were observed for compounds in frozen THF. The blue shift was assigned to the effect of the rigid medium. In THF solution, the fluorescence quantum efficiencies of the compounds were less than 2%, but were very high in a PMMA matrix (ΦFL = 6–61%, τ = 2.1–4.9 ns). The average lifetime of compounds indicates that emission is a singlet emission originating from zwitterionic ESIPT species (Scheme 20). Douhal et al. reported134 that zwitterionic ESIPT species further undergo twisted conformation along with the rearrangement of the surrounding medium which may be the reason for red shifted emission with large Stokes shift in the THF solvent.
 | ||
| Scheme 20 Formation of zwitterionic ESIPT species. | ||
In rigid media, zwitterionic ESIPT species retain their planarity, which causes the blue shifted ESIPT emission. In summary, authors concluded that the introduction of electron donor and acceptor groups on the phenyl ring causes blue and red shift, respectively, while reverse observations were observed for the same groups on the imidazopyrine core. The experimental results i.e. the effects of substitution on fluorescence properties were well supported by theoretical results.
In addition to the two-color polymorph dependent ESIPT study of imidazo[1,2-a]pyridine 1, a three-color polymorph dependent luminescence study of ESIPT imidazo[1,2-a]pyridine derivative 2 was reported by Araki et al. recently.55 Three (yellow–orange–red) colors were achieved by controlling the molecular packing of 6-cyano-2-(2-hydroxyphenyl)imidazo[1,2-a]pyridine (Scheme 21).
 | ||
| Scheme 21 6-Substituted-2-(2-hydroxyphenyl)imidazo[1,2-a]pyridines. | ||
Three polymorphs namely 2-yellow (2-Y), 2-orange (2-O) and 2-red (2-R) were obtained by different crystallization methods from hexane, benzene, chloroform and THF solvents. All polymorphic crystals are luminescent under UV lamp (365 nm) Fig. 18.
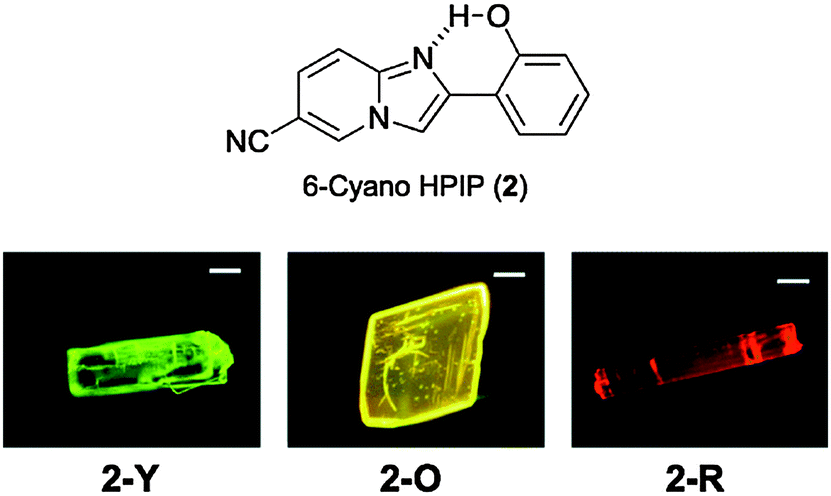 | ||
| Fig. 18 Luminescent images of polymorphic crystals, 2-Y, 2-O, and 2-R. Reproduced with permission.55 Copyright 2014 The Royal Society of Chemistry. | ||
Single emissions were observed for polymorphic crystals (2-Y: λem = 548 nm, ΦFL = 49%, τ = 5.48 ns; 2-O: λem = 570 nm, ΦFL = 25%, τ = 5.51 ns, and 2-R: λem = 585 nm, ΦFL = 10% τ = 2.26 ns) upon excitation at 330 nm. The absorption spectra of three polymorphic crystals were almost identical in terms of shape and absorption maxima wavelength, which indicates that absorption properties of the compound 2 are not sensitive towards polymorph packing. A similar observation was reported56 for the two-color polymorph dependent ESIPT study of imidazo[1,2-a]pyridine 1.
Three polymorphs emit at different wavelengths (yellow–orange–red region) with a significant difference in quantum efficiencies (ΦFL = 10–49%). This difference in luminescence properties of compound 2 was well studied by authors from X-ray single crystal data and theoretical calculations. All polymorphic crystals were monoclinic and had the same space group. The distances between basic nitrogen and acidic hydrogen were 2.582, 2.625, and 2.622 Å, for 2-Y2-O and 2-R respectively indicating favorable geometry for intramolecular hydrogen bonding. The dihedral angle between two aromatic rings is close to zero (θ = 1.4, 2.5 and 1.4° for 2-Y2-O and 2-R respectively) which confirms the planar nature of the polymorphic crystal.
However the molecular packing was different for different polymorphs Fig. 19. 2-Y and 2-R crystals have slip-packed packing, while the 2-O crystal had antiparallel π–π stacking with an interplanar distance of 3.35 Å. In crystal 2-Y, the interplanar distance between two parallel rings was 3.38 Å and the distance between an oxygen atom and the nearest nitrogen atom of the neighboring molecules was 3.64 Å, which prohibits the formation of an intermolecular hydrogen bond. Similar observations were reported for 2-R crystals. In 2-Y and 2-R crystals, the fused the imidazole part overlapped with the phenyl-part of the stacked molecule. However, in 2-O crystals the imidazole part overlapped with phenyl and azole parts of imidazole. This different intermolecular interaction may be the key factor for polymorph dependent luminescence. Different structural packing was supported by DSC and DFT computations and all the results are well in agreement with experimental results.
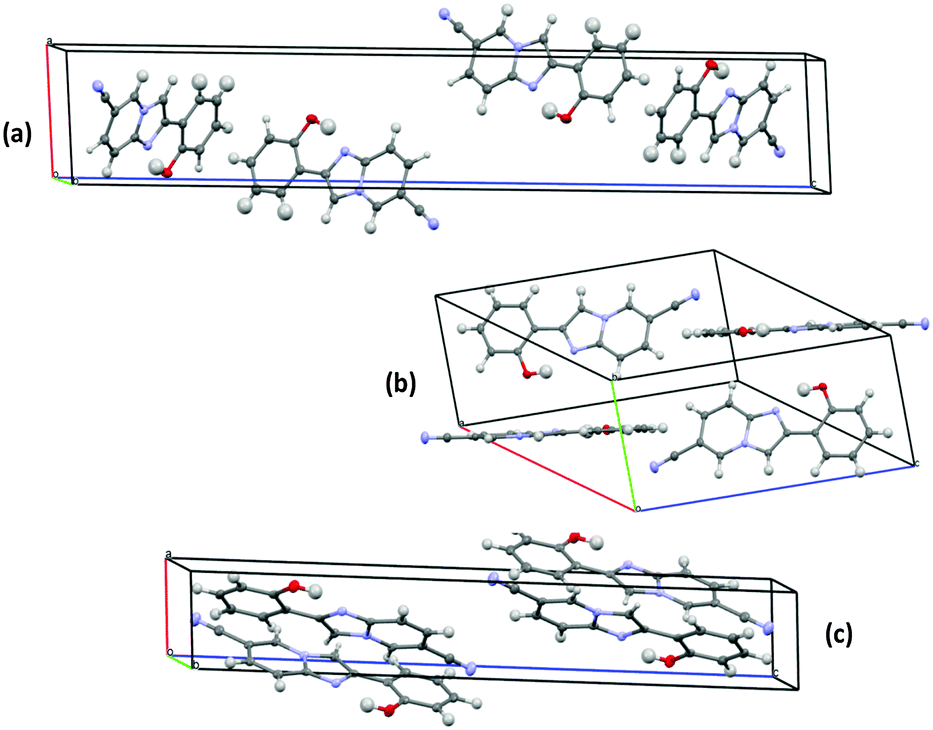 | ||
| Fig. 19 Molecular packing of (a) 2-Y (b) 2-O and (c) 2-R. Reproduced from CIF file.55 | ||
Tunable luminescence properties of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines (Scheme 22) with surrounding medium were also studied by the Araki group.57 Two derivatives 1 and 2 were studied for the influence of matrices on luminescence properties. Polystyrene (PS), poly(vinyl acetate) (PVAC), poly(bisphenol A carbonate) (PC), poly(methyl methacrylate) (PMAA), poly(vinyl alcohol) (PVA) and poly(ethylene glycol) (PEG) were used as matrices for the matrix dependent luminescence study. Spin coated samples were used for absorption and luminescence measurements. Compound 1 showed green to yellow-green luminescence, while compound 2 showed a variety of colors such as purple, yellow and orange. It was reported that compounds 1 and 2 showed single emission in different polymorphic forms with a large Stokes shift. This emission was assigned to ESIPT from intramolecular proton transfer (IPT*) generated from 1a and 2a. In polymer matrices compounds showed dual emission, normal emission (1b* and 2b*) and ESIPT emission (1a* and 2a*).
 | ||
| Scheme 22 2-(2′-Hydroxyphenyl)imidazo[1,2-a]pyridines. | ||
However, in polar solvents the emission was single emission because in polar solvents the dissociation of the intramolecular hydrogen bond between imidazole nitrogen and phenolic hydrogen (1a and 2a) to produce 1b and 2b is more favorable. This may be the reason for dual emission in polymer matrices. The luminescence properties of the compounds were dependent on the polarity of polymer matrices due to the mode of hydrogen bonding between the polymers and the ESIPT unit. The luminescence properties of compound 1 were not strongly dependent on the polarity of matrices Fig. 20, while a significant change in luminescence properties was observed for compound 2 in PS, PVAC, PC, PMMA, PVA and PEG polymer matrices Fig. 20. Further, authors also concluded that the relative intensity of normal and ESIPT emission depends on polymer matrices. The luminescence properties were tuned by varying the concentration of PS, PVAC, PC, PMMA, PVA and PEG polymer matrices. In the case of compound 2, as the concentration of 2 increased, the intensity of normal emission gradually decreased with enhancement of ESIPT emission intensity. In short, the polarity of the surrounding polymer matrices tunes the relative intensities as well as emission bands produced by normal and ESIPT emission. Increasing concentration of compound 2 in polymer matrices changes the emission color from purple-white-green Fig. 20.
 | ||
| Fig. 20 (left) CIE 1931 chromaticity diagram of the luminescence (λex = 330 nm) of spin-coated films containing 0.5 w/w% of 1 (a–f) and 2 (A–F) in PS (a, A) PVAC (b, B), PC (c, C), PMMA (d, D), PVA (e, E), and PEG (f, F). Images in the upper right corner show polymer films luminescing under a UV lamp (365 nm). (right) CIE 1931 chromaticity diagram of the luminescence (λex = 333 nm) of spin-coated PVA films containing 0.5–30% w/w of 2. “N” and “E” represent the color indices of the normal and ESIPT emission bands, respectively. The images below show luminescence of the polymer films and the powder of 2 under a UV lamp (365 nm). Reproduced with permission.57 Copyright 2014 American Chemical Society. | ||
Photophysical properties of compounds 1 (λex = 331–336, λNem = 373–381 nm, λESIPTem = 517–540 nm, ΦFL = 23–40%) and 2 (λex = 347–359, λNem = 402–414 nm, λESIPTem = 540–585 nm, ΦFL = 4–10%) in different polymer matrices concluded that compound 2 is more susceptible to the surrounding medium.
Based on DFT computations and FT-IR analysis results, authors concluded that hydrogen bonding between imidazole –N and phenolic –H is weaker due to electron withdrawing cyano groups present in compound 2. This could be the reason for more susceptibility of compound 2 with different polymer matrices.
After the experimental finding for tuning/enhancement of luminescence properties of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines by different means,54–57 theoretical calculations were also performed by Araki et al. to support all the observations.94,95 They used different computational strategies for this study. Density functional theory (DFT), time-dependent density functional theory (TD-DFT), equation-of-motion coupled-cluster singles and doubles (EOM-CCSD), and our own N-layered integrated molecular orbital and molecular mechanics (ONIOM) calculations were performed on Gaussian 09; complete active space multiconfiguration-SCF (CASSCF), multistate-CASPT2 (MS-CASPT2) calculation on MOLCAS 7.4 and fragment molecular orbital-TDDFT (FMO-TDDFT) calculation using GAMESS ver. 2012. Potential energy surface (PESs), geometry optimization, dipole moments, vertical excitations, electronic distribution and energies of different forms were studied for 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridines in the ground and excited state. The experimental results are well in accordance with theoretical results.
