
Liquid phase oxidation chemistry in continuous-flow microreactors
Hannes P. L. Gemoets
a,
Yuanhai Su
a,
Minjing Shang
a,
Volker Hessel
a,
Rafael Luque
b and
Timothy Noël
*ac
aDepartment of Chemical Engineering and Chemistry, Micro Flow Chemistry & Process Technology, Eindhoven University of Technology, Den Dolech 2, 5612 AZ Eindhoven, The Netherlands. E-mail: t.noel@tue.nl
bDepartamento de Quimica Organica, Universidad de Cordoba, Edificio Marie Curie (C-3), Campus de Rabanales, Ctra. Nnal. IV-A, Km 396, E14014 Cordoba, Spain
cDepartment of Organic Chemistry, Ghent University, Krijgslaan 281 (S4), 9000 Ghent, Belgium
First published on 23rd July 2015
Abstract
Continuous-flow liquid phase oxidation chemistry in microreactors receives a lot of attention as the reactor provides enhanced heat and mass transfer characteristics, safe use of hazardous oxidants, high interfacial areas, and scale-up potential. In this review, an up-to-date overview of both technological and chemical aspects of liquid phase oxidation chemistry in continuous-flow microreactors is given. A description of mass and heat transfer phenomena is provided and fundamental principles are deduced which can be used to make a judicious choice for a suitable reactor. In addition, the safety aspects of continuous-flow technology are discussed. Next, oxidation chemistry in flow is discussed, including the use of oxygen, hydrogen peroxide, ozone and other oxidants in flow. Finally, the scale-up potential for continuous-flow reactors is described.
 Hannes P. L. Gemoets | Hannes P. L. Gemoets was born in Ghent (1991), Belgium. He received an MSc degree (Industrial Chemical Engineering, cum laude) from the KaHo Sint-Lieven in 2013. Currently he is pursuing a PhD under the supervision of Dr Timothy Noel and Prof. Volker Hessel at the University of Technology Eindhoven (Netherlands). His research focuses on the development of novel continuous-flow methods for C–H functionalization strategies. |
 Yuanhai Su | Yuanhai Su was born in China. He received BE from Tianjin University, and then a PhD degree from the Dalian Institute of Chemical Physics under the supervision of Prof. Guangwen Chen and Prof. Quan Yuan, both in chemical engineering. He conducted his postdoc research with the support of the Alexander von Humboldt Foundation, with the host of Prof. Eugeny Kenig in Paderborn University, Germany. Currently, he is the recipient of the Marie Curie Actions fellowship (host: Assistant Prof. Timothy Noël), and works in the Eindhoven University of Technology, the Netherlands. His research interests are in the areas of process intensification, microreactor technology and continuous-flow chemistry. |
 Minjing Shang | Minjing Shang was born in Shandong Province, China. She received BE and MSc degrees from Dalian University of Technology (China) in 2011, under the supervision of Prof. Yongchun Zhang, on the topic of CO2 hydrogenation to form methanol. Currently, she is a PhD candidate under the supervision of Dr Timothy Noël and Prof. Volker Hessel at the Eindhoven University of Technology (Netherlands). Her research interests focus on process intensification and continuous-flow chemistry. |
 Volker Hessel | Prof. Dr Volker Hessel studied chemistry at Mainz University. In 1999 he was appointed Head of the Microreaction Technology Department at Institut fur Mikrotechnik Mainz (IMM) GmbH. In 2002, he was appointed Vice Director and in 2007 he was appointed Director R&D at IMM. In 2011 he was appointed full Professor for the chair of “Micro-Flow Chemistry and Process Technology” at Eindhoven University of Technology. In 2009, he was appointed Honorary Professor at the Technical University of Darmstadt and is a Fellow of their Cluster of Excellence “Smart Interfaces”. He received the AIChE “Excellence in Process Development Research” award in 2007, and in 2010 he received the ERC Advanced Grant on “Novel Process Windows”. |
 Rafael Luque | Rafael Luque has extensively published (>250 peer-reviewed publications) in the areas of (nano)materials science, heterogeneous (nano)catalysis, microwave and flow chemistry, biofuels and green chemical methods in synthetic organic chemistry, filed 3 patent applications and edited 8 books as well as numerous contributions to book chapters and invited, guest, keynote and plenary lectures in scientific events worldwide. Among the recent awards, Rafael has received the TR35 Spain from Technology Review and MIT as one of the top 10 young entrepreneurs in Spain (2012) and the RSC Environment, Sustainability and Energy Early Career Award (2013) from the Royal Society of Chemistry UK. |
 Timothy Noël | Timothy Noël obtained his MSc (Industrial Chemical Engineering) in 2004. In 2009, he received his PhD at the University of Ghent. He then moved to the Massachusetts Institute of Technology as a Fulbright Postdoctoral Fellow with Professor Stephen L. Buchwald (MIT-Novartis Center for Continuous Manufacturing). In 2012, he accepted a position as an assistant professor at the Eindhoven University of Technology. In 2011, he received an Incentive Award for Young Researchers. In 2012, he received a Veni grant (NWO) and was nominated for the European Young Chemist Award. Since 2014, he has been serving as an associate editor of the Journal of Flow Chemistry. Recently, he received a prestigious VIDI grant (NWO) and he coordinates the Marie Skłodowska-Curie ETN program “Photo4Future”. |
1. Introduction
Liquid phase oxidation processes are some of the main pathways to convert petroleum-based hydrocarbon raw materials into important commodity chemicals used in the fine chemical industry.1–3 Throughout the years, there have been some well-known liquid phase oxidation processes realized in an industrial setting, such as the preparation of terephthalic acid, cumene peroxide, adipic acid, acetaldehyde, and propylene oxide. Despite these landmark achievements in the past, new reaction routines and techniques are still highly required due to increasingly stringent government regulations concerning the emission of organic and inorganic wastes and the related environmental and safety impact.4Several issues are associated with liquid phase oxidation chemistry. Hydrocarbon molecules possess several reactive sites, and thus various products/by-products can be formed in liquid oxidation processes. These selectivity issues are some of the main reasons why aerobic oxidation chemistry is often avoided in the pharmaceutical industry. Thus, the design of new and improved catalysts and oxidants to increase the selectivity remains a vigorous research discipline.5–11 Furthermore, reaction mechanisms are often complex for oxidation processes.12–15 Their complexity is further aggravated by complex mass transport processes in e.g., multiphase oxidation processes using oxidants such as oxygen, ozone or hydrogen peroxide.16–18 This is more pronounced in fast oxidation processes as the reaction rate is in such cases greatly influenced by the mass transfer rate. Unclear reaction mechanisms and complex transport phenomena make modelling of the reactor and the process control design very challenging.
Conventional batch or semi-batch reactors, such as agitator bubbling reactors, bubble columns, trickle-bed reactors, gas-lift and loop reactors, multi-stage trayed column reactors, are widely applied for liquid phase oxidations. However, these reactor types are often avoided at large scale production sites due to several reasons, including insufficient heat- and mass transfer rates, low interfacial areas, back mixing of the liquid phase and potential safety issues. Especially, aerobic oxidations pose the process engineer particular challenges to develop a safe and reliable process. In particular, strict handling protocols of the oxidant–organic reactant mixtures are required to ensure process safety. For example, one should make sure that the oxygen concentration in the gas–liquid disengagement zone, or any other open space existing in the reactor system, is kept below the flammability limit when designing a specific multiphase reactor for liquid phase oxidations with oxygen as an oxidant.
The limitations in process control, reactor scale-up and safety may be overcome by utilizing microreactor technology.19–34 This technology has experienced a spectacular development as one of the most important process intensification technologies on the market.35–38 The advantages of this reactor design for chemical production are in general the potential to work under a continuous operational mode, well-defined flow properties, large surface-to-volume ratios, enhanced heat- and mass transfer, excellent process safety and the ease of increasing throughput by numbering-up. The extremely small characteristic dimensions of microreactors provide a low liquid holdup, which is beneficial to avoid many safety risks associated with the use of hazardous oxidants.