In addition to imidazo[1,2-a]pyridine ESIPT emitters, a polymorph dependent ON–OFF luminescence study of the hydroxy benzimidazole type of ESIPT was also studied by the Araki group.102N-Alkyl substituted hydroxybenzimidazole (HBI) was (Scheme 23) used for this study. The luminescence properties of sterically hindered imidazole were compared with reported luminescent HBI. Methyl and tert-butyl groups were introduced to cause steric hindrance with hydrogen atoms of the phenol ring. 2-(1-Methyl-1H-benzo[d]imidazol-2-yl)phenol (MeHBI) exhibits bright ESIPT luminescence in polymorphic form. The emission spectra of MeHBI were different in different solvents. In polar methanol, MeHBI showed single blue emission (E* emission: λex = 288, λem = 352 nm, ΦFL = 30%), while red shifted single emission was observed in cyclohexane (K* emission: λex = 291 and 322, λem = 484 nm, ΦFL = 4%). In THF, MeHBI showed dual emission (E* and K* emission: λex = 292 and 321, λE*em = 355, ΦFL = 2% and λK*em = 483 nm, ΦFL = 1%). Two polymorphic crystals of MeHBI were developed from ethanol NL (non-luminescent) and from benzene L (luminescent) by crystallization techniques. The polymorph obtained from benzene is highly luminescent (λex = 286, λem = 458 nm, ΦFL = 74%, τ = 4.4 ns) in the solid state in comparison to non-luminescent polymorph obtained from ethanol (λex = 291, λem = 470 nm, ΦFL = 0.7%). The 100 fold enhancement of quantum efficiency for polymorph L was described by hydrogen bonding, planarity and molecular packing of polymorphs. The crystal structure data showed that in polymorph L, the torsion angle between two rings is 15°, while in polymorph NL the torsion angle is 127°. This clearly indicates that intramolecular hydrogen bonding in polymorph L is more favorable as compared to that in polymorph NL. The red emission for polymorph L is assigned to ESIPT emission due to planarity and favorable geometry Fig. 21.
 | ||
| Scheme 23 Structures of HBI derivatives. | ||
 | ||
| Fig. 21 Molecular structure (a) MeHBI-L (b) MeHBI-NL (c) THBI and packing (d) MeHBI-L (e) MeHBI-NL (f) THBI, Expect –OH hydrogen other hydrogen atoms are omitted for clarity. Reproduced from the CIF file.102 | ||
In polymorph NL intermolecular hydrogen bonding occurs instead of intra-molecular hydrogen bonding because of twisted conformation which resulted in a non-ESIPT process. Similar to polymorph NL, 2-(1-tertbutyl-1H-benzo[d]imidazol-2-yl)phenol (THBI) is also non-luminescent because of the twisted molecular structure (θ = 104°) which favors intermolecular hydrogen bonding instead of intramolecular hydrogen bonding. These results conclude that the methyl group in MeHBI decreases the coplanarity of HBI which creates two different modes of molecular packing. Authors also studied the effect of temperature on luminescence properties of MeHBI. DSC data of NL showed only a melting point peak, while a peak at 75 °C was observed for L in addition to the melting point peak. The spin coated sample from benzene on quartz showed bright luminescence, indicating that the spin coated sample is in L form. Upon heating, luminescence decreased dramatically up to 75 °C and vanished completely at 90 °C. The XRD patterns of this spin coated heated samples matched with XRD patterns of NL. This concludes that the highly luminescent ON luminescence state of MeHBI can be transferred to the OFF state upon heating. The reverse process OFF–ON is also possible by dissolution in benzene and rapid crystallization. Further, authors also concluded that luminescence properties of MeHBI were mechanically tunable.
ESIPT inspired HBT, HBI, BHO, and imidazo[1,2-a]pyridine are well known as solid state luminescent chromophores and studied extensively in the past decade. Recently Savarimuthu Anthony has reported quinazolinone based (Scheme 24) polymorph-dependent solid state emitters.104
 | ||
| Scheme 24 Structure of HPQ. | ||
2-(2′-Hydroxyphenyl)-4-(3H)-quinazolinone (HPQ) exhibits solid state fluorescence in two different polymorphs through an ESIPT process. Recrystallized HPQ from ethyl acetate, ethanol, methanol, acetone, DMF, DMSO and chloroform and solid powder obtained from the reaction mixture showed blue-solid state fluorescence. Crystallization of HPQ from THF produced two types of crystals; needle shaped and plate shaped. Needle type crystals showed blue emission (HPQ-B) while plate-like crystals showed blue-green emission in the solid state (HPQ-BG). The different colors in two polymorphs were explained on the basis of molecular packing by single crystal analysis. The analysis showed that both crystal (HPQ-B and HPQ-BG) lattices contain intermolecular (amide–amide) as well as intramolecular interactions (N–OH), however the interaction is rather strong in HPQ-BGFig. 22.
 | ||
| Fig. 22 Molecular packing and selected H-bond interactions in the crystal lattice (a and c) HPQ-B and (b and d) HPQ-BG. Reproduced with permission.104 Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In addition to common interactions, unusual intermolecular interactions such as –CO⋯–CO dipolar and OH⋯HC– were observed between phenolic oxygen atoms and hydrogen atoms of the adjacent aromatic ring for HPQ-BGFig. 23.
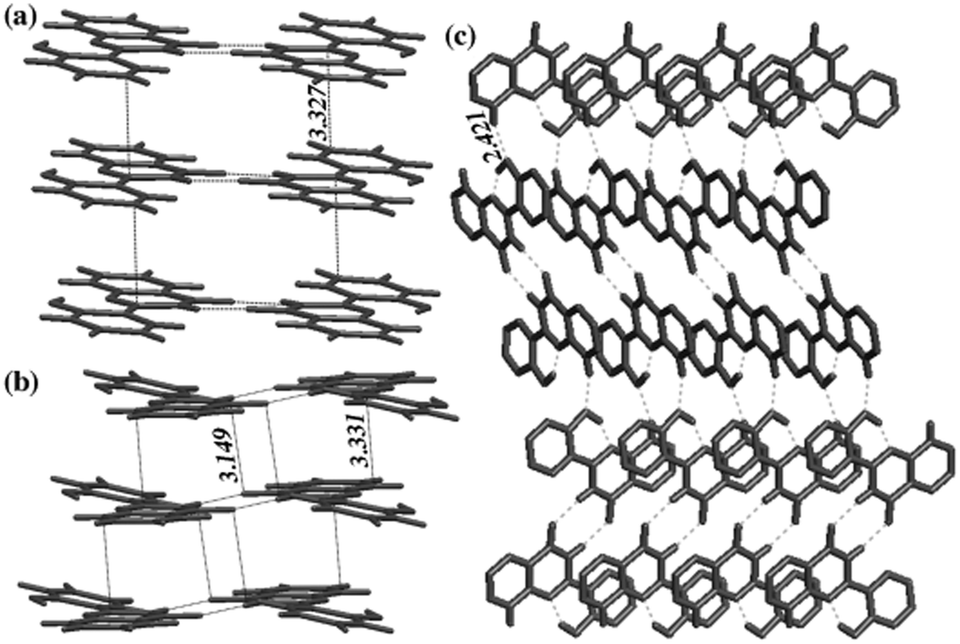 | ||
| Fig. 23 (a) π⋯π interactions in HPQ-B, (b and c) π⋯π, CO⋯CO dipolar and OH⋯HC interactions in HPQ-BG crystal lattices. In (c), only H atoms involved in H-bond interactions are shown. Weak interactions are given as broken lines. Reproduced with permission.104 Copyright 2012 Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim. | ||
The dihedral angle between the quinazolinone and the phenyl ring determines the fluorescence properties of HPQ. In HPQ-B the dihedral angle is 3.68°, while more twisting was observed in HPQ-BG (θ = 9.9°). Polymorphic ability of HPQ leads to tuning of fluorescence properties in the solid state. HPQ-B showed blue shifted emission (λem = 497 nm) in comparison to HPQ-BG (λem = 511 nm). The red shifted emission for HPQ-BG was assigned to additional intermolecular interactions which stabilize the molecular twisting. The emission properties of HPQ were solvent dependent and also reported to have metal chelation ability. The applicability as sensors for Zn2+ and Cd2+ were examined by authors and concluded that the emission is highly selective and sensitive in HPQ towards Zn2+ and Cd2+.
Benzophenone azine (BPA) (Scheme 25) based polymorph dependent ESIPT chromophores exhibiting AIE were reported by Tong et al.99BPA showed switchable solid state fluorescence upon annealing/melting treatment with a large Stokes shift and high fluorescence quantum efficiency. High fluorescence in the solid state was assigned to aggregation, which resulted to strong ESIPT process. This conclusion was supported by red shifted absorption spectra in the aggregate state which is characteristic of compounds showing AIE processes.
 | ||
| Scheme 25 Structure of BPA. | ||
BPA forms two polymorphs namely BPA-1G and BPA-1YG upon crystallization from ethyl acetate. BPA-1G emits in the green (λem = 540 nm, ΦFL = 17%, τ = 1.24 ns) region and BPA-1YG emits in the yellowish-green (λem = 521 nm, ΦFL = 13%, τ = 1.15 ns) region as shown in Fig. 24. This polymorph-dependent fluorescence was explained by the mode of molecular packing of BPA-1 in polymorphs. X-Ray crystal analysis of BPA-1 showed (Fig. 25) the presence of intramolecular hydrogen bonding (YG and G) for the ESIPT process. Two polymorphs were structurally identical, but they were asymmetric in the crystalline form. The torsion angle between phenyl and its attached Schiff base ring part was different for two polymorphs. The torsion is more in polymorph G (θ = 89.3° and 80.4°) in comparison to polymorph YG (θ = 87.5° and 74.7°). The distance between two adjacent layers in polymorph G is higher than that in YG. The difference in molecular packing is a responsible factor for the fluorescence change in two polymorphs. The fluorescence properties of BPA are switchable in polymorphs and powder form after heating or annealing for multiple cycles.
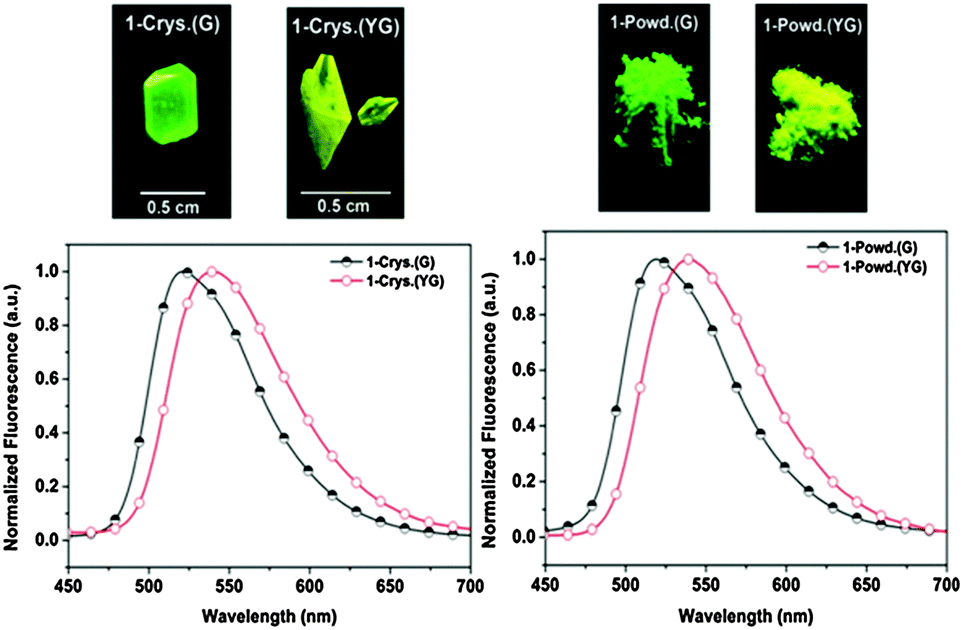 | ||
| Fig. 24 (left) Polymorphic single crystals of BPA-1 (G and YG) under the illumination at 365 nm and their corresponding normalized fluorescence spectra, (right) solid powders of BPA-1 (Powd-G), treated with THF; ((Powd-YG), as prepared) illuminated at 365 nm and their corresponding normalized fluorescence spectra. Reproduced with permission.99 Copyright 2013 American Chemical Society. | ||
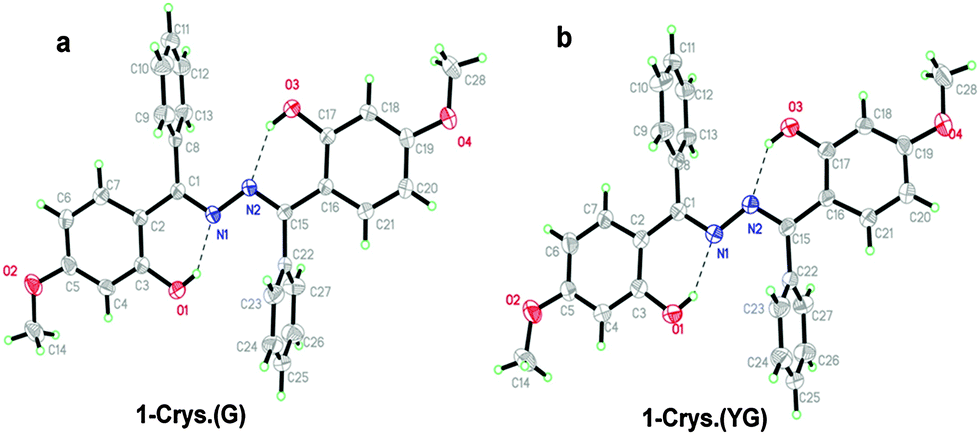 | ||
| Fig. 25 ORTEP drawing with 50% probability ellipsoids (295 K): (a) BPA-1G; (b) BPA-1YG Reproduced with permission.99 Copyright 2013 American Chemical Society. | ||
A short summary of solid-state ESIPT emitters (color tuning) based on polymorph packing, the substituent effect and the type of polymer matrices as dopants is given in Table 7.