It is the goal of this review to provide a comprehensive overview of the potential and the use of microreactor technology for liquid phase oxidation chemistry. A detailed description of the engineering aspects associated with this topic is given. Hereby, we focused on mass- and heat-transfer aspects, which are of importance when designing a suitable oxidation protocol. In addition, process safety aspects associated with this chemistry are vital for a reliable design and are discussed as well. Next, we survey the different oxidation chemistries, which have been performed in continuous-flow microreactors. This includes important fields such as aerobic oxidation, hydrogen peroxide oxidation, and ozonolysis. The results in flow are compared with their batch counterparts where possible. Finally, different scale-up strategies are described. For gas–solid catalytic oxidation chemistry, which is of great importance for the development of micro-fuel processors, we guide the reader to some specialist reviews.39–42 Oxidation processes in supercritical media are not included in this discussion.43–50
2. Fundamental engineering aspects
2.1 Mass transfer limitations in liquid phase oxidation
Many oxidation processes consist of multiphase reaction mixtures (e.g., gas–liquid, liquid–liquid, liquid–solid, and combinations thereof). Herein, it is important to maximize the interfacial area, which causes a reduction of the transport distances. Consequently, the yield of the target compound can be maximized by a careful selection of an appropriate reactor design and a suitable catalyst.51–55 The overall performance of the oxidation reaction is mainly determined by intrinsic kinetics and transport properties inside reactors. The intrinsic reaction rate of liquid phase oxidation can be slow (e.g., toluene oxidation) or fast (e.g., β-isophorone oxidation), mainly depending on the catalytic system and operational conditions.56–58 For fast liquid phase oxidation processes, the reaction rate and the selectivity are significantly influenced by the mass transfer rate, and the key to increase the production rate is to facilitate mass transfer by increasing the interfacial area.59–62 Classical reactors, such as mechanically agitated reactors, bubble columns, and trickle-bed reactors, result in poorly defined interfacial contact areas. The scale-up of such reactors for a large-scale production is not trivial considering the safety hazards associated with oxidation chemistry.63–65 In addition, insufficient heat and mass transfer in these reactors further complicates the scalability.66,67 In contrast, micro-structured reactors can provide higher surface-to-volume ratios and thus higher heat and mass transfer rates for multiphase reaction processes.18,68–74 With known mass transfer characteristics and reaction kinetics in hand, one can judge whether it is justified to use microreactors for a specific liquid phase oxidation process and which microreactor type should be selected.75–78The mass transfer in multiphase liquid phase oxidation processes can be described using a two-film model (Fig. 1). A gaseous component (A, e.g., oxygen) is transferred from the gas to the liquid phase where it can be consumed in the reaction to form products (eqn (1)). The driving force for the gas–liquid mass transfer of A is the concentration gradient, which is governed by thermodynamic and chemical equilibria. In the film theory, the interface zone between the gas and liquid phases can be represented by two stagnant films (i.e., a gas layer and a liquid layer). In this interface zone, a vapor–liquid equilibrium with thickness δint is established. The bulk of the gas and liquid is considered to be well mixed. For only a few absorption-reaction systems, the mass transfer resistance in the gas film should be considered. In these systems, either the liquid phase has a very high solubility for the gaseous reactants or the diffusivity of the gaseous reactants in the gas phase is quite small. For a Taylor flow (slug flow or segmented flow) regime in microreactors, application of the film theory for the calculation of the mass transfer coefficient (kL) is highly feasible. Taylor flow is a very uniform flow pattern and is characterized by gas bubbles, which are separated by liquid slugs and a thin liquid film on the channel walls. In the liquid and the gas bubble, internal circulations are established, which arise from the interfacial friction and the slip velocity. This internal circulation results in homogeneous mixing and facilitates a fast mass transfer between the two phases.79–82 Interestingly, a microreactor operating in the Taylor flow regime shows minimal axial dispersion and provides an excellent residence time distribution, allowing it to behave as an ideal plug flow reactor.82–87
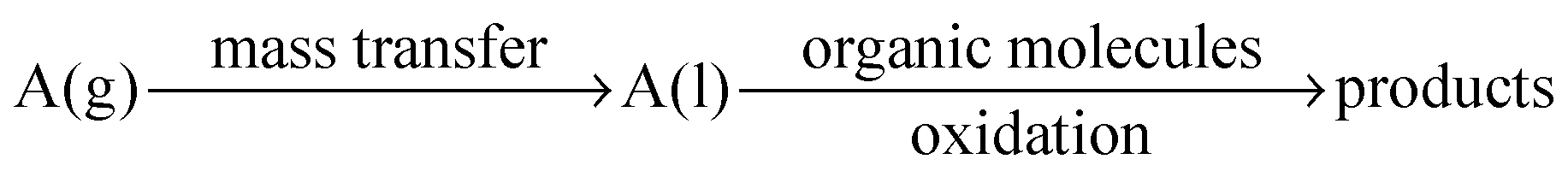 | (1) |
| kL = DA/δint | (1) |
 | ||
| Fig. 1 (a) Physical representation of absorption and reaction, (b) the schematic representation of the two-film model, (c) the sketch of the mass transfer process in a Taylor flow regime in microchannels, and (d) a photo of Taylor flow for gas–liquid selective oxidation of cyclohexane with oxygen in a microreactor. Reproduced from ref. 82 with permission from The Royal Society of Chemistry. | ||
The selection of a suitable reactor for liquid phase oxidation processes is also highly dependent on the characteristics of the intrinsic reaction kinetics. A detailed description of the kinetics and mechanisms of liquid phase oxidation processes has been reviewed.94–97 Most liquid phase oxidation processes involve a free radical chain mechanism, which includes four elemental steps, i.e., initiation, propagation, branching and termination. The intrinsic reaction rate constant (kr) is dependent on the initiation rate constant, the propagation rate constant and the termination rate constant. With both specific parameters of mass transfer and reaction kinetics in hand, the Hatta number can be calculated, which allows one to evaluate the extent of mass transfer limitations:98
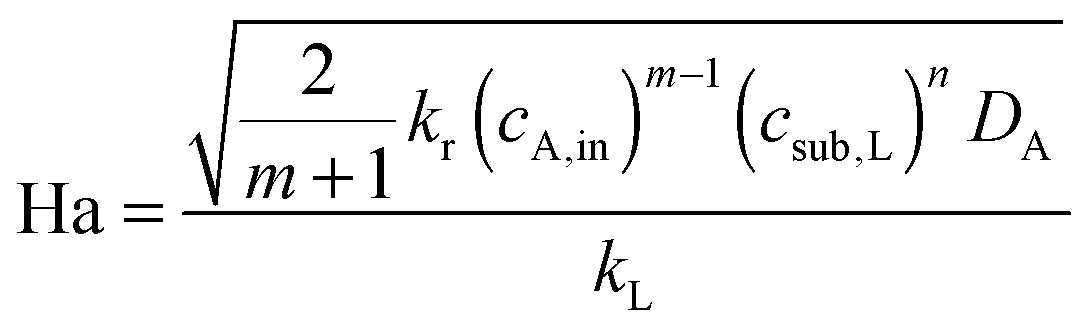 | (2) |
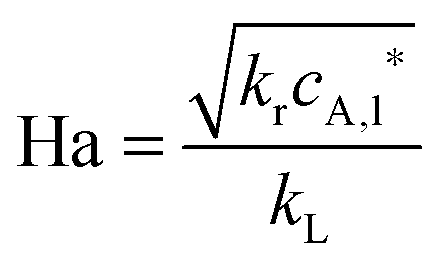 | (3) |
 | ||
| Fig. 2 Hatta number versus mass transfer coefficient with kr = 9.49 × 10−1 s−1, D = 3.08 × 10−8 m2 s−1, kL for bubble columns in the range of 3.7 × 10−5 to 8.2 × 10−5 m s−1, and kL for capillary microreactors larger than 2.5 × 10−4 m s−1. Reproduced from ref. 99 with permission from Elsevier. | ||
For catalyzed liquid phase oxidation processes and gas–liquid–solid catalyzed oxidation processes, mechanisms, such as the Mars–van Krevelen mechanism and the classical Langmuir–Hinshelwood mechanism, can be applied to describe the reaction rate expression. In these cases, the calculation of the intrinsic reaction rate constant is different from those reactions with a free radical chain mechanism. A full description of these catalyzed reaction mechanisms is outside the focus of this review and we direct the reader to some relevant references for more details.101
2.2 Heat transfer in liquid phase oxidation processes
Any chemical reaction can be fully characterized by its reaction mechanism, thermal (exothermic/endothermic) properties, thermodynamic equilibrium and chemical kinetic equilibrium. Many liquid phase oxidation reactions are highly exothermic and thus inefficient heat management in a reactor possibly leads to a low selectivity and yield. Moreover, some intermediates/products of oxidation reactions are highly thermally unstable (e.g., peroxides) and can undergo vigorous decomposition at elevated temperatures.101 In conventional reactors, explosive mixtures can be easily produced in the reactor head space due to volatilization. When heat dissipation is insufficient, local hot spots are formed which can cause an ignition of the explosive mixtures. In contrast to conventional reactors, heat management in microreactors is better due to the high surface to volume ratio. This allows a wider operational zone providing higher selectivity and yield.19,102–104For exothermic reactions, the heat equilibrium in a reaction system is dependent on several factors including the rate of heat generated from the reaction, the heat absorption rate by the reaction mixture and the rate of heat removed from the reactor wall through conduction or convection. In order to increase the heat transfer rate in microreactors for reaction processes, one can reduce the reactor dimensions to obtain a higher specific interfacial area, place active heat exchange or mixing elements (e.g., heat pin-fins, channel curvature and static mixers) and use materials or heat-exchange fluids with high heat conductivity.
To improve heat transfer in microchannels it is crucial to understand the theoretical basis behind it. It should be noted that the literature on heat transfer phenomena in microchannels is contradictory. This is due to the high complexity as there is a strong correlation between heat transport phenomena and microchannel characteristics. Scaling effects (e.g., entrance effect, surface roughness, and channel geometry), which are negligible on a macro-scale, cannot be ignored on a micro-scale. A complete description is beyond the scope of this review and is discussed in great detail in the following ref. 105–107. In this review, we provide some general trends and guidelines, which can help the reader to understand the underlying principles of heat transfer.
The classical energy balance equation can be used to describe any heat transfer process without considering the effect of axial dispersion:
 | (4) |
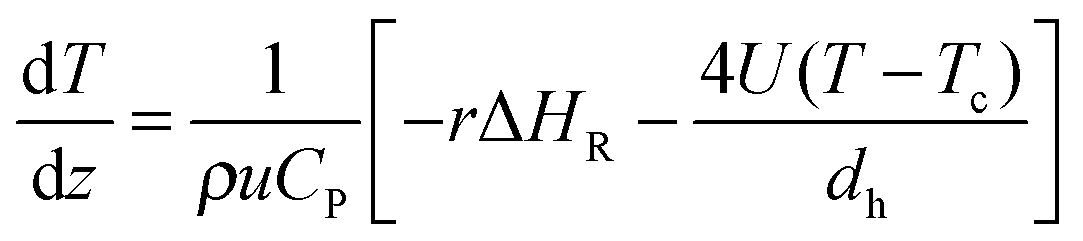 | (5) |
 | ||
| Fig. 3 Experimental and simulated adiabatic temperature and concentration profiles in a microchannel reactor with five orifices as transport intensification units for exothermic biphasic synthesis of tert-butyl peroxypivalate (experimental data is indicated using a point diagram; simulated data is indicated with a solid line; the position of each orifice is marked with a dashed line; pivaloylchloride (PivCl); tert-butylhydroperoxide (TBHP); tert-butylperoxide (KTBP)). Reproduced from ref. 109 with permission from The Royal Society of Chemistry. | ||
Furthermore, the adiabatic temperature rise can be calculated according to the following equation:
 | (6) |
 | (7) |
 | (8) |
| Material | Thermal conductivity (k) [W m−1 K−1] |
|---|---|
| PFA | 0.195 |
| FEP | 0.19–0.24 |
| Glass | 1 |
| Stainless steel | 12–45 |
| Silicon | 149 |
| Aluminium (pure) | 237 |
| Aluminium (alloy) | 120–180 |
| Silicon carbide | 120–490 |
| Copper | 401 |
In order to perform reactions under near isothermal conditions and to avoid exothermal runaways, it is necessary to ensure that the heat removal rate is at least equal to the heat generation rate. That is, the ratio of the heat generation rate to the heat removal rate should be lower than 1. This principle can be applied to the heat management in microreactors.75,113
 | (9) |
The importance of good heat management is convincingly demonstrated in the highly exothermic preparation of tert-butyl peroxypivalate. In a batch process, a large amount of solvent is required to provide a sufficient temperature buffer to control the reaction temperature and to avoid thermal runaway.4,75,114,115 In flow, a compact microreactor assembly consisting of a microchannel with orifices and a heat exchanger plate was employed (Fig. 4a).109,116 This reactor design allows for the two-step synthesis of tert-butyl peroxypivalate without adding additional solvents as a temperature buffer. The first step constitutes a nearly isothermal deprotonation step, while the second step involves a strongly exothermic reaction between potassium tert-butylperoxide and pivaloylchloride. The temperature profile was measured in situ by using an infrared camera. As can be seen from Fig. 4b, the temperature profile in the absence of cooling fluid showed a very steep temperature rise (ΔT = 63 K) in the section where the second reaction takes place. In the presence of active cooling, smooth temperature profiles along the microreactor could be obtained (Fig. 4c and d).
 | ||
| Fig. 4 (a) Exploded view of the microreactor assembly consisting of an orifice microchannel and a heat exchanger plate for exothermic biphasic synthesis of tert-butyl peroxypivalate, and infrared pictures indicating the in-line temperature in the reaction plate (flow rate of reaction mixture is 18.5 mL min−1) using different flow rates of the cooling fluid: (b) 0 mL min−1, (c) 21 mL min−1 and (d) 9 mL min−1. Reproduced from ref. 109 with permission from The Royal Society of Chemistry. | ||
2.3 Safety aspects in liquid phase oxidation processes
Liquid phase oxidation processes often combine highly reactive species (e.g., oxygen, ozone, and peroxide) with organic solvents which can lead to combustion.117–119 Ignition of explosive reaction mixtures results in a significant increase in temperature and pressure inside the reactor, possibly leading to rupture of the reactor and explosions. Therefore, pure oxygen or oxygen-enriched air is rarely used even though it is known that the reaction rate and selectivity can be enhanced significantly with increasing oxygen concentrations. Typically, nitrogen diluted oxygen mixtures are used, which are well below the limiting oxygen concentration (LOC) of organic solvents or reagents. The LOC is defined as the limiting concentration of oxygen below which combustion of gas mixtures is impossible. Table 2 shows an overview of LOC data for frequently applied organic solvents in the pharmaceutical industry. The use of inert gas largely increases the process complexity in conventional reactors. For example, it is necessary to continuously refresh the gas mixture in bubble columns to reach full conversion.120| Solvent | T (°C) | Limiting oxygen concentration (vol%) | ||
|---|---|---|---|---|
| 1 bar | 10 bar | 20 bar | ||
| Acetic acid | 200 | 10.6 | — | 9.6 |
| NMP | 200 | 8.1 | — | 7.6 |
| DMSO | 200 | 3.9 | — | — |
| DMSO | 100 | 6.4 | — | — |
| tert-Amyl alcohol | 100 | 9.6 | — | 10.1 |
| Ethyl acetate | 100 | 9.4 | — | 9.9 |
| 2-MeTHF | 100 | 9.4 | — | 9.1 |
| Methanol | 100 | 7.6 | — | 6.9 |
| Acetonitrile | 100 | — | 12.1 | 11.9 |
| Toluene | 100 | 10.4 | 10.3 | 9.9 |
| Methanol | 25 | 8.6 | — | — |
| Acetonitrile | 25 | 12.7 | — | — |
| Toluene | 25 | 11.6 | — | — |
The power of an explosion is proportional to the mass of the explosive mixture with the power of 1/3.121,122 This relation immediately explains why microreactors are safe to use when employing hazardous reaction conditions.123 However, while the total volume of a microreactor is small and thus contains only small amounts of potentially explosive material, one should take great care that the explosion is not propagated to nearby storage vessels, containing larger amounts of flammable starting materials or products.124,125 Hereto, it is important that the reaction is immediately quenched upon the exiting microreactor, so that explosion propagation is avoided. Continuous-flow processing also allows the generation of hazardous intermediates in situ, which are subsequently reacted away in a follow-up reaction. This allows one to minimize the total amount of hazardous material holdup in the laboratory or chemical plant.
The safety issues in liquid phase oxidation processes are also highly associated with the hydrodynamics and transport phenomena in the reactors. The occurrence of local hot spots due to insufficient heat and mass transfer rates serves as an ignition source for explosive reaction mixtures (thermal explosions). Safety problems may be avoided by transforming the batch process into a continuous-flow protocol. Siccama and Westerterp have investigated via experimental and numerical methods the ignition of ethylene–air mixtures inside a tube (50 mm ID) using a hot surface.126,127 It was found that the explosion region was reduced under continuous-flow conditions as compared to batch processing (Fig. 5). Higher oxygen concentrations can be used in flow when gas is flowing at a higher flow rate, thus avoiding the occurrence of dead zones, which are often the cause of the actual explosion. The explosion limits for reactive gas mixtures in flow were found to be influenced by many parameters such as the flow rate, pressure and temperature, but also by the shape and the size of the tube and by the characteristics of the ignition source. In general, an increase in the flow rate and a decrease in the pressure or the temperature will increase the minimum critical oxygen concentration above which flame propagation becomes possible. Continuous-flow processing lowers these upper explosion limits and thus widens the operational zone for liquid phase oxidation processes with a higher reliability than batch processing.128
 | ||
| Fig. 5 Critical oxygen concentration in the explosion point in the 50 mm tube. Reproduced from ref. 128 with permission from John Wiley and Sons. | ||
The high surface-to-volume ratios encountered in microreactor technology demonstrate that the reactor material is an important design parameter.129 We have already demonstrated the importance of the conductivity of the material for efficient heat dissipation in exothermic oxidation processes (so-called thermal quenching of the reaction). In addition, the material itself can play a catalytic role. For example, stainless steel reactors can catalyze the decomposition and isomerization of organic peroxides.130,131 Moreover, a steel reactor wall can initiate the radical formation of hydroperoxide intermediates in the oxidation of saturated hydrocarbons using molecular oxygen as the terminal oxidant and acetaldehyde as the sacrificial co-reductant.132 This demonstrates the heterogeneous initiation effect of steel for the production of radicals. In addition, Veser has described an interesting radical quenching effect in the platinum-catalyzed oxidation of hydrogen.133 Kinetic explosions can occur in such situations when radical species are uncontrollably formed. Quenching of the explosion can be achieved by radical chain termination, in which two radicals combine to give one stable molecule. This occurs typically at the catalytic reactor walls. Given the large surface-to-volume ratios in microreactors, reactants can diffuse rapidly to the reactor walls, which quenches efficiently the potential kinetic explosion and thus increases the explosion limit. This effect allows one to utilize higher reaction temperatures in microreactors. As an example, explosive behavior was observed at 420 °C and atmospheric pressure in a conventional reactor with 1 m diameter, while higher temperatures (up 750 °C) were feasible in a microreactor with a diameter of 1 mm.
The choice of reactor materials and reactor types also depends on the operational conditions. Especially at high temperatures and elevated pressures, one should carefully select the reactor material in order to withstand these harsh conditions. Stainless steel microreactors can resist high temperature and high pressure, and provide a high heat conductivity.134,135 However, stainless steel is not compatible with acidic media except for those with special surface treatments. PFA, FEP or PTFE microreactors have the advantages of easy fabrication, low cost and high flexibility. They can resist strong acidic and alkaline media at low to moderate temperatures and pressures. Such polymeric materials are transparent which allows one to study the hydrodynamics inside the microchannel. This is beneficial for improving the reactor design and the overall process.77 A high transparency is also crucial for photochemical processes (e.g., singlet oxygen generation); PFA and FEP are transparent to both UV and visible light. Nevertheless, PFA microreactors cannot resist high temperatures and the limiting pressure decreases significantly with increasing temperatures. Importantly, these polymers have a low thermal conductivity, which can be a problem for highly exothermic reactions. Silicon microreactors have similar transparency properties and better thermal conductivity than polymer-based reactors. It can withstand high temperatures and high pressures, and provide chemical resistance to a broad range of chemicals.136 However, they are not always resistant to strong alkaline reaction mixtures, especially at elevated temperatures. Surface treatments can help to overcome this limitation.137 Silicon carbide has an even higher thermal conductivity and combines it with an exceptional chemical stability. However, this material is quite expensive and is also brittle.
3. Aerobic oxidation processes in continuous flow
In this section, aerobic oxidation processes carried out in continuous-flow microreactors, are discussed. The chapter is divided into five sub-chapters according to the catalysis mode.3.1 Transition metal-catalyzed aerobic oxidation processes in continuous flow
Transition metal-catalyzed oxidation reactions are known as one of the fundamental transformations in chemistry. Their ability to use mild and selective chemistry is what makes this topic appealing for today's applications. In oxidative transformations, the use of oxygen is of high interest, since molecular oxygen is completely ecological, highly abundant and the cheapest source of the oxidant. Moreover, because of its gaseous nature, work up procedures are made effortless and allow the use of super-stoichiometric amounts to boost reaction rates. In recent years, the use of microreactors has become increasingly popular for aerobic oxidation processes, since microreactors offer superior mass transfer characteristics under biphasic reaction conditions and safeguard high reproducibility and safety over the process. ).
).
|
|
||||
|---|---|---|---|---|
| Entry | Reference (publication year) | Microreactor design | Reaction conditions | Conversion (X), Selectivity (S) |
| 1 | Plucinski et al.138–140 (2005) | Stainless steel multichannel micro-packed-bed reactor – 5 parallel channels – V = 900 μL (3 mm × 3 mm) per channel |
Ru/Al2O3 (0.9 wt% Ru) c = 1 M, T = 115 °C, pO2 = 8 bar |
X = 55%, S = 99% |
| 2 | Gavriilidis et al.141 (2006) | Silicon-glass micro-packed-bed reactor (MPBR) – Single serpentine shaped channel – V = 26 μL (0.6 mm × 0.3 mm) |
TRAP/Al2O3 (2 wt% TRAP) Particle size: 50–90 μm c = 0.24 M, T = 75 °C, pO2 = atm |
X = 40% |
| 3 | Gavriilidis et al.142 (2011) | Silicon-glass micro-packed-bed reactor (MPBR) – Single serpentine shaped channel – V = 34 μL (0.6 mm × 0.3 mm) |
(Au–Pd)/TiO2 (1 wt% Au–Pd, molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) Particle size: 53–63 μm Neat, T = 120 °C, pO2 = 5 bar |
X = 95%, S = 78% |
| 4 | Kappe et al.143 (2013) | H-Cube Pro™ microreactor – V = 500 μL (catalyst cartridge) |
Fe/Al-SBA15 (1 wt% Fe) Particle size: 1–2 μm c = 0.1 M, T = 140 °C, pO2 = 25 bar |
X = 100%, S = 95%, Recirculation |
| 5 | Gavriilidis et al.144 (2015) | Teflon AF-2400 tube-in-tube microreactor – V = 150 μL – Inner tube: Teflon AF-2400 (ID = 0.8 mm, OD = 1.0 mm) – Outer tube: PTFE (ID = 1.59 mm, OD = 3.18 mm) |
(Au–Pd)/TiO2 (1 wt% Au–Pd, 1:19) Particle size: 63–75 μm Neat, T = 120 °C, pO2 = 7 bar |
X = 44%, S = 73% |
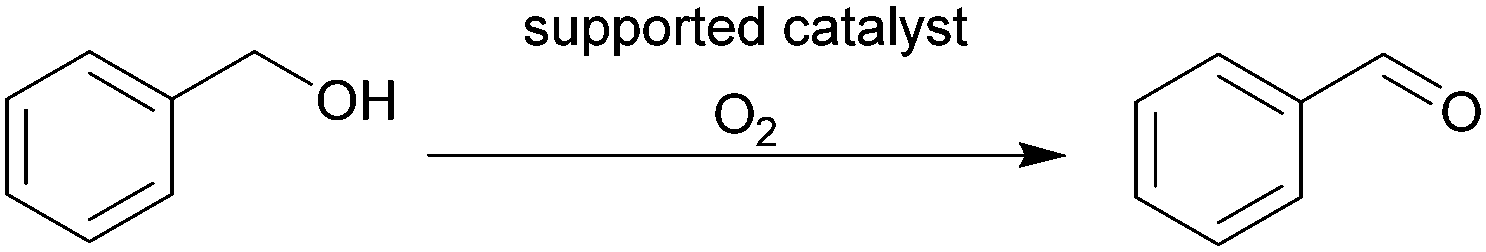
The continuous-flow aerobic oxidation of benzyl alcohol by supported tetrapropylammonium perruthenate (TRAP) in a silicon-glass micro-packed-bed reactor (MPBR) (0.6 mm × 0.3 mm, V = 26 μL) was reported by Gavriilidis et al.141 For the multiphase (gas–liquid–solid) reaction, a conversion up to 40% was obtained within a residence time of approximately 1 min (Table 3, entry 2). The same group reported a solvent free aerobic oxidation process of benzyl alcohol with pure oxygen.142 A similar silicon-glass MPBR (0.6 mm × 0.3 mm, V = 34 μL) was used, packed with 1% (Au–Pd)/TiO2 catalyst. The reaction progress was monitored using in situ Raman spectroscopy. A conversion of 95% was measured with a selectivity of 78% to benzaldehyde, under optimized conditions (catalyst particles: 53–63 μm, 120 °C and 5 bar) (Table 3, entry 3).