| ESIPTs structure | Spectral and photophysical data | Ref. |
|---|---|---|
| Φ FLs: fluorescence quantum efficiency in the solid state, ΦFLpm: fluorescence quantum efficiency in polymorphs, ΦFLp: fluorescence quantum efficiency in polymer matrices, Fl: fluorescence, τ: fluorescence lifetime, CIE: CIE 1931 chromaticity diagram. | ||
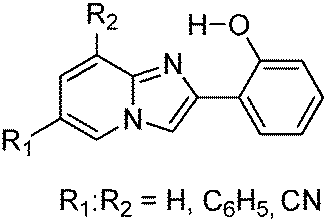
|
White luminescence; ΦFLs = 31–37%; λem = 529–536 nm; CIE (0.36–0.55, 0.41–0.50); combination of ESIPT and non-ESIPT generates white light; white luminescence is a summation of luminescence of ESIPT and non-ESIPT units. | 19 |
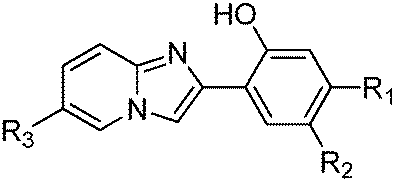
|
Dual emission; ΦFLpm = 6–61%, τ = 2.1–4.9 ns; λem = 378–571 nm, singlet emission from zwitterionic ESIPT species; single as well as dual emission in PMMA; Fl depends on nature and position of substitution; electron donor and acceptor groups on hydroxy phenyl ring causes blue and red shift respectively. | 54 |
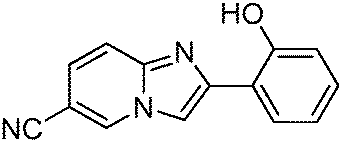
|
Three solid-state luminescence colors; yellow–orange–red; ΦFLp = 10–49%; λem = 548–585 nm; τ = 2.26–5.48 ns; slip and π–π stacking; polymorph dependent ESIPT emission; different molecular packing for three polymorph. | 55 and 57 |
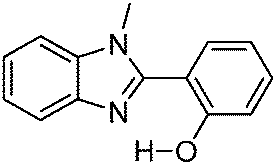
|
Polymorph dependence emission; ΦFLs = 74%; λem = 458 nm; τ = 4.4 ns; coplanar conformation; Fl depends on H-bonding, planarity and molecular packing; two polymorphs, Luminescent and non-luminescent; ON–OFF type of luminescent ESIPT; Fl mechanically tuneable. | 102 |
|
Polymorph dependence emission; λem = 497–511 nm; amide–amide interaction; unusual interactions (CO–CO dipolar and OH⋯HC–); two polymorphs (blue and blue-green). |
104 |
|
Polymorph dependence emission; ΦFLag = 13–17%; λem = 521–540 nm; τ = 1.15–1.24 ns; non-planar conformation; thermochromism; two polymorph (green and yellow-green); polymorph dependence ESIPT; reversible Fl tuning by heating/annealing; thermally stable ∼236 °C. | 99 |
7. Solid state ESIPT emitters through the AIE/AIEE mechanism
In 2001, first report describing the enhancement of fluorescence emission in the aggregate state was reported by Tang and coworkers.136 AIE chromophores are highly emissive in the aggregate state and weakly or non-emissive in solution.70 In recent years, an AIE or an AIEE process has been utilized for the generations of tunable solid state luminescent materials.105,137 In the AIE process, molecules pack efficiently through π–π stack interactions, which blocks the non-radiative deactivation pathways through the restriction of the intramolecular rotation effect (RIR).70 This helps in improving the luminescence properties in the aggregate state. It was reported that H-aggregates cause fluorescence quenching,3,138 while J-aggregates of conjugated molecules enhance the luminescence properties (AIEE).91 AIE and AIEE responsive materials also exhibit multiple color emission due to the mode of packing or different conformation of molecules in the aggregated state. Since 2001 many research groups have reported the design, synthesis and mechanism of solid state chromophores by AIE/AIEE processes for optoelectronic devices.70 However, the AIE and AIEE processes for ESIPT chromophores are rare. The structures of ESIPT solid state chromophores with AIE mechanism reported till 201191,96,139–145 have been summarized in Table 8 and their photophysical properties have been summarized by Park et al.3 This section considers the subsequent report on solid state ESIPT fluorophores with AIE or AIEE properties.| ESIPTs structure | Spectral and photophysical data | Ref. |
|---|---|---|
| Φ FLs: fluorescence quantum efficiency in the solid state, Fl: fluorescence, τ: fluorescence lifetime. | ||
|
AIEE; ΦFL = ∼13%; λem = ∼513–570 nm; τ = 2.32–3.09 ns; green to red emission depending on nature of substituents; J- & H-aggregation; intramolecular rotation allowed only for N–N bond; AIEE pH dependent. | 145 |
|
AIE; ΦFL = ∼6%; λem = ∼546 nm; τ = 2.1 ns; 60 fold enhancement of quantum efficiency in aggregate state; J-aggregates; orange crystal in aggregate state. | 140 |
|
Gelation-induced enhanced emission; ΦFL = ∼3%; λem = 520 nm; helical J-type packing; cholesterol groups as a gelator moieties; photochromism; photoinduced isomerization. | 141 and 142 |
|
AIEE; ΦFL = ∼29%; λem = ∼510 nm; J- & H-aggregation; restricted intramolecular/intermolecular motion in aggregated state; 21 fold enhancement of quantum efficiency in aggregated state; red shifted emission in aggregate state; J-aggregates causes enhanced emission; H-aggregates would induce a decreased emission; molecular packing depended on nature of alkyl group. | 91 |
|
Blue-green K* emission; ΦFL = ∼47%; λem = ∼486 nm; τ = 3900 ps; slip-stacking; planar; intramolecular H-interaction; dual emission in solvents. | 139 |
|
Orange K* emission; ΦFL = ∼13%; λem = ∼573 nm; τ = 650 ps; slip-stacking; dual emission in solvents. | 139 |
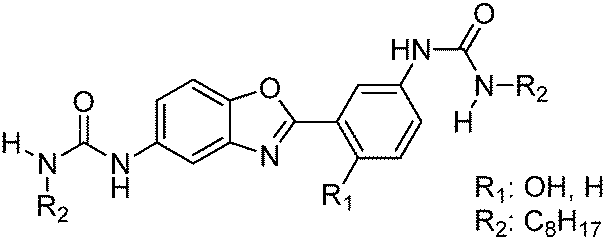
|
Gelation-induced enhanced emission; ΦFL = ∼35%; λem = ∼402 and 519 nm; τ = 0.5–7.2 ns; π–π stacking; planar conformation; H-bonding interaction; abnormal AIE; AIE causes blue shifted emission. | 143 |
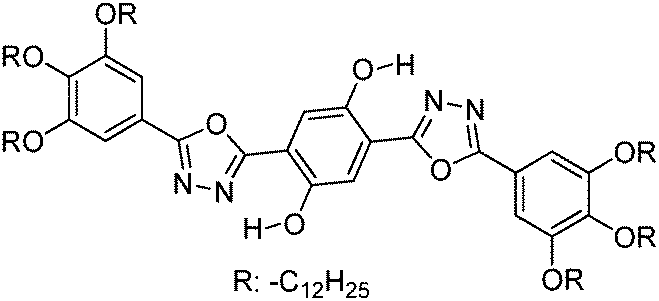
|
Liquid crystals; ΦFL = ∼34%; λem = ∼422 and 576 nm; J-stacking; two mesophases (columnar hexagonal and rectangular); temperature dependent emission. | 144 |
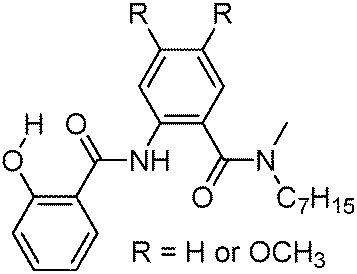
|
Gelation-induced enhanced emission; ΦFL = ∼13%; λem = ∼470 nm; rigidochromism; π-stacking/columnar hexagonal packing; 10–50 folds fluorescence enhancement in gel state; gelation due to intramolecular H-interaction; restriction of TICT. | 96 |
AIE/AIEE characteristics of the reported chromophores are due to the restriction of the intramolecular rotation effect (RIR) through organic ion formation or hydrogen bond formation. Recently, AIE processes through the restriction of charge transfer (RCT)146 or the restriction of twisted intramolecular charge transfer (RTICT)85 and restriction of cis–trans isomerization have been reported.84 Huang and colleagues synthesized HBT based chromophores84N-(3-(benzo[d]thiazol-2-yl)-4-hydroxyphenyl)phenyl-carboxamide (TA) and N-(3-(benzo[d]thiazol-2-yl)-4-hydroxyphenyl)anthracene-9-carboxamide (TAA) (Scheme 26), which are analogues of DHIA have been reported by Yang and coworkers.91
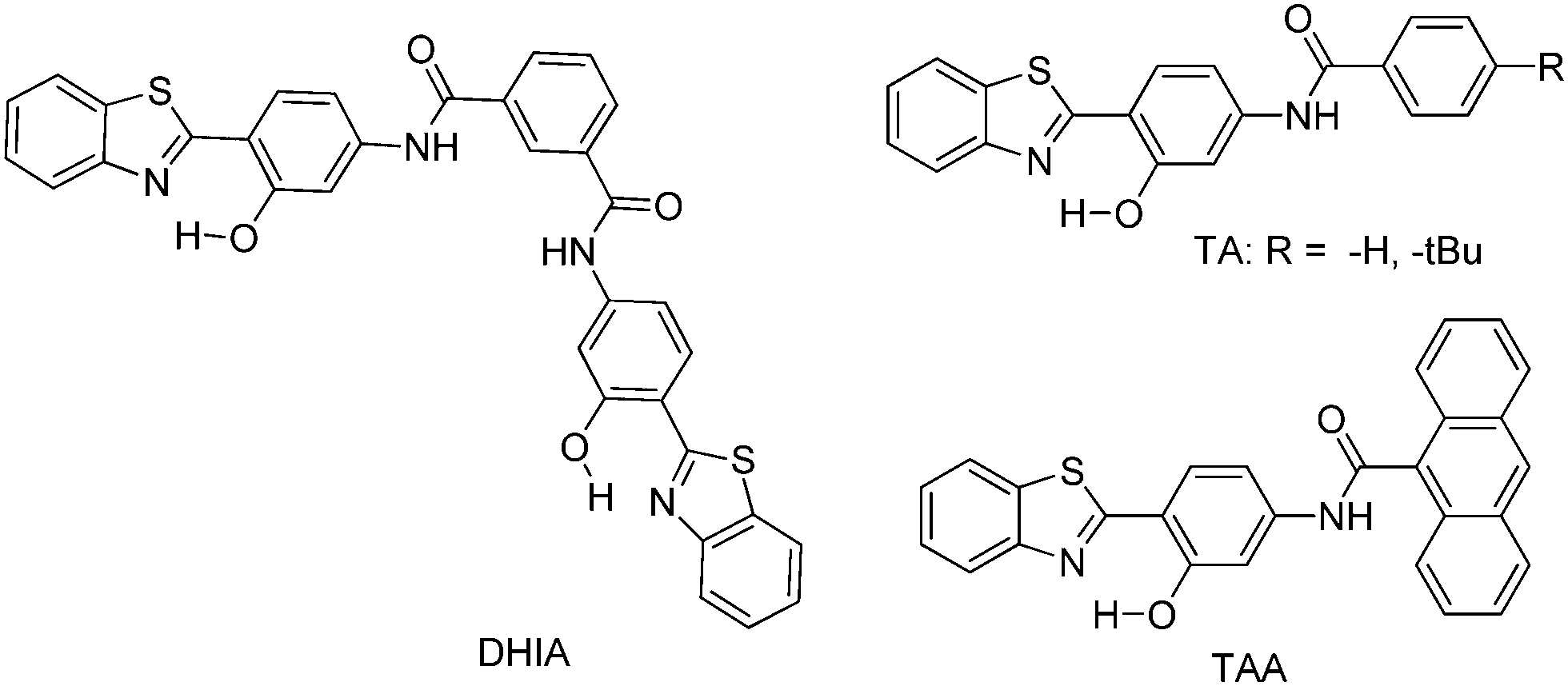 | ||
| Scheme 26 Structures of TA, TAA and DHIA. | ||
TA and TAA showed very weak fluorescence in solvents of different polarities, but the fluorescence intensity was significantly enhanced upon aggregation as demonstrated in Fig. 26. The absorption maxima for TA and TAA aggregates were red shifted compared to dilute solution (THF). This red shift can be assigned to rigid crystalline packing of molecules in the aggregated state. It was reported that the AIEE mechanism is more efficient in non-planar structures.70 In crystalline TAA, the dihedral angle between HBT and the anthracene unit is 80.7° which prevents quenching by blocking face-to-face interactions in the aggregate state. In TAA, molecular packing is head-to-tail between two units which can be stabilized by N–π stacking between nitrogen atoms of two thiazole units Fig. 27. The bathochromic shift in absorption indicates the formation of J-type aggregates which were supported by π–π interactions between two adjacent anthracene units and Ar–H⋯OC– hydrogen bond between two adjacent TAA. Yang and coworkers also reported that AIEE for the DHIA type of chromophores was due to J- or H-type aggregation which restricts the non-radiative TICT of the enol excited state. A similar process occurs in TA for AIEE processes. In HBT derivatives, cis–trans isomerization of the keto form can effectively deactivate the excited state which causes quenching of fluorescence emission in solution. In the aggregate state, cis–trans isomerization is effectively blocked through tight molecular packing. This conformational change significantly suppresses the deactivation pathways and populates the radiative decay. The fluorescence lifetime of TAA in THF is less than 100 ps, indicating significant nonradiative decay. In the aggregate state, the fluorescence lifetime is significantly enhanced to 2.55 ns. Based on these results Huang et al. concluded that the AIEE mechanism for the TAA chromophore is not only because of restricted TICT, but also from the effective restriction of cis–trans isomerization in the keto excited state. TAA showed bright fluorescence in the solid state (ΦFL = 78.1%, τ = 2.55 ns) which was 27 fold higher than DHIA.
 | ||
| Fig. 26 Photographs of TAA under UV illumination at 365 nm in THF, water and powder state from left to right, respectively. Reproduced with permission.84 Copyright 2012 PCCP Owner Societies. | ||
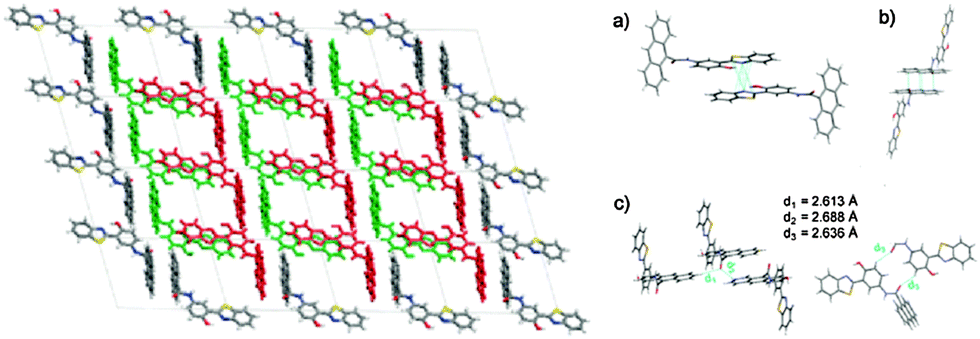 | ||
| Fig. 27 (left) Molecular packing of TAA in single crystals viewed along the b–c plane (right) (a) N⋯π interaction of HBT subunits. (b) π–π interaction of anthracene subunits. (c) Multiple aromatic Ar–H⋯OC hydrogen bonds. Reproduced with permission.84 Copyright 2012 PCCP Owner Societies. | ||
The OLED device was fabricated using TAA as the emissive layer, with a device configuration [ITO/NPB (60 nm)/TAA (10 nm)/TPBI (50 nm)/LiF (1 nm)/Al (80 nm)]. NPB and TPBI were used as hole and electron transport layers. The device showed a maximum luminescence of 5616 cd m−2 at 11.85 V. The device offers an external quantum efficiency of 0.46% with a current density of 0.50 mA cm−2, equivalent to a current efficiency of 1.18 cd A−1. The red shifted electroluminescence (λel = 560 nm) peak was observed in comparison to fluorescence emission (λem = 552 nm). This red shift may be caused by a more ordered molecular arrangement.147
Gelators functionalized by the ESIPT unit are known for solid state emission for use in photoelectrical and nano-electrical devices. Few reports based on ESIPT organogelators (Table 8) which contain long alkyl chains or dendrimeric units have been reported in the past.96,142,143 Recently, Yang and coworkers148 synthesized highly emissive ESIPT organogelators based on 2-hydroxybenzoxazole (HBO). N,N-Bis(4-(benzo[d]oxazol-2-yl)-3-hydroxyphenyl)isophthalamide (BHPIA) and N,N-bis(4-(benzo[d]oxazol-2-yl)-3-hydroxyphenyl)-5-tert-butylisophthalamide (BHPBIA) contain small alkyl chains (Scheme 27). These gelators are structural analogues of DHIA, reported by the same group.91 The gelators BHPIA and BHPBIA are highly emissive in the solid state with a large Stokes shift of 137 nm. This characteristic was assigned to AIEE due to J-aggregation and the ESIPT process. Hydrogen and tert-butyl groups at the central ring were incorporated purposefully to study the effect of slip-stacking on the ESIPT process as well as to study the substituent effect for organogel formation. BHPBIA forms an organogel effectively in comparison to BHPIA because the tert-butyl group reduces tight molecular stacking and intermolecular interactions. Optical properties, molecular stacking, and FT-IR studies indicate that four types of interactions (N–π, O–π stacking of the oxazole rings, π–π stacking of phenol rings and consecutive H-bonding) interplayed in BHPBIA. These interactions help in the formation of long-range-1-D nanostructures.
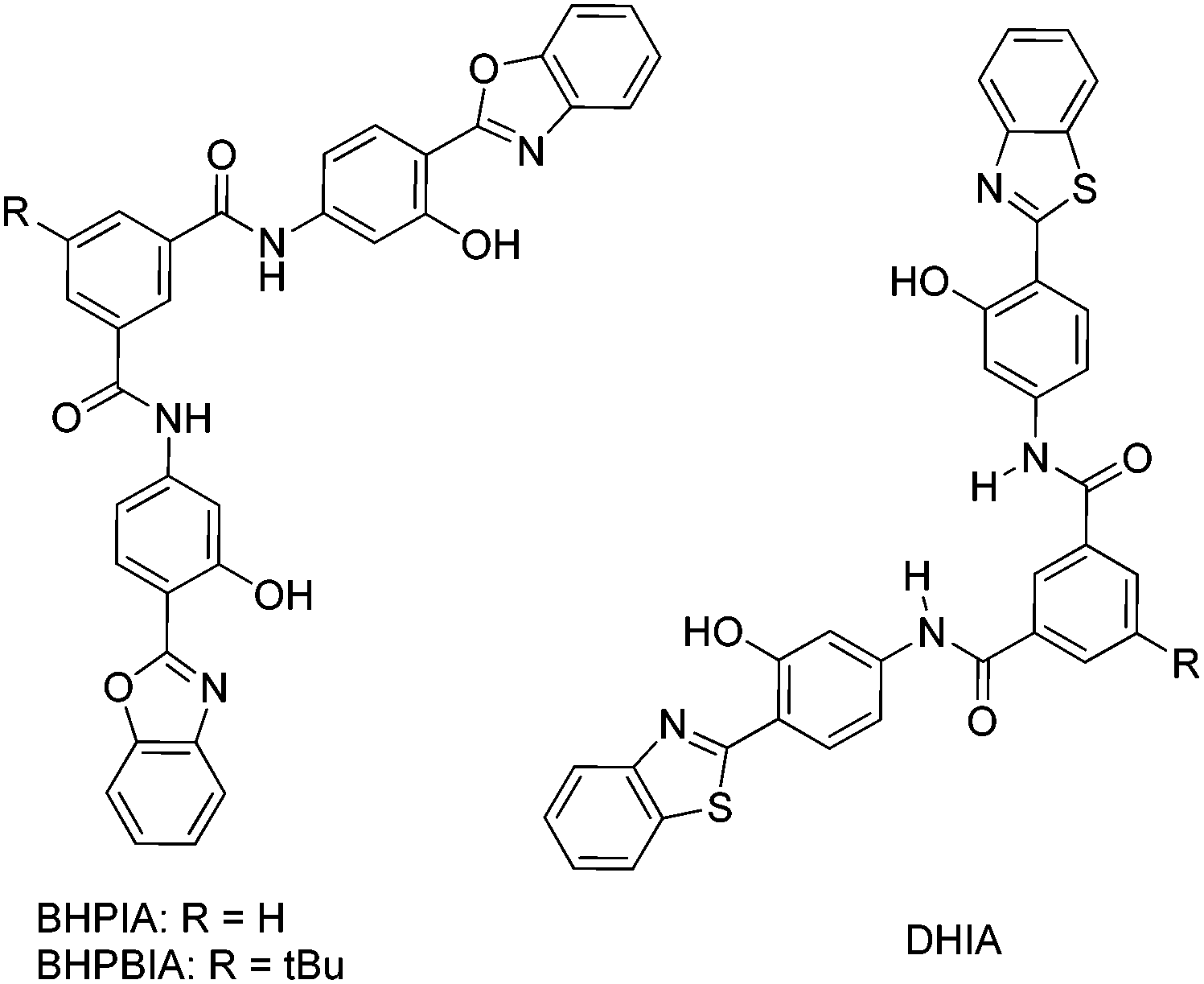 | ||
| Scheme 27 ESIPT organogelators based on hydroxy benzimidazoles (BHPIA and BHPBIA), and their analogue DHIA. | ||
Xia et al. synthesized a novel class of ESIPT fluorophores based on the quinoxalinone core which showed aggregation induced emission.100 Various substituted (Z)-3-(quinolin-2-ylmethylene)-3,4-dihydroquinoxaline-2(1H)-one (Q1–Q5) derivatives (Scheme 28) were synthesized and their photophysical properties studied in the solid state and in solution.
 | ||
| Scheme 28 Substituted (Z)-3-(quinolin-2-ylmethylene)-3,4-dihydroquinoxaline-2(1H)-one derivatives. | ||
Compounds showed intense orange-red fluorescence in the solid state with a large Stokes shift similar to emission spectra in THF. In the aggregated state, compounds showed ∼50 fold enhancement of fluorescence quantum efficiencies in comparison to that in solution. In both the solid state and solution, the compounds had been predicted to take planar geometry from DFT computations. Although a bathochromic shift in absorption or appearances of a new band in the red region with respect to original bands are well known characteristics of J-aggregated chromophores. These observations were not seen at various THF–water ratios. As a consequence, Xia et al. concluded that AIE for Q1–Q5 was not due to planarity or the RIR effect in the solid state. However AIE is due to the restraint TICT process in the aggregated molecules in the excited state which was predicated by theoretical calculations.
Subsequently, the Xia group reported combined RTICT and RIR processes for AIE in the ESIPT system92 (Scheme 29). They synthesized two chromophores containing ESIPT units. A tetraphenylethene (TPE) group was introduced on the N atom (Q6) of dihydroquinoxalinone and on the phenyl ring of the quinoline core (Q7). The AIE process due to RIR and RTICT units or RIR and multiple RTICT units has been reported in the past for π-conjugated chromophores.149–152 However, the AIE mechanism for ESIPT Q6 and Q7 was different in comparison to reported AIE systems. The absorption and emission properties of Q (λabs = 430, 452 nm, λem = 486, 544, 583 nm), Q6 (λabs = 432, 455 nm, λem = 543, 585 nm), and Q7 (λabs = 444, 460 nm, λem = 555, 595 nm) suggest that the introduction of a TPE unit on the parent core will not change the ESIPT characteristics. However it affects the AIE mechanism.
 | ||
| Scheme 29 TICT and IR mechanisms of Q, Q6 and Q7. | ||
In the aggregate state, fluorescence intensity increases significantly with red shifted emission and an enhancement of 2.7 and 11.5-fold emission intensity in Q6 and Q7 respectively due to the introduction of a TPE unit which is responsible to block radiativeless pathways effectively. The higher emission intensity for Q7 was assigned to the crystallization induced emission enhancement (CIEE) which was supported by molecular morphology. Based on theoretical calculation, Xia et al. concluded that the AIE mechanism in parent Q was due to RTICT around the exocyclic bond, while AIE in Q6 and Q7 was due to RIR of TPE phenyl rings (Scheme 29). Authors also claimed that this is the first report for such a type of AIE.
Recently, tunable luminescence and white-light emission from single gelator molecules through the ESIPT coupled AIEE process were reported by Das et al.105 In this article, authors claim that this is the first report describing the ESIPT coupled AIEE process for single molecules in the generation of tunable luminescence. Two poly(aryl ether) dendron based gelators D1 and D2 were synthesized (Scheme 30) by the Das group.