In a later study, the disproportionation of benzyl alcohol to toluene was confirmed experimentally using 1% (Au–Pd)/TiO2 and 1% (Au–Pd)/MgO as supported catalysts.145
The use of first-row transition metals for catalysis is of great interest as these metals are abundant and cheap. Kappe et al. demonstrated that the aerobic oxidation of benzyl alcohol to benzaldehyde could be carried out with heterogeneous iron nanoparticles in micro-flow.143 Iron oxide (Fe2O3) nanoparticles were covalently bound to an aluminosilicate support (Al-SBA15). The Fe/Al-SBA15 (1 wt% Fe) catalyst was diluted with silica gel and loaded into a cartridge. A commercially available H-Cube Pro™ module microreactor was used. With 2,2,6,6-tertramethylpiperidine-1-oxyl (TEMPO) as the co-catalyst, a yield of 42% was acquired for a single pass reaction. Full conversion was achieved (95% selectivity) by recirculation of the reaction mixture through the microreactor (Table 3, entry 4). In addition, no leaching of the catalyst was detected.
The tube-in-tube reactor developed by the Ley group is an operationally simple membrane microreactor consisting of two concentric capillaries.146,147 The inner capillary functions as a semipermeable membrane (Teflon AF-2400) and allows gas diffusion from the gas channel to the liquid channel. Recently, Gavriilidis et al. demonstrated the use of this reactor for the solvent-free aerobic heterogeneous oxidation of benzyl alcohol.144 The continuous supply of oxygen through the reactor wall offered significant improvement in both conversion and selectivity as compared to a non-permeable reactor design with a pre-saturated feed. The highest conversion achieved was 44% with 73% selectivity to benzaldehyde (Table 3, entry 5).
The continuous-flow oxidation of vanillyl alcohol to vanillin was reported by Semenov et al.148 Yields up to 98% were obtained within the first hour of run time (Scheme 1). The mesoreactor consisted of quartz tubing (7 mm diameter, 1 mL) and was loaded with the palladium catalyst in subunits (a commercially available supporting matrix). The catalyst retained activity up to 100 hours of run time and could be regenerated.
 | ||
| Scheme 1 Oxidation of vanillyl alcohol to vanillin under flow reaction conditions.148 | ||
Kobayashi et al. described the first quantitative aerobic oxidation process of both primary and secondary alcohols to their respective aldehydes and ketones in a microreactor.149,150 The reactor consisted of a wall coated, gold-immobilized, capillary column. The three-phase system proved to be highly applicable for a wide scope of benzylic, aliphatic and allylic alcohols. Yields ranged between 89–99% and up to 92% in the case of benzyl alcohol using a bimetallic Au/Pd-immobilized column (Scheme 2). No leaching of gold was observed after four days of continuous running.
 | ||
| Scheme 2 Oxidation of alcohols with molecular oxygen in a gold-immobilized capillary microreactor.149 | ||
Hii et al. reported a highly practical process for the aerobic oxidation of alcohols to aldehydes or ketones.151 A commercially available XCube™ flow reactor was used. The cartridge was packed with readily available Ru/Al2O3. Recirculation of the reaction mixture was maintained until full conversion was achieved. The system proved to be highly selective (>95%) in the case of allylic and (hetero)benzylic alcohols, including substrates with bulky substituents and/or heteroatoms at the ortho-position. Primary and secondary aliphatic alcohols proved to be more challenging (conversion 64–78%). However, by the addition of a desiccant cartridge (MgSO4), activity of the catalyst could be retained, and in the case of 2-hexanone, the yield could be improved significantly from 75 up to 91% (Scheme 3).
 | ||
| Scheme 3 Aerobic oxidation of alcohols in a XCube™ microreactor.151 | ||
A multistep synthesis of amides from alcohols and amines using a continuous-flow microreactor system was reported by Jensen et al.135 The reaction system consisted of three sections: (1) a packed-bed microreactor for the heterogeneous aerobic oxidation of alcohols, (2) a membrane separator for the removal of residual O2 and (3) a spiral-channel microreactor for the oxidative amidation reaction. For the first step, Ru/Al2O3 (5 wt% Ru) was found to be the most efficient heterogeneous catalyst. As oxidants, molecular oxygen and urea hydrogen peroxide (UHP) are used for the first and the third section, respectively. The integrated setup was suitable for a wide scope of benzylic (69–94%) and heterocyclic aromatic alcohols (79–97%), displaying good to excellent yields (Scheme 4). In addition, this process can be applied to secondary amines, including piperidine (81%), pyrrolidine (81%) and 1,2,3,4-tetrahydroisoquinoline (69%). However, aliphatic alcohols were not reactive within this system.
 | ||
| Scheme 4 Multistep synthesis of amides from benzylic or heterocyclic aromatic alcohols and secondary amines in continuous flow.135 | ||
Stahl et al. reported a continuous-flow setup for the aerobic oxidation of diverse benzylic and heterobenzylic alcohols.152 The mesoscale reactor consisted of stainless steel tubing (6 mm OD, 76 mm length) and was packed with the commercially available Ru(OH)x/Al2O3 (2.3 wt% Ru) catalyst. A diluted source of oxygen (8% in N2) was used as the oxidant to stay below the flammability threshold of the solvent. The packed-bed reactor was operated in an up-flow orientation to ensure optimal liquid–solid interaction and was submerged in a heating bath. Partial deactivation of the catalyst was noticed during initial benzyl alcohol oxidation. Accumulation of trace amounts of benzoic acid was found to poison the active sites of the catalyst. However, after more than 50 turnovers, the activity stabilized, which allowed steady-state operation. High to excellent yields (88–99%) for single pass runs were obtained for a variety of substrates. In addition, a continuous 72 hour run, with 2-(hydroxymethyl)thiophene as a substrate, was conducted (Scheme 5). Steady state yields of 2-thiophene carboxaldehyde remained >99% throughout the whole run and minimal leaching of the catalyst was detected (5 ppm). Furthermore, the flow system also promoted the oxidative dehydrogenation of indoline, yielding 95% indole. Asao et al. also employed a similar continuous-flow mesoreactor for the aerobic oxidation of various alcohols using a nanoporous gold catalyst.153
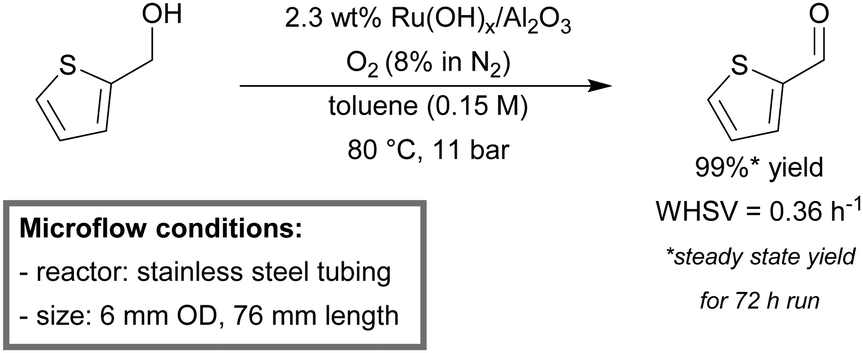 | ||
| Scheme 5 Continuous flow aerobic alcohol oxidation using Ru(OH)x/Al2O3 as the catalyst and O2 (8% in N2) as the source of the oxidant.152 | ||
One major advantage of microreactors (compared to batch) is the increased heat-transfer characteristics.19,154,155 This is attributed to the high surface-to-volume ratios of microchannels. The conventional method of heating microreactors is done using convectional heating instruments (e.g., oil baths). Alternatively, more efficient heating can be achieved by direct heating methods, such as microwave heating and induction.
Inductive heating was demonstrated by Kirschning et al. for the continuous-flow oxidation of both allylic and benzylic alcohols.156 The magnetic particles were packed into a mesofluidic PEEK (polyether ether ketone) reactor (3 mL) and diluted with MAGSILICA® to facilitate inductive heating. A medium frequency (25 kHz) external inductor was used to rapidly heat the magnetic particles. A Teflon AF-2400 tube-in-tube reactor (0.8 mm AD, 0.6 mm ID) was placed in front of the inductively heated PEEK reactor in order to saturate the reaction mixture with molecular oxygen. A scope of both primary and secondary alcohols was investigated. In almost all cases, full conversion was observed in a single pass reaction, providing a good to excellent yields (63–98%). In order to demonstrate the practicality of this catalytic system, a scale-up study was conducted; 4-bromobenzyl alcohol could successfully be oxidized to its corresponding aldehyde (96%) on a gram scale (Scheme 6).
 | ||
| Scheme 6 Aerobic oxidation of allylic and benzylic alcohols under inductively-heated flow conditions with gold-doped superparamagnetic nanostructured particles.156 | ||
Recently, Uozumi et al. developed a highly practical, fast and clean continuous-flow procedure for the aqueous aerobic flow oxidation of primary and secondary alcohols to their corresponding carboxylic acids and ketones.157 Platinum nanoparticles dispersed on an amphiphilic polystyrene-poly(ethylene glycol) resin (APR-Pt) were used as an active heterogeneous catalyst. The catalyst was loaded into a cartridge (4 mm ID × 70 mm) and fitted to a commercial XCube™. A broad scope of alcohols, including aliphatic, aromatic and heteroaromatic alcohols were efficiently oxidized (63–99%) within a short contact time (44 to 73 seconds). Furthermore, benzyl alcohol could be partially oxidized to benzaldehyde (90%) in the presence of triethylamine (Et3N). In addition, a gram-scale production of surfactants (dodecyl{oligo(oxyethylene)}acetic acids) was performed by the oxidation of a mixture of oligo(ethylene glycol) monododecyl ethers in the aqueous continuous-flow setup (Scheme 7). These results show that the flow setup is robust for long duration processing (113 hours of runtime).
 | ||
| Scheme 7 Aerobic oxidation off oligo(ethylene glycol) ethers to the desired surfactant carboxylic acid in an aqueous continuous-flow procedure.157 | ||
Favre-Réguillon et al. used high-throughput screening (HTS) to develop a more selective protocol for the room temperature, aerobic homogeneous oxidation of aldehydes.163 A microreactor was built from PFA tubing (1 mm ID, 4 mL). Screening for active catalytic systems was done via sequential pulse experimentation, using a sample loop to introduce a different reaction mixture in each slug. The liquid slugs were separated by oxygen slugs (Taylor flow). Two parallel HTS experiments were conducted. The first set of experiments was to find a suitable metal catalyst in order to accelerate reaction kinetics, while retaining selectivity. 100 ppm of Mn(II) was found to increase the conversion of aerobic oxidation from 80% (uncatalyzed) to full conversion for a residence time of 6 minutes in continuous flow. For the second screening, the residence time was set to 15 minutes in order to ensure full conversion for the uncatalyzed reaction (selectivity 77%). Different salts were evaluated for their ability to increase the selectivity. 2 wt% of sodium 2-ethylhexanoate was found to increase the selectivity up to 98%. The last step was then to incorporate both the catalyst (Mn(II) 100 ppm) and the additive (sodium 2-ethylhexanoate 2 wt%) into the same reaction for a 6 minute flow run. As predicted, the combination of both resulted in a synergistic effect, achieving near quantitative conversion (98%) of the aldehyde, while maintaining the selectivity of 98% towards its carboxylic acid (Scheme 8).
 | ||
| Scheme 8 Room temperature aerobic oxidation of 2-ethylhexanal catalyzed by Mn(II) in micro-flow.163 | ||
Kappe et al. developed a continuous-flow system for the direct aerobic oxidation of 2-benzylpyridines, catalyzed by homogeneous FeCl3.164 By the use of a high T/p continuous-flow process, the group was able to enhance the reaction kinetics significantly to a minute instead of hour scale.165 Molecular oxygen could be replaced by air and propylene carbonate (PC) was used as a green solvent. At high concentration (1.2 M) and low catalyst loading (5 mol%), a yield of 81% phenyl(pyridin-2-yl)methanone was obtained within 13 minutes of residence time (Scheme 9).
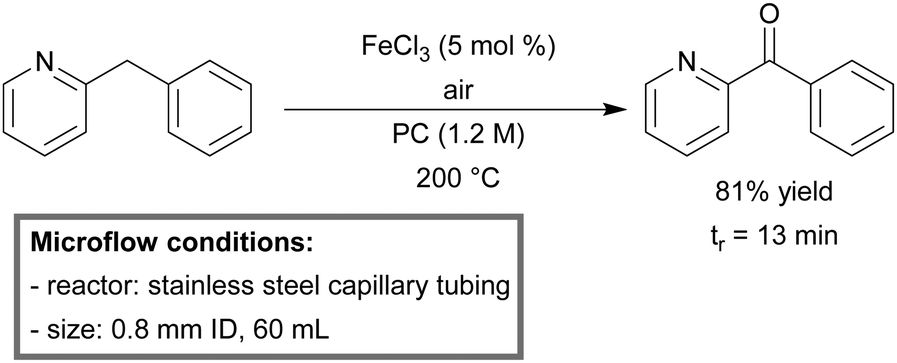 | ||
| Scheme 9 Aerobic oxidation of 2-benzylpyridines in a gas–liquid continuous-flow reactor.164 | ||
Molecular oxygen is by far the choice of oxidant considering its low price and green character. However, in the case of partial oxidation, chemists tend to use alternative oxidants, since over-oxidation can be a significant problem. One such process is the aerobic anti-Markovnikov Wacker oxidation of styrene compounds into phenylacetaldehydes. Ley et al. succeeded in making this aerobic oxidation possible by the use of microreactors.166 They developed an efficient anti-Markovnikov oxidation procedure for various styrenes utilizing a Teflon AF-2400 tube-in-tube microreactor (0.7 mL), coupled to a stainless steel coil (30 mL). With (MeCN)2PdCl2 (5 mol%) and CuCl2 (5 mol%) as catalysts and molecular oxygen (8 bar) as the oxidant, the scope of functionalized styrenes could successfully be oxidized to the corresponding aldehydes (56–80%) with minimal over-oxidation and within 60 minutes of residence time (Scheme 10). Due to the relative purity of the product stream, simple work up was possible via crystallization in the presence of sodium bisulfite.
 | ||
| Scheme 10 A continuous-flow aerobic anti-Markovnikov Wacker oxidation of styrenes using molecular oxygen as the oxidant.166 | ||
Kappe et al. constructed a continuous-flow reactor for the preparation of acetophenone and benzoic acid from ethyl benzene.167 The combination of CoBr2 (2.5 mol%) in combination with Mn(OAc)2 (2.5 mol%) in acetic acid was used as an active catalytic system. This system is known in the literature as the ‘MC-system’, and is one of the most active and selective catalytic systems available for aerobic, homogeneous liquid phase oxidation processes.168,169 In addition, synthetic air could be utilized as the oxidant. The microreactor protocol was shown to be superior compared to its batch counterpart. In the case of acetophenone, the reaction time could be reduced significantly from 150 min in batch to 6 minutes of residence time in flow, while maintaining the selectivity (77%). A microfluidic system was built from PFA capillary tubing (0.8 mm ID, 25 mL). At 120 °C, essentially 66% of acetophenone was isolated without the need for chromatography (Scheme 11). For the full oxidation of ethyl benzene to benzoic acid, a second flow setup was built, consisting of a stainless steel capillary tubing (0.8 mm ID, 60 mL) in combination with a higher reaction temperature (150 °C). An isolated yield of 71% was obtained within 16 minutes.
 | ||
| Scheme 11 Aerobic oxidation of ethyl benzene to acetophenone and benzoic acid in a continuous-flow microreactor.167 | ||
A continuous-flow homogeneous aerobic oxidation method for primary alcohols, with Cu(I)/TEMPO as the catalyst and N-methylimidazole (NMI) as the base was reported by Stahl et al.170 This was an improvement to their previous pioneering report on palladium(II)-catalyzed aerobic oxidation of alcohols in flow.171 No catalyst poisoning from substrates containing sulfur or nitrogen heteroatoms was noticed. In addition, faster catalytic rates were observed, which allowed minimal exposure time of the substrate to oxygen, and therefore avoided over-oxidation. This system was a modification of the existing flow system. The microreactor was made from stainless steel (1/8′′ OD, 66 mL) tubing. The temperature was set at 100 °C or 60 °C for benzylic alcohols or aliphatic alcohols, respectively. The quantitative yield (99%) for a variety of benzylic alcohols was obtained within 5 minutes of residence time (Scheme 12). For aliphatic alcohols near quantitative yields (95–99%) were obtained at longer residence times (30–45 minutes).
 | ||
| Scheme 12 Aerobic oxidation of primary alcohols by the Cu(I)(OTf)/TEMPO catalyst in continuous flow.170 | ||
An improved protocol for the selective aerobic oxidation of primary alcohols by Cu(I)/TEMPO catalysis in micro-flow was reported by Favre-Réguillon.172 Kinetic studies revealed that oxygen showed a first order relationship, which indicates that a higher oxygen concentration could be beneficial. Therefore, the mixture of 9% O2 in N2 was exchanged for a pure oxygen feed. Furthermore, calculations of activation energy for dioxygen binding with copper revealed low activation values (4–12 kJ mol−1), indicating that the use of high temperature will have a limiting effect on the rate of oxidation. As a consequence, room temperature was utilized instead of 60–100 °C as reported by Stahl previously. In addition, lower relative concentrations of TEMPO to copper (1:10) gave better results in screening experiments. Cu(I) species were found to be sensitive to aging, thus the Cu(I) species was replaced by a more active and stable Cu(II) source ([(bpy)Cu(II)-(OH)]2(OTf)2). For the reaction setup, a PFA microreactor (1.6 mm ID, 5 mL) was constructed. Benzylic and allylic alcohols could be oxidized successfully to their corresponding aldehydes with an excellent yield (90–99%) (Scheme 13). Aliphatic alcohols proved to be less reactive (conversion < 50%), but higher conversion could be acquired at longer residence times (>5 min).
 | ||
| Scheme 13 Aerobic oxidation of primary alcohols by the [(bpy)Cu(II)-(OH)]2(OTf)2/TEMPO catalyst in continuous flow.172 | ||
3.2 Photosensitized singlet oxygen oxidation processes in continuous flow
Singlet oxygen is well known for its wide spread use in applications, such as synthetic chemistry, biology, medicine, and materials science.173 Singlet oxygen can be produced in situ by the use of dye-based (e.g., rose bengal) or metal-based photosensitizers (e.g., Ru(bpy)3Cl2) and light. These sensitizers absorb electromagnetic waves at certain wavelengths (e.g., visible light) depending on their specific structure. Due to their high electrophilicity, singlet oxygen is found to be much more active than its ground (triplet) state, and is capable of oxidizing substrates that would otherwise be unaffected by triplet oxygen. Therefore, photosensitized singlet oxygen oxidation processes are often associated with mild reaction conditions (e.g., room temperature and visible light). However, certain limitations are associated with the use of singlet oxygen. The lifetime of singlet oxygen is highly solvent dependent (e.g., 9.5 μs in methanol) which results in very low effective concentrations of singlet oxygen. In addition, formation of by-products is common at prolonged irradiation times due to the high activity of singlet oxygen and formation of peroxide intermediates. Furthermore, gas–liquid photoreactions are challenging on larger scale batch procedures, due to the low interfacial area and limited irradiation of the reaction mixture (Lambert–Beer law).Most of these limitations can be overcome by the implementation of micro-flow technology.77,174–178 Due to their high surface-to-volume ratio, microreactors offer homogeneous irradiation of the reaction mixture, resulting in maximum photon absorption and faster reaction kinetics. In addition, high control of reaction time minimizes possible over-oxidation and by-product formation.
 | ||
| Scheme 14 Photosensitized singlet oxygen oxidation of α-terpinene to ascardole in a micro-capillary film (MCF) reactor.180 | ||
Another example is the photosensitized oxidation of citronellol to rose oxide in glass microreactors (1 mm ID, 0.27 mL), performed by Meyer et al.181 A comparison was made between a batch reactor and a microreactor in terms of the space-time yield and photonic efficiency. The photonic efficiency was approximately twice as high as that of the batch reaction. Furthermore, the space-time yield (STY) was about one order of magnitude higher for the microreactor.