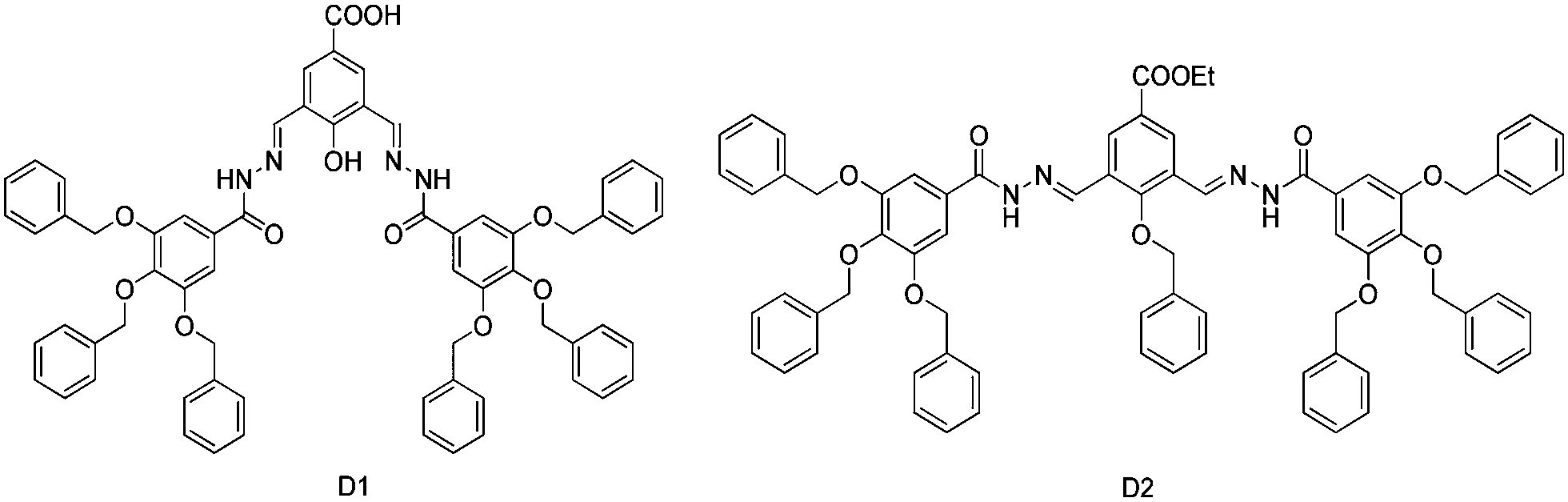 | ||
| Scheme 30 Poly(arylether) dendron based gelators D1 and D2. | ||
Compound D1 contains a basic flexible imine group and acidic phenolic hydrogen suitable for ESIPT. To understand the role of the ESIPT process in the luminescence enhancement, dendron D2 was synthesized for a comparison study. The absorption and fluorescence properties of D1 were studied in pure THF as well as in various ratios of a THF/water mixture. In pure THF, D1 showed two absorption bands (λabs = 306 and 355 nm), however, the intensity of absorption bands decreases upon addition of water due to the formation of aggregates. Dendron D1 (–OH group) has one hydroxyl group which can undergo ESIPT and showed dual emission in THF (λex = 365 nm; λem = 454 and 487 nm). In a THF–water mixture (1:1) along with dual emission, a new emission band (λem = 551 nm) was observed for D1. In higher percentage of water, the emission band at 487 nm was disappeared and intensity of 551 nm band increased. This emission band (λem = 551 nm) with a large Stokes shift of 186 nm was assigned to the AIEE band. After various combinations of water/THF, both ESIPT and AIEE processes were concluded to be operational at a 1:1 water/THF ratio with dual emission (λem = 468 and 551 nm). At a higher concentration of water the intensity of the 551 nm band increases, which indicates that the AIEE process dominates over the ESIPT process. The simultaneous ESIPT and AIEE processes in D1 are due to the lock conformer Fig. 28.
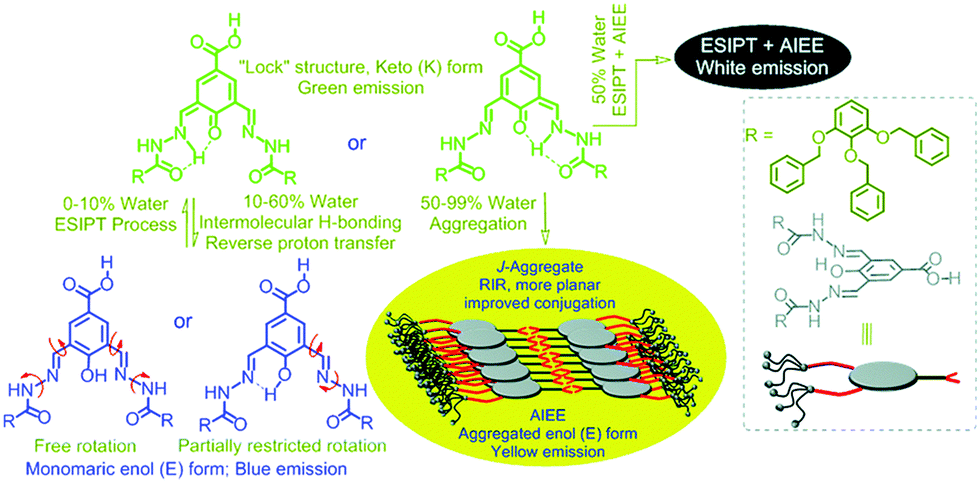 | ||
| Fig. 28 Changes in the configuration of D1 from the twisted ground state to a more planar excited state, and finally J-aggregated rigid structure through ESIPT coupled AIEE processes with the variation of the water fraction to its THF solution. Reproduced with permission.105 Copyright 2015 The Royal Society of Chemistry. | ||
In the case of D2 a single broad emission band (λem = 481 nm) was observed under similar conditions, this was assigned to the AIE process. This clearly indicates that D1 undergoes ESIPT as well as AIEE processes, while D2 is only susceptible to AIE. The fluorescence properties of the D1 depend on the ratio of THF/water, at a 1:1 ratio D1 covered the entire visible spectrum (400–700 nm) resulting in white-light emission with CIE co-ordinates (x, y) = (0.31, 0.37). Field emission scanning electron microscopy (FESEM) study results clearly indicate that conformational changes occur after varying the amount of water. The aggregation was J-type one confirmed by electronic properties and X-ray powder diffraction (XPRD). The Fourier transform infrared spectroscopy (FT-IR) study also supported the observation of a locked structure of D1 over D2. The compound D1 is highly emissive in the powder form and on thin films, while D2 is unable to form a gel under similar conditions.
Recently, the AIE phenomenon has been demonstrated to be an efficient tool for monitoring the activities of various biomolecules. Huang et al. prepared a small ESIPT molecule 2-((4-(benzo[d]thiazol-2-yl)-3-hydroxyphenyl)carbamoyl)benzoic acid (TIA) (Scheme 31) exhibiting AIE for fast and photostable turn-on bioimaging.98TIA is an analogue of TA, TAA and DHIA and is soluble in organic solvents like THF, DMF and DMSO and also poorly soluble in water. TIA showed weak dual emission in THF. However it is strongly emissive in the aggregate form and in the solid state. A 52 fold enhancement of fluorescence quantum efficiency (ΦFL = 4%) was observed in the solid state in comparison to dilute solution. The emission bands between 320–420 nm and 470–600 nm were assigned to enol and keto emission respectively. This chromophore was used for the sensing of HeLa cells and it was concluded that TIA is very sensitive to HeLa cells.
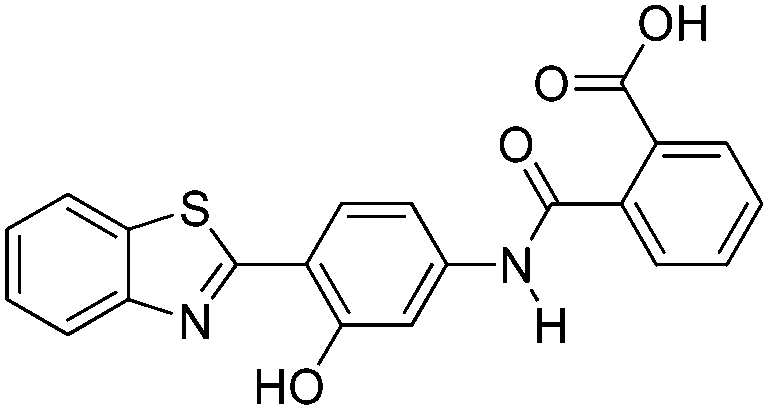 | ||
| Scheme 31 Structure of TIA. | ||
Solid state ESIPT emitters based on AIE–AIEE are summarized in Table 9.
| ESIPT chromophore structure | Spectral and photophysical data | Ref. |
|---|---|---|
| Φ FLag: quantum efficiencies in the aggregated state, τ: fluorescence lifetime, ΦFLs: quantum efficiencies in the solid state. | ||
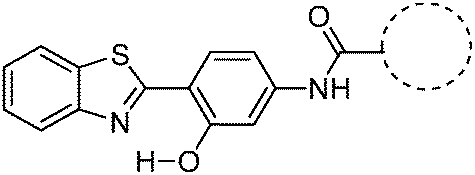
|
AIEE; ΦFLag = ∼78.1%; τ = 2.55 ns; J-type aggregation; N–π stacking; AIEE due to RTICT and restriction of cis–trans isomerization; non-planer conformation. | 84 and 91 |
|
AIE; ΦFLs = 4%; λem = 470–600 nm; 52 fold enhancement Fl intensity after aggregation. | 98 |
|
AIEE; λem = ∼360–420 and ∼477 nm; Δλ = ∼137 nm; slip-stacking; J-aggregation; RIR; thermo-reversible gel; alkyl group prohibits tight molecular packing and intermolecular interaction. low-molecular-weight-mass organogelators with AIEE and ESIPT properties. | 148 |
|
AIE; AIEE; λem = ∼500 and ∼600 nm; Many fold (∼36) enhancement of Fl intensity upon aggregation; J-aggregation; AIE/AIEE due to RIR, RTICT and CIEE effects; morphology dependent emission; first report for solid state emission due to RIR and CIEE in one molecule. | 92 and 100 |
|
AIEE; blue–green–white–yellow emission; λem = ∼487 and ∼551 nm; J-aggregation; RIR; intramolecular H-interaction; AIEE coupled with ESIPT; first report for AIEE-ESIPT for single molecule for tuneable color. | 105 |
In addition to the above solid-state ESIPT emitters caused by the AIE/AIEE process, ESIPT chromophores exhibiting the AIE/AIEE mechanism in aqueous/solution were also reported in the last few years.52,153–157 These types of chromophores were used for biosensing applications, but solid-state properties of these fluorophores were not explored in detail by respective groups. Different types of ESIPT chromophores52,153–157 enhancing the fluorescence by AIE or AIEE mechanisms are summarized in Table 10.
| ESIPT chromophore structure | Spectral and photophysical data | Ref. |
|---|---|---|
| λ ems: emission in the solid state, λemag: emission in the aggregated state, ΦFLag: quantum efficiency in the aggregated state, Np: data not available. | ||
|
AIE; ΦFLag = 54%; λem = ∼531 nm; J-aggregation; yellow emission; AIE due to RIR; fluorescence enhancement is due to aggregation induced planarization and J-aggregate formation; thermally stable ∼300 °C. | 52 |
|
AIE; ΦFLag = 82%; λem = ∼570 nm; J-aggregation; reddish-orange emission; AIE due to RIR; thermally stable ∼300 °C. | 52 |
|
AIEE; ΦFLag = 45%; λemag = 441 nm; J-aggregation. | 153 |
|
AIE; λemag = 532 nm; planar, rigid, intramolecular H-bonding; 14 fold enhancement of emission in aggregated state. | 154 |
|
AIE; ΦFLag = 10%; λemag = 517 nm, H-aggregation; two-photon absorption. | 155 |
|
AIE; ΦFLag = 6%; λemag = 575 nm, J-aggregation; two-photon absorption. | 155 |
|
AIE; ΦFLag = ∼26%; λemag = ∼547 nm, J-or H-aggregation; co-planar. | 155 |
|
AIE; ΦFL = 18%; λem = 640 nm; 60 fold enhancement of fluorescence intensity in aggregate state; absence of π–π stacking. | 156 |
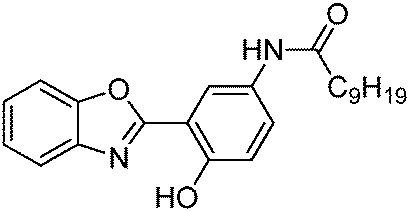
|
AIEE; λem = 500 nm. | 157 |
Chromophores containing ESIPT units with an enhancement of fluorescence properties in the solid state or the aggregate state through AIE/AIEE have also been reported in recent years.158–160 The solid state properties of these chromophores were also studied for different applications. However, the role of the ESIPT unit in the enhancement of fluorescence efficiency was not considered for these fluorophores. The reported representative examples158–160 are summarized in Table 11.
| ESIPT chromophore structure | Spectral and photophysical data | Ref. |
|---|---|---|
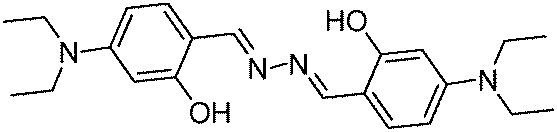
|
Donor–acceptor chromophore; ΦFL = 8–33%; λem = 529–553 nm; π–π interaction; planar conformation; intramolecular H-interaction; piezo/thermo responsive emission. | 158 |
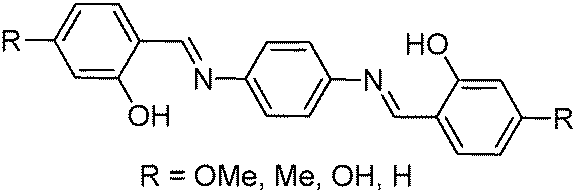
|
AIE; ΦFL = 8–33%; λem = 503–543 nm; morphology controlled emission; tuneable three color emission (green, yellow, orange); thermally stable ∼319 °C. | 159 |
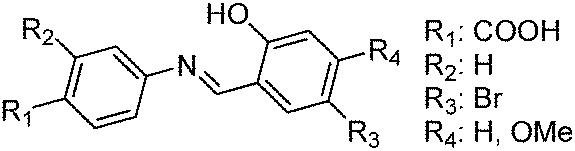
|
AIEE; ΦFL = 5–17%; λem = 512–556 nm; AIEE due to restriction of intramolecular rotation of C–N bond; thermochromism. | 160 |
8. Summary and outlooks
This article summarizes the progress towards the development of solid state ESIPT emitters mainly focusing on photophysical properties in the solid state. The summarized solid state ESIPT fluorophores are derivatives of carbazole, benzothiazole, imidazole, indanone, phenanthroimidazoles, benzofuranbenzoxazoles, julidine, bis-benzothiazole, imidazopyridine, indolizines, quinazolinone, polyimide, benzophenoneazines, quinoxalines and azine having red/white-light emission. As summarized, ESIPT fluorophores are emissive (i) in the polymorphic forms (ii) in the gel (iii) in the polymer matrices (v) in the KBr matrix (vi) in the aggregate state and (vii) in the solution.Most of them are white-light emitters in the solid state and in the solution. A short summary of various approaches for the generation of white-light emission from ESIPT chromophores is listed below.
• White-light-emission through a single molecule dyad.
• Fine tuning the energetics of ESIPT.
• Based on the solvatochromism properties.
• Mixture of proton-transfer and nonproton-transfer fluorophores.
• Mixture of two ESIPT luminophores having different Stokes shift.
• Mixture of two complementary, blue- and orange-emitting chromophores exhibiting ESIPT processes.
• Panchromatic emission via modulating the substitution patterns of ESIPT chromophores.
• Tuning of acidic and basic centers of ESIPT chromophores.
• Altering the surrounding medium (e.g. polymer matrices or rigid media) of ESIPT chromophores.
• Controlling molecular packing of ESIPT chromophores (e.g. polymorph dependence emission).
• AIE and AIEE processes generate white light emission of ESIPT chromophores.
• Based on the halochromism properties of ESIPT chromophores.
In reported ESIPT systems, the fluorescence properties are superior (high fluorescence quantum efficiencies, red emission etc.) in solid/rigid media in comparison to solution. The enhancement of fluorescence quantum efficiencies in the solid state has been realized by many approaches such as restricted intramolecular rotation (RIR), hydrogen bonding, the restriction of twisted intramolecular charge transfer (RTICT), flipping of flexible alicyclic components, J-aggregations, cross packing, and dimer/excimer stacking.
Based on the different design strategies and photophysical data of the summarized ESIPT fluorophores in the solid state/polymer matrices/rigid media/solution, here we have summarized a few points which suppress the other deactivation pathways occurring in the excited state resulting in the enhancement of fluorescence quantum yields and red shifted emission.
• Planar geometry in the excited state helps to enhance the fluorescence properties over non-planar geometry.
• Strong intra-molecular hydrogen bonding favors the ESIPT process over weak H-bonding.
• RIR, RTICT and RCT enhance the fluorescence properties.
• J-aggregation with a head-to-tail arrangement greatly blocks the nonradiative pathways and effectively suppresses the self quenching which helps in the enhancement of fluorescence properties over the H-type of aggregation (face-to-face arrangement).
• Intramolecular stacking interactions in a crystal (face-to-face intermolecular interactions between π-conjugated chromophores) quench the fluorescence expect in a few cases causing a fluorescence enhancement via a restricted intramolecular rotation (RIR) effect.
• Introduction of steric groups on the ESIPT chromophores could improve the fluorescence properties by limiting the non-radiative pathways via vibronic and rotational relaxation.
• Two molecular freely rotatable joints in solution (free state) can be locked in the aggregate state by the formation of a strong intramolecular H-bond with basic atoms (locked state i.e. the RIR effect) which causes the enhancement of quantum efficiency.
• In polar/protic solvents generally quenching of keto emission is caused because of intermolecular hydrogen bonding between solvents and acidic hydrogen instead of intramolecular hydrogen bonding.
• pH of the medium determines the red/blue shift of ESIPT chromophores (generally basic media red shift and acidic media blue shift).
• Restricted cis–trans isomerization of the excited keto form causes the enhancement of fluorescence quantum efficiencies by suppressing the deactivation pathway (restricted cis–trans isomerization is more effective in rigid media).
• π–π interplanar distance between two units determines the fluorescence behavior of ESIPT chromophores.
• Dendrimer units on the ESIPT core help to enhance fluorescence quantum efficiencies via inhibition of fluorescence quenching due to intermolecular interactions (bulky dendrimeric units enhance the fluorescence quantum efficiencies by suppressing the isomerization process).
• The twisted intramolecular charge transfer (TICT) pathway in the excited state keto form quenches the fluorescence.
• 5-Membered (generally) ring ESIPT fluorophores exhibit higher fluorescence quantum efficiencies than 6-membered ESIPT fluorophores (planar conformation is more stable in 5-membered ring ESIPT molecules).
• Fluorescence quantum efficiencies depend on the nature and position of substituted groups.
• Tuning/enhancement of fluorescence depends on the extension of conjugation, the nature of heterocyclic rings, the nature of electron donor/acceptor groups and supported matrices.
• In polymer matrices, the polarity of the surrounding matrix controls the fluorescence emission as well as fluorescence quantum efficiencies.
• Mode of molecular packing helps to tune the color of ESIPT chromophores (ESIPT chromophores give varying emissions in different polymorphic forms).
• AIE and AIEE processes enhance the quantum efficiencies of the ESIPT fluorophores in the solid state (generally).
• In imidazopyridine, extensions of π-conjugation cause a red shift in emission but lowering in the fluorescence quantum efficiencies.
• Energy-level mismatch between the LUMOs of the enol and the keto tautomer is a key factor for the suppression of non-radiative pathways (large mismatch of their molecular orbital's energy levels) which causes the fluorescence enhancement.
• Nodal plane model is a simple but informative tool for the prediction of fluorescence properties of known fluorophores as well as for designing fluorophores as per emission requirements.
The above mentioned strategies will guide in designing or tuning the properties of solid state ESIPT emitters.
The reported ESIPT inspired fluorescent materials were used for the fabrication of organic light emitting diodes (OLEDs), field-effect transistors, white-light emitters for optoelectronic devices, metal sensors and bioimaging devices. Tremendous interdisciplinary research efforts over the last five years have made significant progress in the designing; synthesis and applications of ESIPT chromophores. We believe that the innovative design, efficient synthesis and improved fluorescence properties will be some of the prime challenges on the path of developing highly emissive solid state ESIPT chromophores. We hope that this review will provide sufficient information to design new strategies for the development of ESIPT materials which will emit in the red region with very high fluorescence quantum efficiencies.
Abbreviations
| ESIPT | Excited state intramolecular proton transfer |
| λ abs | Absorption maxima wavelength |
| λ em | Emission maxima wavelength |
| λ ex | Excitation wavelength |
| Φ FL | Fluorescence quantum efficiency |
| E* | Enol emission |
| K* | Keto emission (ESIPT emission) |
| τ | Fluorescence lifetime |
| Δλ | Stokes shift |
| EL | Electroluminescence |
| B | Maximum brightness |
| WLE | White light emission |
| EQE | External quantum efficiency |
| ACQ | Aggregation caused quenching |
| AIE | Aggregation induced emission |
| AIEE | Aggregation induced enhanced emission |
| RIR | Restricted intramolecular rotation |
| TICT | Twisted intramolecular charge transfer |
| RTICT | Restriction of twisted intramolecular charge transfer |
| IPT | Intramolecular proton transfer |
| ICT | Intramolecular charge transfer |
| RCT | Restriction of charge transfer |
| CIEE | Crystallization induced emission enhancement |
| OLED | Organic light emitting diodes |
| WOLEDs | White organic light emitting diodes |
| GFP | Green fluorescent protein |
| CIE | International commission on illumination |
| HBI | 2-(2′-Hydroxyphenyl)benzimidazole |
| HBO | 2-(2′-Hydroxyphenyl)benzoxazole |
| HBT | 2-(2′-Hydroxyphenyl)benzothiazole |
| PIP | 2-Phenylimidazo[1,2-a]pyridine |
| TPBI | 1,3,5-Tri(1-phenyl-1H-benzo[d]imidazol-2-yl)phenyl |
| NPD | 4,4′-Bis[N-(1-naphthyl)-N-phenylamino]biphenyl |
| CPB | 4,4′-N,N-Dicarbonyl-1,1-biphenyl |
| NPB | 4,4′-Bis[N-(1-naphthyl)-N-phenylamino]biphenyl |
| TCTA | 4,4′,4-Tri(N-carbazolyl)triphenylamine |
| CBP | 4,4′-Bis-(9-carbazolyl)biphenyl |
| PMAA | Poly(methyl methacrylate) |
| PS | Polystyrene |
| PVAC | Poly(vinyl acetate) |
| PC | Poly(bisphenol A carbonate) |
| PVA | Poly(vinyl alcohol) |
| PEG | Poly(ethylene glycol) |
| PE-DOT | Poly(ethylene dioxythiophene) |
| PSS | Poly(styrene sulfonate) |
| HPhI | 2-(1H-Phenanthro[9,10-d]imidazol-2-yl)phenol |
| HPPhI | 2-(1-Phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol |
| TsPPhI | 4-Methyl-N-(2-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)benzenesulfonamide |
| HBDI | (Z)-4-(4-Hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one (HBDI) |
| HBBO | 2-2′-(Hydroxybenzofuran)benzoxazole |
| TA | N-(3-(Benzo[d]thiazol-2-yl)-4-hydroxyphenyl)phenyl-carboxamide |
| TAA | N-(3-(Benzo[d]thiazol-2-yl)-4-hydroxyphenyl)anthracene-9-carboxamide |
| TIA | 2-((4-(Benzo[d]thiazol-2-yl)-3-hydroxyphenyl)carbamoyl)benzoic acid |
| 2,4-DBTP | 2,4-Dibenzothiazolylphenols |
| 2,6-DBTP | 2,6-Dibenzothiazolylphenols |
| MeHBI | 2-(1-Methyl-1H-benzo[d]imidazol-2-yl)phenol |
| TBHI | 2-(1-tert-Butyl-1H-benzo[d]imidazol-2-yl)phenol |
| HPQ | 2-(2′-Hydroxyphenyl)-4-(3H)-quinazolinone |
| BPA | Benzophenone azine |
| BHPIA | N,N-Bis(4-benzo[d]oxazol-2-yl)-3-(hydroxyphenyl)isophthalamide |
| BHPBIA | N,N-Bis(4-benzo[d]oxazol-2-yl)-3-(hydroxyphenyl)-5-tert-butylisophthalamide |
| TPE | Tetraphenylethene |
| NHPI | 1-(1,4,5-Triphenyl-1H-imidazo-2-yl)phenol |
| HPIC | 2-(1-Phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol |
| HBPI | 3-(1,4,5-Triphenyl-1H-imidazol-2-yl)-[1,1′-biphenyl]-4-ol |
| HBPIC | 3-(1-Phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-[1,1′-biphenyl]-4-ol |
| HPO | 2-(4,5-Diphenyloxazol-2-yl)phenol |
| HPNI | 3-(1,4,5-Triphenyl-1H-imidazol-2-yl)naphthalene-2-ol |
| HPNIC | 3-(1-Phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-naphthalen-2-ol |
| HPNO | 3-(4,5-Diphenyloxazol-2-yl)naphthalen-2-ol |
| DFT | Density functional theory |
| TD-DFT | Time dependent density functional theory |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| EOM-CCSD | Equation-of-motion coupled-cluster singles and doubles |
| ONIOM | Our own N-layered integrated molecular orbital and molecular mechanics |
| CASSCF | Complete active space multiconfiguration-SCF |
| MS-CASPT2 | Multistate-CASPT2 |
| FMO-TDDFT | Fragment molecular orbital-TDDFT |
| DMF | Dimethylformamide |
| KBr | Potassium bromide |
| THF | Tetrahydrofuran |
| DMSO | Dimethyl sulfoxide |
| DMF | Dimethylformamide |
| XPRD | X-ray powder diffraction |
| DSC | Differential scanning calorimetry |
| FESEM | Field emission scanning electron microscopy |
| FT-IR | Fourier transform infrared spectroscopy |
| UV | Ultraviolet |
Acknowledgements
The authors acknowledge KAKENHI and NEXT grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (No. 26102011, 26249145, and 26620104,). V. S. P. thanks the Japan Society for the Promotion of Science (JSPS) for research fellowship (No. 2604063).Notes and references
- J. Goodman and L. E. Brus, J. Am. Chem. Soc., 1978, 100, 7472–7474 CrossRef CAS.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- A. Weller, Naturwissenschaften, 1955, 42, 175–176 CrossRef CAS.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1–20 CrossRef CAS.