Kim et al. developed a dual channel PDMS microreactor for the (−)-citronellol oxidation (Fig. 6).182,183 The upper channel was coated with polyvinylsilazane (PVSZ) in order to protect the reactor from solvent swelling. The bottom channel was used for gaseous oxygen feed. The two channels were separated by a PDMS gas permeable membrane. The photosensitized oxidation of (−)-citronellol was chosen as a model reaction. The dual-channel microreactor performance was compared using a conventional batch setup and a single channel microreactor. Both microreactors provided a higher yield (95–97%) of rose oxide as compared to the batch setup (89%). With a residence time of only 3 minutes and an internal volume of 285 μL, the dual-channel provided a daily output of 45.49 mmol, outperforming the single channel (the same reaction volume) and the batch reactor (50 mL) by more than 10 and 2.6 times, respectively.
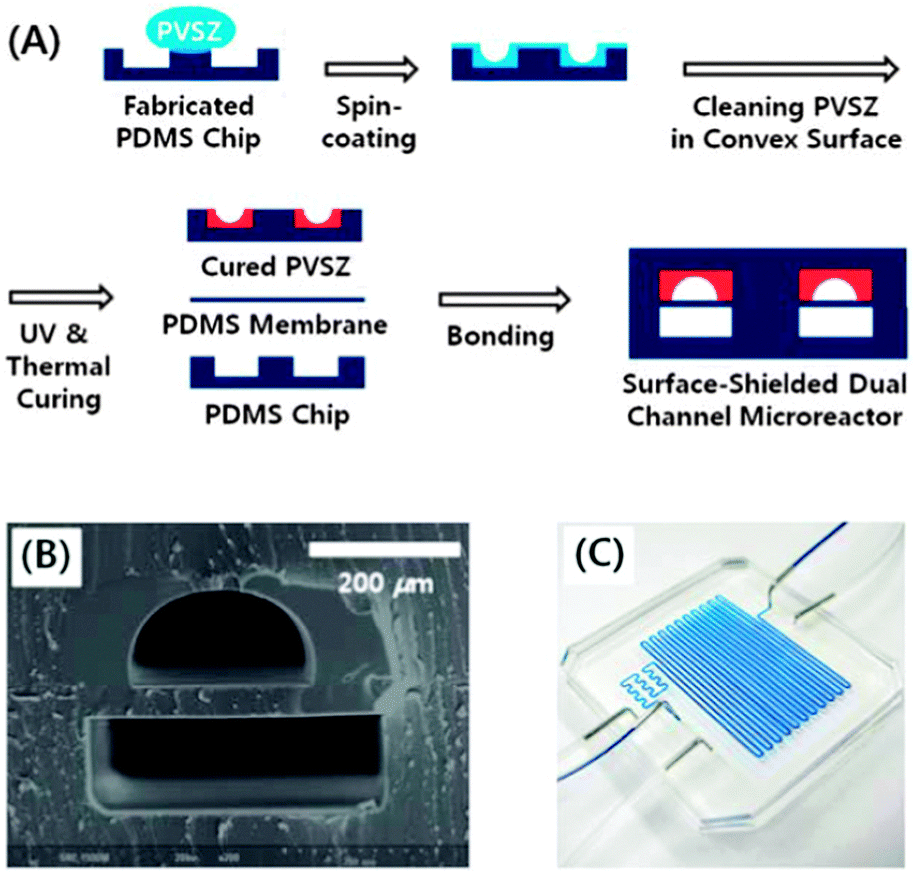 | ||
| Fig. 6 (A) Schematic illustration for the fabrication of the dual-channel microreactor with the PVSZ coated upper channel. (B) Cross-sectional view of the dual-channel microreactor. (C) Dual-channel microreactor under operating conditions. Reproduced from ref. 182 with permission from The Royal Society of Chemistry. | ||
The importance of the light source and microreactor geometry optimization was shown by Lapkin et al.184 The oxidation of α-pinene to pinocarvone under photochemical conditions was used as a model reaction. Various flow reactors, a recirculating annular reactor and a microreactor chip (500 μm × 240 μm, V = 240 μL) were investigated. Combined with each flow setup, different light sources (75 W Xe arc lamp, 48 W LED array and 250 W metal halide lamp) were utilized. A conventional immersed-well batch photoreactor, equipped with a 125 W mercury lamp, was used as reference. In the case of the annular recirculating flow reactor, a much better efficiency was obtained (23 times increase) due to higher photon flux and more efficient mixing. In the case of the microreactors, actinometric measurements revealed that the 416 nm LED array was most efficient, both in lower energy consumption and the largest amount of absorbed photon flux. This can be attributed to the match between microscale LEDs and the dimensions of the microreactor. Space time yield (STY) calculations favored microreactors (3–7 times higher STY), mostly because of better light utilization. In order to find the window of maximum performance/productivity, a cost-analysis model was derived for the homogeneous singlet oxygen oxygenation of α-pinene in microreactors. The model that described the optimal operating costs ($ per mol pinocarvone produced) as a function of the microchannel depth; 240 μm was considered as ideal.
Park et al. developed a general continuous-flow design for the photooxidation of monoterpenes (e.g., α-pinene, β-pinene, δ-limonene, α-terpinene and (−)-citronellol).185,186 They evaluated two photochemical reactors, i.e., a monochannel reactor and a tube-in-tube reactor. The monochannel microreactor (PEEK tubing, 500 μm ID, 26 μL) was used for small scale optimizations and rapid screening of the reaction scope. The tube-in-tube reactor (AF-2400 tubing, 600 μm ID, 7.92 mL) was tested for possible scale up. Photooxidation of representative terpenes was carried out and daily output (DOP = reagent concentration (M) × reactor volume (mL) × yield (%) × 1440 min/reaction time (min)) was used as a benchmark parameter. The DOP results concluded that the tube-in-tube microreactor clearly outperforms the conventional batch procedures (e.g., DOP 67.9 fold greater in the case of (−)-citronellol). To further increase the eco-friendly character of microreactors, an experiment with natural sunlight was conducted. During midday, β-pinene in methanol (0.5 M), together with methylene blue (5 wt%), was merged with oxygen and injected into the monochannel microreactor in a segmented flow regime. Natural sunlight was concentrated using a convex lens (d = 270 mm). At 3 bar backpressure, a yield of up to 62% was obtained within 30 minutes of residence time (Scheme 15).
 | ||
| Scheme 15 Continuous-flow photooxygenation of β-pinene under sunlight.185 | ||
Seeberger et al. used a capillary FEP reactor (750 μm ID, 14 mL) for photosensitized singlet oxygen oxidation chemistry in flow.187 The tubing was wrapped tightly around a quartz immersion well (cooled by a thermostat), containing a 450 W medium pressure mercury lamp and a Pyrex filter. Tetraphenylporphyrin (TPP) was used as a stable photosensitizer. Under steady state conditions, 27 mL of citronellol could be oxidized within one hour, resulting in a productivity of 2.5 mmol min−1. To further demonstrate the utility of the photoreactor, substrates with various functionalities, including alkenes, 1,3-dienes, furan and thioethers, were oxidized to their key intermediates. As example, 2-methylfuran could be oxidized successfully to 3-acetylacrylic acid, a key substrate to produce drug substances,188,189 with a yield of 68% within 0.8 minutes of residence time (Scheme 16).
 | ||
| Scheme 16 Singlet oxygen oxidation of 2-methylfuran to 3-acetylacryl acid in continuous flow.187 | ||
A multi-step flow reactor designed for the synthesis of artemisinin from dihydroartemisinic acid (DHAA) was developed by Seeberger et al.190 Artemisinin is known as the most effective antimalarial drug today, but due to its complex structure (Scheme 17), the total synthesis of artemisinin is considered inapplicable for large scale production. (<5% yield over 15 steps). The alternative is to start from the precursor DHAA, which is found in the same plant (Artemisia annua) as where artemisinin is isolated from. However, DHAA can be isolated in much higher yields. The following steps to yield artemisinin from DHAA are: singlet oxygen photooxidation, Hock cleavage, triplet oxygen oxidation and multiple ring closure condensation. Especially, the first step, singlet oxygen photooxygenation, was considered to be highly beneficial in micro-flow. The multi-step micro-flow system was capable of producing 200 g (39% yield) of artemisinin per day. Recent modifications resulted in an improved artemisinin production in terms of efficiency.191 For the first step, the former 450 W medium pressure mercury lamp was replaced by high-power LEDs of 72 W in total and the temperature was set to −20 °C. Tetraphenylporphyrin (TPP) was replaced by 9,10-dicyanoanthracene (DCA), a much more stable and efficient photocatalyst. Finally, the chlorinated solvent (DCM) was exchanged for toluene, as being more environmental benign. With a total residence time of 11.5 minutes, a production of 165 g (65% yield) of artemisinin per day could be obtained (Scheme 17).
 | ||
| Scheme 17 Multi-step continuous-flow setup for the artemisinin synthesis from dihydroartemisinic acid (DHAA).191 | ||
A similar photo-microreactor was used for the direct oxidative cyanation of primary and secondary amines.192 Amines could be successfully oxidized to their corresponding imines by the presence of singlet oxygen. Consequently, the in situ generated imines were trapped by trimethylsilyl cyanide (TMSCN) to yield the corresponding α-aminonitriles in good to excellent yields. In the case of dibenzylamine, the quantitative yield (99%) was obtained within 90 second residence time (Scheme 18). The reactor could be operated at room temperature with low loading of TPP (0.1–0.3 mol%) as the photosensitizer. By lowering the temperature to −50 °C and adding sub-stoichiometric amounts of tetra-n-butylammoniumfluoride (TBAF), they were able to produce, for the first time, α-aminonitriles directly from primary amines. Recently, the method was extended to allow for the preparation of unnatural amino acids193 and selective cross-condensation of amines.194
 | ||
| Scheme 18 Continuous-flow aerobic oxidative cyanation of primary and secondary amines using singlet oxygen.192 | ||
Itoh and et al. demonstrated the use of photo-microreactors for the improvement of the aerobic oxidation of toluene derivatives.195 A microreactor setup was built, consisting of a glass microreactor chip (500 μm × 300 μm, and 360 μL) and 375 nm LED light (11.4 W). The chip was fed with a liquid phase and molecular oxygen, generating a segmented flow regime. The liquid phase contained the substrate (0.5 M) and photosensitizer 2-tert-butylanthraquinone (10 mol%) in toluene. A yield of 48% was obtained for the photooxidation of toluene into benzoic acid, within 2 hour residence time. As compared to batch, 24 hours were necessary in order to complete the reaction.
Recently, Noël et al. reported a metal-free batch and a continuous-flow protocol to access disulfide linkages through photooxidation of thiols.196 With Eosin Y (1 mol%) as the metal-free photosensitizer and a 24 W compact fluorescent light bulb they were able to obtain a quantitative yield of disulfides for a variety of (hetero)aromatic and aliphatic thiols in an open flask procedure. It was observed that the mixing rate greatly influenced the rate of oxidation. Therefore, a biphasic microreactor setup was developed in order to overcome mass transfer limitations. A PFA capillary microreactor (750 μm, 950 μL) was constructed and exposed to LED light (3.12 W). Segmented gas–liquid flow was used to further increase the mass transfer efficiency. A variety of thiophenol substrates was evaluated and resulted in near quantitative yields (93–99%) within 20 minutes of residence time. Notably, the substrate furfuryl mercaptan could be exclusively dimerized under micro-flow conditions with a yield of 87%. To demonstrate the mild character of this procedure, an intramolecular disulfide bond formation was conducted in flow to prepare oxytocin, a peptide hormone. Full conversion was obtained within 200 seconds of residence time (Scheme 19). The same reactor was used for kinetic studies.100 1st order for thiophenol and 2nd order for molecular oxygen were observed. Furthermore, the calculated Hatta number (Ha = 0.06) suggested that gas–liquid mass transfer limitations were completely eliminated by the use of microreactors.
 | ||
| Scheme 19 Metal-free oxidative S–S coupling of thiols in continuous flow.196 | ||
5-Hydroxymethylfurfural (5-HMF) is recognized as a new platform molecule from the bio-based industry.197,198 A potential compound of interest derived from the HMF-platform is 5-hydroxy-5-(hydroxymethyl)-furan-2(5H)-one (H2MF). This scaffold is directly available from 5-HMF by means of singlet oxygen oxidation, but was only accessible in low to moderate yields and selectivities.199 However, Kappe et al. recently developed a continuous-flow procedure for the H2MF production to form 5-HMF.200 The microreactor was built from commercially available PFA tubing (1 mm ID, 10 mL) and wrapped around a glass cylinder. As a light source, a 60 W compact fluorescent light bulb was employed. After optimization (0.2 M 5-HMF, 0.5 mol% rose bengal, 17 bar BPR and iPrOH:H2O (1:1) solvent), H2MF could be isolated in an excellent yield (93%) within 40 minutes of residence time (Scheme 20). The micro-flow setup was applicable for a variety of bio-based furfural substrates. The synthetic method was considered sustainable, since only visible light, oxygen, catalytic amounts of photosensitizer and an environmental friendly solvent mixture were used.
 | ||
| Scheme 20 Singlet oxygen oxidation of 5-HMF in continuous flow.200 | ||
Lammertink et al. developed a new concept of microreactors, based on membrane technology.201 A porous photocatalytic membrane microreactor was built and applied for various photooxidation reactions. The reactor was microfabricated (CNC milling) from porous aluminium oxide (α-Al2O3). A layer of TiO2 was immobilized onto the channel walls via UV treatment and selective hydrophilization of the photocatalyst. The hydrophilic character of the TiO2 layer allowed efficient contact of the aqueous reaction mixture inside the microchannels, while molecular oxygen was fed through the hydrophobic membrane (Fig. 7). The photocatalytic oxidative degradation of methylene blue and phenol was used as a benchmark.
 | ||
| Fig. 7 The use of a porous photocatalytic membrane microreactor for the oxygenation of methylene blue and phenol. Reproduced from ref. 150 with permission from Elsevier. | ||
A thiolene-based microreactor was loaded with an immobilized fullerene catalyst.202 Fullerenes were used as photosensitizers for the singlet oxygen oxidation of α-terpinene and methionine. In both cases, quantitative conversions could be obtained within 40–50 seconds of residence time.
Boyle et al. functionalized a commercially available, 16 channel, glass microreactor with photo-active porphyrins.203 The oxidation of three model substrates (i.e., cholesterol, α-terpinene and citronellol), with molecular oxygen, was performed and compared to their batch equivalent. Although overall yields were low in the microreactor, space time yields were higher because of increased efficiency in singlet oxygen production.
Several other papers describe the application of microreactors in the photocatalytic degradation of organic compounds, employing immobilized TiO2,204,205 silicon nanoporous silicon,206 cyanoaromatics encapsulated on porous silica207 and sulfonated zinc phthalocyanine (ZnPcSmix).208
3.3 Biocatalytic aerobic oxidation processes in continuous flow
Oxidation reactions can also be carried out by enzymes. These biocatalysts often outperform conventional catalysts (e.g., transition metal catalysts) in terms of selectivity. Enzymes used for oxidation (e.g., glucose oxidase (GOx)), often use air as the oxidant and are therefore mostly limited by gas–liquid mass transport of oxygen. Consequently, enzymatic oxidative reactions could highly benefit from microreactor technology.The first miniaturized enzymatic reaction systems have already been developed decades ago for high-throughput screening analysis, and could be regarded as predecessors of the current continuous-flow microreactors.209 Recently, research on the combination of biocatalysis and microscale technology is increasing.210–213 In addition, efforts have been made to develop enzyme microreactors as biosensors for biomedical applications (e.g., glucose detection).214–216
Plazl et al. developed a two-phase liquid–liquid microreactor for the selective aerobic oxidation of cholesterol to 4-cholesten-3-one, catalyzed by cholesterol oxidase.219,220 In a Y-shaped mixer the aqueous phase (containing the enzyme) and the organic phase (containing the substrate) were merged, forming a parallel aqueous-organic two-phase flow pattern. Under optimal conditions, 67% yield was obtained with a residence time of 62 seconds.
The same group reported a laccase-catalyzed 3,4-dihydroxy-L-phenylalanine (L-DOPA) oxidation, performed in an identical microreactor (220 μm × 50 μm, and 3.6 μL). A two-dimensional mathematical model was developed and was found to be in good agreement with experimental data (87% conversion of L-DOPA at a residence time of 100 seconds).
Zelić et al. demonstrated the laccase-catalyzed oxidation of catechol and L-DOPA in microreactors.221 In the case of catechol, oxidation rates were up to 167-fold higher than for the microreactor.
A falling film microreactor (FFMR) was used for the GOx-catalyzed oxidation of β-D-glucose (Scheme 21).222 The FFMR utilizes gravity to establish a thin liquid film which is brought into contact with the gas phase. The FFMR ensures permanent oxygen saturation, even at high reaction rates. A 12-fold and 588-fold faster reaction kinetics as compared to the bubble column or batch reaction, respectively, were obtained for the microreactor. The STY increased by a factor of 300 compared to the batch reactor (at 30% conversion), demonstrating the more efficient use of the enzyme activity. Scalability was demonstrated by the use of a larger FFMR model (50 channels, 1200 μm × 400 μm).
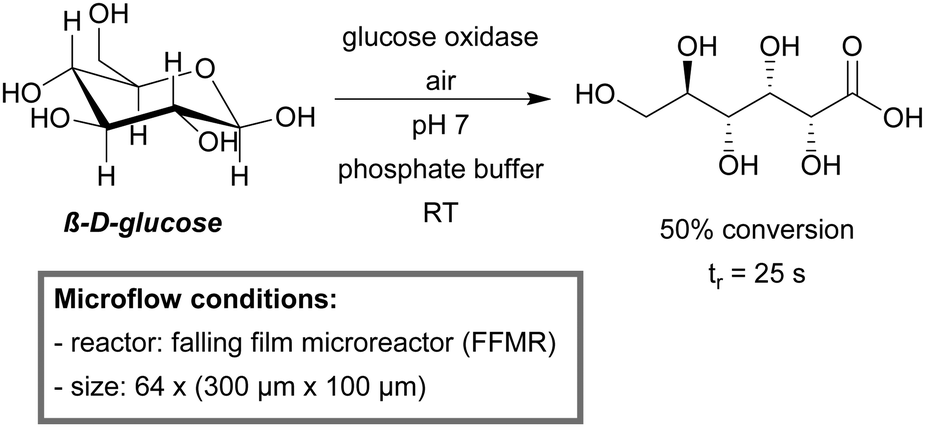 | ||
| Scheme 21 Aerobic oxidation of β-D-glucose catalyzed by glucose oxidase in a FFMR.222 | ||
Schmid et al. used a Teflon AF-2400 tube-in-tube microreactor (0.8 mm ID, 0.5 mL) to administer oxygen through the reactor wall continuously for the enzyme-catalyzed oxidation of 2-hydroxybiphenyl.223 2-Hydroxybiphenyl 3-monooxygenase (HbpA) was used to enable the oxidation of the substrate. Formate dehydrogenase (FDH) was added to ensure constant recycling of the cofactor NADH. The productivity achieved in the tube-in-tube microreactor was 4 times higher than that in a three-phase aqueous–organic–air segmented flow microreactor.
With the use of a sequential tube-in-tube reactor setup, Buehler et al. were able to produce 3-phenylcatechol on a preparative scale (1 gram) with a STY of 14.5 g L−1 h−1 (Scheme 22).224
 | ||
| Scheme 22 Enzyme mediated aerobic oxidation of 2-hydroxybiphenyl to 3-phenylcatechol in a tube-in-tube microreactor.224 | ||
Several immobilization techniques have been developed in flow.225–228 However, loss of activity due to blockage of the active sites, denaturation of enzymes, fouling and leaching still limits its application.229,230
Ho et al. developed a stable capillary enzyme bioreactor for the determination of blood glucose.231 Glucose levels could be monitored in a wide range of concentrations with high sensitivity (detection limit 10 μM) and demonstrated high reproducibility. Enzymatic activity could be retained for over 120 days.
Yang et al. demonstrated the successful immobilization of GOx on polyamidoamine dendrimer (PAMAM).228 Because of its unique dendritic structure, PAMAM could be used as a flexible spacer between the solid support (capillary wall) and the active enzyme. The dendritic structure allowed higher amounts of enzyme to be immobilized onto the reactor wall without constraining the availability of the active sites. The microreactor proved to be stable, whereas the activity decreased by less than 10% over 5 days of operation.