- P. F. Barbara, P. K. Walsh and L. E. Brus, J. Phys. Chem., 1989, 93, 29–34 CrossRef CAS.
- A. Douhal, F. Lahmani and A. H. Zewail, Chem. Phys., 1996, 207, 477–498 CrossRef CAS.
- C.-C. Hsieh, C.-M. Jiang and P.-T. Chou, Acc. Chem. Res., 2010, 43, 1364–1374 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- B. Dick and N. P. Ernsting, J. Phys. Chem., 1987, 91, 4261–4265 CrossRef CAS.
- T. Iijima, A. Momotake, Y. Shinohara, T. Sato, Y. Nishimura and T. Arai, J. Phys. Chem. A, 2010, 5, 1603–1609 CrossRef PubMed.
- J. Cheng, D. Liu, L. Bao, K. Xu, Y. Yang and K. Han, Chem. – Asian J., 2014, 9, 3215–3220 CrossRef CAS PubMed.
- G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21–48 CrossRef CAS.
- F. Laquai, Y.-S. Park, J.-J. Kim and T. Basché, Macromol. Rapid Commun., 2009, 30, 1203–1231 CrossRef CAS PubMed.
- K.-Y. Chen, C.-C. Hsieh, Y.-M. Cheng, C.-H. Lai and P.-T. Chou, Chem. Commun., 2006, 4395–4397 RSC.
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed.
- H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654–657 CAS.
- J. Cheng, D. Liu, W. Li, L. Bao and K. Han, J. Phys. Chem. C, 2015, 119, 4242–4251 CAS.
- K. Benelhadj, J. Massue, P. Retailleau, G. Ulrich and R. Ziessel, Org. Lett., 2013, 15, 2918–2921 CrossRef CAS PubMed.
- K. Furukawa, N. Yamamoto, T. Nakabayashi, N. Ohta, K. Amimoto and H. Sekiya, Chem. Phys. Lett., 2012, 539–540, 45–49 CrossRef CAS.
- H. Konoshima, S. Nagao, I. Kiyota, K. Amimoto, N. Yamamoto, M. Sekine, M. Nakata, K. Furukawa and H. Sekiya, Phys. Chem. Chem. Phys., 2012, 14, 16448–16457 RSC.
- V. S. Padalkar, P. Ramasami and N. Sekar, J. Fluoresc., 2013, 23, 839–851 CrossRef CAS PubMed.
- S. Park, J. E. Kwon and S. Y. Park, Phys. Chem. Chem. Phys., 2012, 14, 8878–8884 RSC.
- S. Park, O.-H. Kwon, S. Kim, S. Park, M.-G. Choi, M. Cha, S. Y. Park and D.-J. Jang, J. Am. Chem. Soc., 2005, 127, 10070–10074 CrossRef CAS PubMed.
- V. S. Padalkar, P. Ramasami and N. Sekar, J. Lumin., 2014, 146, 527–538 CrossRef CAS.
- V. Padalkar, A. Tathe, V. Gupta, V. Patil, K. Phatangare and N. Sekar, J. Fluoresc., 2012, 22, 311–322 CrossRef CAS PubMed.
- W. Chen, E. B. Twum, L. Li, B. D. Wright, P. L. Rinaldi and Y. Pang, J. Org. Chem., 2012, 77, 285–290 CrossRef CAS PubMed.
- A. Ohshima, A. Momotake, R. Nagahata and T. Arai, J. Phys. Chem. A, 2005, 109, 9731–9736 CrossRef CAS PubMed.
- V. Padalkar, P. Ramasami and N. Sekar, Procedia Comput. Sci., 2013, 18, 797–805 CrossRef.
- V. S. Patil, V. S. Padalkar, A. B. Tathe and N. Sekar, Dyes Pigm., 2013, 98, 507–657 CrossRef CAS.
- D. Yao, S. Zhao, J. Guo, Z. Zhang, H. Zhang, Y. Liu and Y. Wang, J. Mater. Chem., 2011, 21, 3568–3570 RSC.
- J. Ma, J. Zhao, P. Yang, D. Huang, C. Zhang and Q. Li, Chem. Commun., 2012, 48, 9720–9722 RSC.
- F. S. Rodembusch, L. F. Campo, V. Stefani and A. Rigacci, J. Mater. Chem., 2005, 15, 1537–1541 RSC.
- P. Majumdar and J. Zhao, J. Phys. Chem. B, 2014, 119, 2384–2394 CrossRef PubMed.
- V. S. Padalkar and N. Sekar, J. Lumin., 2014, 155, 58–64 CrossRef CAS.
- P. Chou, Y. Chen, W. Yu, Y. Chou and C. Wei, J. Phys. Chem. A, 2001, 105, 1731–1740 CrossRef CAS.
- M. W. Chung, T. Y. Lin, C. C. Hsieh, K. C. Tang, H. Fu, P. T. Chou, S. H. Yang and Y. Chi, J. Phys. Chem. A, 2010, 114, 7886–7891 CrossRef CAS PubMed.
- M. T. Ignasiak, C. Houée-Levin, G. Kciuk, B. Marciniak and T. Pedzinski, ChemPhysChem, 2015, 16, 628–633 CrossRef CAS PubMed.
- P.-T. Chou and M. L. Martinez, Radiat. Phys. Chem., 1993, 41, 373–378 CrossRef CAS.
- D. Ghosh, S. Batuta, S. Das, N. A. Begum and D. Mandal, J. Phys. Chem. B, 2015, 119, 5650–5661 CrossRef CAS PubMed.
- X. Jin, C. Liu, X. Wang, H. Huang, X. Zhang and H. Zhu, Sens. Actuators, B, 2015, 216, 141–149 CrossRef CAS.
- S. Nagaoka, H. Endo and K. Ohara, J. Phys. Chem. B, 2015, 119, 2225–2232 CrossRef PubMed.
- S. J. Schmidtke, D. F. Underwood and D. A. Blank, J. Am. Chem. Soc., 2004, 126, 8620–8621 CrossRef CAS PubMed.
- M. J. Paterson, M. A. Robb, L. Blancafort and A. D. DeBellis, J. Am. Chem. Soc., 2004, 126, 2912–2922 CrossRef CAS PubMed.
- F. F. D. Oliveira, D. C. B. D. Santos, A. A. M. Lapis, J. R. Corrêa, A. F. Gomes, F. C. Gozzo, P. F. Moreira Jr., V. C. de Oliveira, F. H. Quina and B. A. D. Neto, Bioorg. Med. Chem. Lett., 2010, 20, 6001–6007 CrossRef CAS PubMed.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- Y. Nakane, T. Takeda, N. Hoshino, K. Sakai and T. Akutagawa, J. Phys. Chem. A, 2015, 119, 6223–6231 CrossRef CAS PubMed.
- R. Das, G. Duportail, A. Ghose, L. Richert, A. Klymchenko, S. Chakraborty, S. Yesylevskyy and Y. Mely, Phys. Chem. Chem. Phys., 2014, 16, 776–784 RSC.
- S. Barman, S. K. Mukhopadhyay, M. Gangopadhyay, S. Biswas, S. Dey and N. D. P. Singh, J. Mater. Chem. B, 2015, 3, 3490–3497 RSC.
- H. Xiao, K. Chen, D. Cui, N. Jiang, G. Yin, J. Wang and R. Wang, New J. Chem., 2014, 38, 2386–2393 RSC.
- L. Wilbraham, M. Savarese, N. Rega, C. Adamo, I. Cio, S. Chimiche, N. Federico, C. Universitario and M. S. Angelo, J. Phys. Chem. B, 2015, 119, 2459–2466 CrossRef CAS PubMed.
- T. Mutai, H. Sawatani, T. Shida, H. Shono and K. Araki, J. Org. Chem., 2013, 78, 2482–2489 CrossRef CAS PubMed.
- T. Mutai, H. Shono, Y. Shigemitsu and K. Araki, CrystEngComm, 2014, 16, 3890–3895 RSC.
- T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 9522–9524 CrossRef CAS PubMed.
- S. Furukawa, H. Shono, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2014, 6, 16065–16070 CAS.
- V. Bhalla, Roopa and M. Kumar, Org. Lett., 2012, 14, 2802–2805 CrossRef CAS PubMed.
- M. K. Bera, C. Chakraborty, P. K. Singh, C. Sahu, K. Sen, S. Maji, A. K. Das and S. Malik, J. Mater. Chem. B, 2014, 2, 4733–4739 RSC.
- L. Geng, X.-F. Yang, Y. Zhong, Z. Li and H. Li, Dyes Pigm., 2015, 120, 213–219 CrossRef CAS.
- H.-M. Lv, Y. Chen, J. Lei, C. Au and S.-F. Yin, Anal. Methods, 2015, 7, 3883–3887 CAS.
- M. L. Martinez, W. C. Cooper and P.-T. Chou, Chem. Phys. Lett., 1992, 193, 151–154 CrossRef CAS.
- D. Santa María, R. M. Claramunt, V. Bobosik, M. C. Torralba, M. R. Torres, I. Alkorta and J. Elguero, Tetrahedron, 2013, 69, 3027–3038 CrossRef.
- V. I. Tomin, A. P. Demchenko and P.-T. Chou, J. Photochem. Photobiol., C, 2015, 22, 1–18 CrossRef CAS.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed.
- K. Sakai, T. Ishikawa and T. Akutagawa, J. Mater. Chem. C, 2013, 1, 7866–7871 RSC.
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621–630 CrossRef CAS.
- F. Liang, L. Wang, D. Ma, X. Jing and F. Wang, Appl. Phys. Lett., 2002, 81, 4–6 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- T. Mutai and K. Araki, Kokagaku, 2009, 40(3), 184–187 CAS.
- V. S. Padalkar, S. B. Chemate, S. K. Lanke and N. Sekar, J. Lumin., 2015, 168, 114–123 CrossRef CAS.
- V. S. Patil, V. S. Padalkar, K. R. Phatangare, V. D. Gupta, P. G. Umape and N. Sekar, J. Phys. Chem. A, 2012, 116, 536–545 CrossRef CAS PubMed.
- V. Patil, V. Padalkar and N. Sekar, J. Lumin., 2015, 158, 243–251 CrossRef CAS.
- K. R. Phatangare, V. D. Gupta, A. B. Tathe, V. S. Padalkar, V. S. Patil, P. Ramasami and N. Sekar, Tetrahedron, 2013, 69, 1767–1777 CrossRef CAS.
- R. Rao, C.-W. Liao, W.-L. Su and S.-S. Sun, J. Mater. Chem. C, 2013, 1, 5491–5501 RSC.
- Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. Jiang, J. Phys. Chem. C, 2007, 111, 6544–6549 CAS.
- B. Li, J. Lan, D. Wu and J. You, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201507272.
- T. Zhou, F. Li, Y. Fan, W. Song, X. Mu, H. Zhang and Y. Wang, Chem. Commun., 2009, 3199–3201 RSC.
- A. Douvali, G. S. Papaefstathiou, M. P. Gullo, A. Barbieri, A. C. Tsipis, C. D. Malliakas, M. G. Kanatzidis, I. Papadas, G. S. Armatas, A. G. Hatzidimitriou, T. Lazarides and M. J. Manos, Inorg. Chem., 2015, 54, 5813–5826 CrossRef CAS PubMed.
- M. Kasha, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2379–2392 RSC.
- S. Kim, J. Seo and S. Y. Park, J. Photochem. Photobiol., A, 2007, 191, 19–24 CrossRef CAS.
- M. I. Knyazhansky, A. V. Metelista, A. J. Bushkov and S. M. Aldoshin, J. Photochem. Photobiol., A, 1996, 97, 121–126 CrossRef CAS.
- M. Cai, Z. Gao, X. Zhou, X. Wang, S. Chen, Y. Zhao, Y. Qian, N. Shi, B. Mi, L. Xie and W. Huang, Phys. Chem. Chem. Phys., 2012, 14, 5289–5296 RSC.