3.4 Uncatalyzed aerobic oxidation processes in continuous flow
The increasing environmental awareness demands for more atom efficient, mild and sustainable chemical processes. Conventional oxidation processes however, are often undesired because of their excessive need for heavy metals and hazardous oxidants, which lead to laborious waste treatment and inevitable pollution. Over the last decade chemists are striving to develop oxidation processes, which guarantee high selectivity under metal-free and uncatalyzed reaction conditions. One such example is the uncatalyzed, aerobic oxidation of cyclohexane to cyclohexanol and cyclohexanone. This mixture of products, also called KA oil, is produced on a large scale for Nylon production.232 However, the process suffers from selectivity problems and therefore conversion is kept low.101 In order to enhance the process selectivity, several research groups have employed continuous-flow microreactors. Bellefon et al. developed a chip microreactor setup to selectively oxidize cyclohexane under explosive conditions.82 Jevtic134 and Fischer99 both utilized capillary microreactors. Fraulin used microreactors and studied the progress of the reaction by in situ Raman spectroscopy studies.233Jensen et al. developed a continuous-flow metal-free oxidation of picolines by using air as the oxidant.234 In a 240 μL chip microreactor, 2-, 3- and 4-picoline could be selectively oxidized within 5 minutes of reaction time. In the case of 4-picoline, 100% HPLC yield was obtained after 1 minute of residence time at room temperature (Scheme 23). Jia et al. reported another aerobic benzylic C–H oxidation procedure.235 Microreactor technology was used to safely produce molecular oxygen in situ, by merging tert-butyl hydroperoxide (t-BuOOH) and sodium hypochlorite (NaOCl). Due to high heat dissipation, the reactor could be operated at room temperature.
 | ||
| Scheme 23 Continuous-flow metal-free oxidation of picolines by using air as the oxidant.234 | ||
A mild and straightforward aerobic flow oxidation procedure for aliphatic aldehydes to their corresponding carboxylic acids was developed by de Bellefon et al.236 The reactor operated at room temperature was built from PFA capillary tubing (1 mm ID, 11.7 mL). On-line GC-analysis was conducted using a sample valve. Mass transfer efficiency between the gaseous (oxygen) and the liquid phase was assessed by changing the superficial velocity of the segmented flow while maintaining a constant residence time. An optimum was reached for superficial velocities >0.5 m min−1, indicating that the chemical limited regime was achieved. 2-Ethylhexanal was converted quantitatively to its corresponding carboxylic acid at high concentration (3.2 M) and with a selectivity of 75% (Scheme 24). The residence time was approximately 15 minutes.
 | ||
| Scheme 24 Uncatalyzed aerobic oxidation of 2-ethylhexanal in a PFA capillary microreactor.236 | ||
The continuous-flow synthesis of ortho-functionalized phenols by aerobic oxidation of aryl Grignard reagents has been demonstrated by Jamison et al.237 The integrated micro-flow system consisted of a three-step setup. In the first capillary reactor, a sulfur or nitrogen containing nucleophile was exposed to isopropylmagnesium chloride lithium chloride (i-PrMgCl·LiCl) at room temperature, in order to deprotonate the substrate. The resulting mixture was then mixed with 1,2-dihalobenzenes, at elevated temperatures (80–120 °C), for the in situ formation of the reactive benzyne intermediate, followed by a nucleophilic addition, obtaining the ortho-functionalized organomagnesium intermediate. Finally the reaction stream was cooled to −25 °C and mixed with compressed air in order to produce the desired ortho-functionalized phenol compound. The overall residence time was 14 minutes with yields varying between 33–55% for nucleophiles, including thiophenols, N-containing heterocycles and secondary anilines (Scheme 25).
 | ||
| Scheme 25 Integrated three-step preparation of ortho-functionalized phenols in continous-flow.237 | ||
3.5 Aerobic coupling chemistry processes in continuous flow
Since its introduction in the 1970s, cross-coupling chemistry has played an important role in organic chemistry.238–241 Cross-coupling allows for the efficient formation of carbon–carbon and carbon–heteroatom bonds in the presence of a transition metal catalyst, and has found substantial application in the pharmaceutical industry and materials science.242 Recently, with the introduction of microreactor technology, several research groups reported the use of micro-scale devices to conduct two-phase aerobic cross-coupling reactions.243–247An example of a continuous-flow oxidative Heck reaction, performed in a dual-channel microreactor, was reported by Kim et al.243 A three-layered PDMS microreactor was built with a dual-channel structure (Fig. 8). The upper channel was fed with a solution containing the substrate and the catalyst. The bottom channel was fed with gaseous molecular oxygen. Between the two channels, a thin gas permeable PDMS layer (45 μm) ensured oxygen dosing to the reaction mixture. Due to the intimate contact between the gas and the solution phase, a significant enhancement was obtained for both yield (72–82%) and selectivity (87–93%). Because of the high availability of oxygen, the palladium catalyst could easily be reoxidized to its activated Pd(II) format, thus accelerating reaction kinetics. As a result the reaction time was reduced from 12 hours in batch to 30 minutes in flow.
 | ||
| Fig. 8 Dual-channel microreactor cross-section. Microchannel dimensions: 85 μm × 300 μm × 120 cm. Membrane: 45 μM thickness. Reprinted with permission from ref. 243. Copyright (2010) American Chemical Society. | ||
Noël et al. realized the aerobic cross-dehydrogenative Heck coupling of indoles in micro-flow.245 The reactor was built from commercially available FEP capillary tubing (750 μm ID, 3.8 mL). During optimization studies, it was found that a segmented flow regime (Taylor flow) with molecular oxygen was crucial for the successful reoxidation of the palladium catalyst. Moreover, catalyst degradation was completely prevented due to high surface-to-volume ratios between the two phases. The high degree of control over both mass- and heat-transfer, enabled the acceleration of reaction kinetics from hour to minute scale. As an example, the coupling between indole and 2,2,2-trifluoroethyl acrylate could be completed within a residence time of 10 minutes, yielding 82% of coupled product (Scheme 26). This was a significant improvement in yield and time as compared to its batch counterpart (58% yield after 4 hours of batch reaction). The setup was effective in the preparation of a wide range of 3-vinylindoles (27–92% yield), including the preparation of methyl-(E)-3-(6-fluoro-1H-indol-3-yl)acrylate (67% yield), a potential anticancer agent.248
 | ||
| Scheme 26 Aerobic cross-dehydrogenative Heck reaction of indoles in continuous-flow.245 | ||
Ley et al. demonstrated the implementation of a tube-in-tube microreactor for the synthesis of 1,3-butadiynes, via aerobic Glaser–Hay acetylene coupling.246 The tube-in-tube reactor was used to saturate an acetonitrile solvent stream with oxygen before merging with the catalyst and substrate stream. The resulting mixture entered a 20 mL heating coil (100 °C). After that the reaction coil and in line purification methods (packed-bed cartridges) were utilized to scavenge the copper catalyst and the TMEDA base. This allowed product isolation of various 1,3-butadiynes in high purity (49–99% yield), without the need for additional chromatographic purification. As an example, the coupling reaction of 3-ethynylanisole could be carried out on a gram scale (2.3 grams), yielding 84% of pure product without further need for additional chromatography.
4. Oxidation processes in flow by means of hydrogen peroxide
Besides oxygen, hydrogen peroxide constitutes another important oxidant in organic synthetic chemistry. However, the utilization of H2O2 in batch is normally only possible in low concentration and under mild reaction conditions due to its intrinsic hazardous properties. Due to the advantages of microreactor technology, i.e., high mass and heat transfer efficiency and easy scale-up, the application of H2O2 in a microreactor has received more and more attention.Gavriilidis et al. used hydrogen peroxide as an oxidant to facilitate epoxidation of 1-pentene in a microchannel coated with titanium silicalite-1 zeolite (TS-1, Scheme 27).249,250 Reaction rates increased with increasing amounts of Ti in the zeolite framework. It was also observed that the crystal size largely affected the reaction rate. A smaller crystal size resulted in a higher surface area and an improved mass transfer efficiency, providing higher reaction rates. The catalyst deactivated under the reaction conditions via a short-term reversible deactivation mechanism and a long-term irreversible deactivation mechanism. It was hypothesized that the short-term deactivation was caused by the deposition of the by-product and H2O, while the long-term deactivation was the result of leaching.
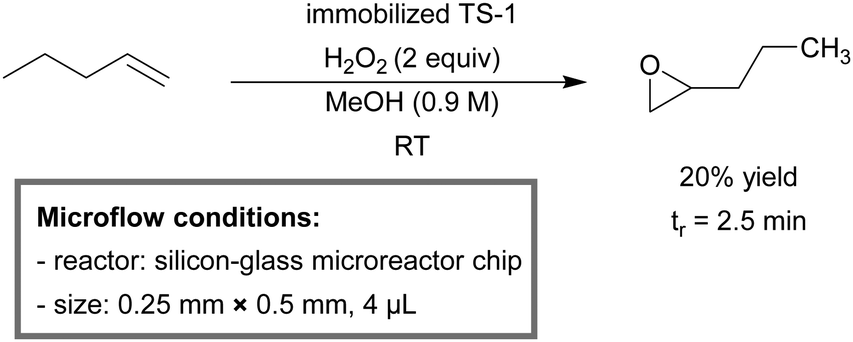 | ||
| Scheme 27 Epoxidation of 1-pentene with H2O2 in a TS-1 immobilized microreactor.249 | ||
Epoxidized soybean oil (ESO) is an important chemical intermediate, which can be used as a plasticizer in polyvinyl chloride plastics and a stabilizer for plastics.251 Kinetic modelling of soybean oil epoxidation at elevated temperatures in a microreactor demonstrated that this would lead to much shorter reaction times.252 Increasing the temperature also led to an increased decomposition of H2O2, which eventually became the dominant reaction. Santacesaria et al. employed a glass tubular reactor (1 cm ID, 22 or 38 mL) packed with stainless steel spheres (2.2 mm ∅) to carry out the soybean oil epoxidation under biphasic reaction conditions.253 They found that it was difficult to reach the industrial target (oxirane number ≈ 6.5 and residual iodine number ≤ 2) with only a single packed-bed reactor. A two reactor system allowed one to overcome these limitations. The first reactor provided a high surface area and a short residence time, which facilitated the initial conversion of soybean oil and reduced the ring-opening side reaction. The second reactor had a lower surface area and an extended residence time. According to the simulation results, the two reactor strategy met the industrial requirement within 45 min of residence time while the industrial process needed 6–8 h. Guo et al. investigated the epoxidation of soybean oil in a Bayer sandwich microreactor and found that the reaction could be improved by using in situ peracids.254
Dordick et al. have reported the first continuous-flow polyphenol synthesis by means of biocatalysis.255 Soybean peroxidase (SBP) was used as a biocatalyst in the presence of H2O2 and the substrate. Various phenolic substrates (e.g., p-cresol, p-methoxyphenol, and p-hydroxyphenyl alcohol) were introduced into the biochip (15 μm × 200 μm, 90 nL) and could be polymerized successfully.
The poly-L-leucine (PLL)-catalyzed oxidation of chalcone by hydrogen peroxide was carried out in a specially designed plate reactor, which consisted of two staggered herringbone micromixer-microchannel sections in series.256 A conversion of 87% and 88% ee could be obtained for the target compound (Scheme 28). A mathematical model was used to aid the reactor design and the results from the model were closely