- Y. Qian, M. M. Cai, L. H. Xie, G. Q. Yang, S. K. Wu and W. Huang, ChemPhysChem, 2011, 12, 397–404 CrossRef CAS PubMed.
- S. H. Kim, S. Park, J. E. Kwon and S. Y. Park, Adv. Funct. Mater., 2011, 21, 644–651 CrossRef CAS.
- S. Park, E. K. Ji, H. K. Se, J. Seo, K. Chung, S. Y. Park, D. J. Jang, B. M. Medina, J. Gierschner and Y. P. Soo, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS PubMed.
- C. C. Hsieh, P. T. Chou, C. W. Shih, W. T. Chuang, M. W. Chung, J. Lee and T. Joo, J. Am. Chem. Soc., 2011, 133, 2932–2943 CrossRef CAS PubMed.
- M. Ikegami and T. Arai, J. Chem. Soc., Perkin Trans. 2, 2002, 1296–1301 RSC.
- W.-T. Chuang, C.-C. Hsieh, C.-H. Lai, C.-H. Lai, C.-W. Shih, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Org. Chem., 2011, 76, 8189–8202 CrossRef CAS PubMed.
- Y. Qian, S. Li, G. Zhang, Q. Wang, S. Wang, H. Xu, C. Li, Y. Li and G. Yang, J. Phys. Chem. B, 2007, 111, 5861–5868 CrossRef CAS PubMed.
- D.-E. Wu, Q.-C. Yao and M. Xia, Phys. Chem. Chem. Phys., 2015, 17, 3287–3294 RSC.
- Q. Wu, Q. Peng, Y. Niu, X. Gao and Z. Shuai, J. Phys. Chem. A, 2012, 116, 3881–3888 CrossRef CAS PubMed.
- Y. Shigemitsu, T. Mutai, H. Houjou and K. Araki, J. Phys. Chem. A, 2012, 116, 12041–12048 CrossRef CAS PubMed.
- Y. Shigemitsu, T. Mutai, H. Houjou and K. Araki, Phys. Chem. Chem. Phys., 2014, 16, 14388–14395 RSC.
- M. K. Nayak, B. H. Kim, J. E. Kwon, S. Park, J. Seo, J. W. Chung and S. Y. Park, Chem. – Eur. J., 2010, 16, 7437–7447 CrossRef CAS PubMed.
- A. Nano, M. P. Gullo, B. Ventura, N. Armaroli, A. Barbieri and R. Ziessel, Chem. Commun., 2015, 51, 3351–3354 RSC.
- Z. Tu, M. Liu, Y. Qian, G. Yang, M. Cai, L. Wang and W. Huang, RSC Adv., 2015, 5, 7789–7793 RSC.
- R. Wei, P. Song and A. Tong, J. Phys. Chem. C, 2013, 117, 3467–3474 CAS.
- Q.-C. Yao, X.-L. Lu and M. Xia, New J. Chem., 2014, 38, 2693–2700 RSC.
- K. Kanosue, T. Shimosaka, J. Wakita and S. Ando, Macromolecules, 2015, 48, 1777–1778 CrossRef CAS.
- T. Shida, T. Mutai and K. Araki, CrystEngComm, 2013, 15, 10179–10182 RSC.
- K. Sakai, H. Kawamura, N. Kobayashi, T. Ishikawa, C. Ikeda, T. Kikuchi and T. Akutagawa, CrystEngComm, 2014, 16, 3180–3185 RSC.
- S. P. Anthony, Chem. – Asian J., 2012, 7, 374–379 CrossRef CAS PubMed.
- A. Maity, F. Ali, H. Agarwalla, B. Anothumakkool and A. Das, Chem. Commun., 2015, 51, 2130–2133 RSC.
- W. Sun, S. Li, R. Hu, Y. Qian, S. Wang and G. Yang, J. Phys. Chem. A, 2009, 113, 5888–5895 CrossRef CAS PubMed.
- H. C. Peng, C. C. Kang, M. R. Liang, C. Y. Chen, A. Demchenko, C. T. Chen and P. T. Chou, ACS Appl. Mater. Interfaces, 2011, 3, 1713–1720 CAS.
- K. Benelhadj, W. Muzuzu, J. Massue, P. Retailleau, A. Charaf-Eddin, A. D. Laurent, D. Jacquemin, G. Ulrich and R. Ziessel, Chem. – Eur. J., 2014, 20, 12843–12857 CrossRef CAS PubMed.
- S. Kim, J. Seo, H. K. Jung, J. J. Kim and S. Y. Park, Adv. Mater., 2005, 17, 2077–2082 CrossRef CAS.
- P. Coppo, M. Duati, V. N. Kozhevnikov, J. W. Hofstraat and L. De Cola, Angew. Chem., Int. Ed., 2005, 44, 1806–1810 CrossRef CAS PubMed.
- A. H. Shelton, I. V. Sazanovich, J. A. Weinstein and M. D. Ward, Chem. Commun., 2012, 48, 2749–2751 RSC.
- M. Mazzeo, V. Vitale, F. Della Sala, M. Anni, G. Barbarella, L. Favaretto, G. Sotgiu, R. Cingolani and G. Gigli, Adv. Mater., 2005, 17, 34–39 CrossRef CAS.
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS PubMed.
- M. K. Nayak, J. Photochem. Photobiol., A, 2012, 241, 26–37 CrossRef CAS.
- V. Thanikachalam, J. Jayabharathi, A. Arunpandiyan and P. Ramanathan, J. Fluoresc., 2014, 24, 377–387 CrossRef CAS PubMed.
- K.-I. Sakai, S. Takahashi, A. Kobayashi, T. Akutagawa, T. Nakamura, M. Dosen, M. Kato and U. Nagashima, Dalton Trans., 2010, 39, 1989–1995 RSC.
- J. Huang, Q. Liu, J. H. Zou, X. H. Zhu, A. Y. Li, J. W. Li, S. Wu, J. Peng, Y. Cao, R. Xia, D. D. C. Bradley and J. Roncali, Adv. Funct. Mater., 2009, 19, 2978–2986 CrossRef CAS.
- S. Nakazono, Y. Imazaki, H. Yoo, J. Yang, T. Sasamori, N. Tokitoh, T. Cédric, H. Kageyama, D. Kim, H. Shinokubo and A. Osuka, Chem. – Eur. J., 2009, 15, 7530–7533 CrossRef CAS PubMed.
- Y.-T. Lee, C.-L. Chiang and C.-T. Chen, Chem. Commun., 2008, 217–219 RSC.
- M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 48, 3653–3656 CrossRef CAS PubMed.
- A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed.
- A. J. Stasyuk, M. Banasiewicz, B. Ventura, M. K. Cyrański and D. T. Gryko, New J. Chem., 2014, 38, 189–197 RSC.
- S. Nagaoka, U. Nagashima, N. Ohta, M. Fujita and T. Takemura, J. Phys. Chem., 1988, 92, 166–171 CrossRef CAS.
- H. Ma, B. Chen, T. Sassa, L. R. Dalton and A. K. Y. Jen, J. Am. Chem. Soc., 2001, 123, 986–987 CrossRef CAS PubMed.
- V. S. Padalkar, D. Sakamaki, N. Tohnai, T. Akutagawa, K. Sakai and S. Seki, RSC Adv., 2015, 5, 80283–80296 RSC.
- M. Tasior, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem. – Asian J., 2012, 7, 2656–2661 CrossRef CAS PubMed.
- L. Xie, Y. Chen, W. Wu, H. Guo, J. Zhao and X. Yu, Dyes Pigm., 2012, 92, 1361–1369 CrossRef CAS.
- L. Cui, W. Zhu, Y. Xu and X. Qian, Anal. Chim. Acta, 2013, 786, 139–145 CrossRef CAS PubMed.
- M. A. Satam, R. D. Telore, A. B. Tathe, V. D. Gupta and N. Sekar, Spectrochim. Acta, Part A, 2014, 127, 16–24 CrossRef CAS PubMed.
- M. A. Satam, R. D. Telore and N. Sekar, Dyes Pigm., 2015, 123, 274–284 CrossRef CAS.
- M. Avadanei, V. Cozan, S. Shova and J. A. Paixão, Chem. Phys., 2014, 444, 43–51 CrossRef CAS.
- K. F. Byth, J. D. Culshaw, S. Green, S. E. Oakes and A. P. Thomas, Bioorg. Med. Chem. Lett., 2004, 14, 2245–2248 CrossRef CAS PubMed.
- C. J. R. Fookes, T. Q. Pham, F. Mattner, I. Greguric, C. Loc, X. Liu, P. Berghofer, R. Shepherd, M. Gregoire and A. Katsifis, J. Med. Chem., 2008, 51, 3700–3712 CrossRef CAS PubMed.
- A. Douhal, F. Amat-Guerri and A. U. Acuna, J. Phys. Chem., 1995, 99, 76–80 CrossRef CAS.
- A. Douhal, F. Amat-Guerri and A. U. Acuna, Angew. Chem., Int. Ed., 1997, 36, 1514–1516 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- C. Y. K. Chan, Z. Zhao, J. W. Y. Lam, J. Liu, S. Chen, P. Lu, F. Mahtab, X. Chen, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 378–389 CrossRef CAS.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- J. Seo, S. Kim, Y. S. Lee, O. H. Kwon, K. H. Park, S. Y. Choi, Y. K. Chung, D. J. Jang and S. Y. Park, J. Photochem. Photobiol., A, 2007, 191, 51–58 CrossRef CAS.
- S. Li, L. He, F. Xiong, Y. Li and G. Yang, J. Phys. Chem. B, 2004, 108, 10887–10892 CrossRef CAS.
- P. Chen, R. Lu, P. Xue, T. Xu, G. Chen and Y. Zhao, Langmuir, 2009, 25, 8395–8399 CrossRef CAS PubMed.
- P. Xue, R. Lu, G. Chen, Y. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem. – Eur. J., 2007, 13, 8231–8239 CrossRef CAS PubMed.
- T. H. Kim, M. S. Choi, B.-H. Sohn, S.-Y. Park, W. S. Lyoo and T. S. Lee, Chem. Commun., 2008, 2364–2366 RSC.
- J. Seo, S. Kim, S. H. Gihm, C. R. Park and S. Y. Park, J. Mater. Chem., 2007, 17, 5052–5057 RSC.
- W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163–2166 CrossRef CAS PubMed.
- B.-R. Gao, H.-Y. Wang, Y.-W. Hao, L.-M. Fu, H.-H. Fang, Y. Jiang, L. Wang, Q.-D. Chen, H. Xia, L.-Y. Pan, Y.-G. Ma and H.-B. Sun, J. Phys. Chem. B, 2010, 114, 128–134 CrossRef CAS PubMed.
- J. Chen, B. Xu, K. Yang, Y. Cao, H. H. Y. Sung, I. D. Williams and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 17086–17093 CrossRef CAS PubMed.
- Y. Qian, S. Li, Q. Wang, X. Sheng, S. Wu, S. Wang, J. Li and G. Yang, Soft Matter, 2012, 8, 757–764 RSC.
- E. Wang, J. W. Y. Lam, R. Hu, C. Zhang, Y. S. Zhao and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 1801–1807 RSC.
- J. Zhang, B. Xu, J. Chen, L. Wang and W. Tian, J. Phys. Chem. C, 2013, 117, 23117–23125 CAS.
- E. Lager, J. Liu, A. Aguilar-Aguilar, B. Z. Tang and E. Peña-Cabrera, J. Org. Chem., 2009, 74, 2053–2058 CrossRef CAS PubMed.
- M. Yang, D. Xu, W. Xi, L. Wang, J. Zheng, J. Huang, J. Zhang, H. Zhou, J. Wu and Y. Tian, J. Org. Chem., 2013, 78, 10344–10359 CrossRef CAS PubMed.
- S. Sharma, A. Dhir and C. P. Pradeep, Sens. Actuators, B, 2014, 191, 445–449 CrossRef CAS.
- M. Gao, Q. Hu, G. Feng, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 3438–3442 RSC.
- H. Deng, B. Liu, C. Yang, G. Li, Y. Zhuang, B. Li and X. Zhu, RSC Adv., 2014, 4, 62021–62029 RSC.
- Z. Song, R. T. K. Kwok, E. Zhao, Z. He, Y. Hong, J. W. Y. Lam, B. Liu and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17245–17254 CAS.
- G. Li, D. Zhu, Q. Liu, L. Xue and H. Jiang, Org. Lett., 2013, 15, 924–927 CrossRef CAS PubMed.
- X. Chen, R. Wei, Y. Xiang, Z. Zhou, K. Li, P. Song and A. Tong, J. Phys. Chem. C, 2011, 115, 14353–14359 CAS.
- C. Niu, L. Zhao, T. Fang, X. Deng, H. Ma, J. Zhang, N. Na, J. Han and J. Ouyang, Langmuir, 2014, 30, 2351–2359 CrossRef CAS PubMed.
- X. T. Chen, Y. Xiang, P. S. Song, R. R. Wei, Z. J. Zhou, K. Li and A. J. Tong, J. Lumin., 2011, 131, 1453–1459 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |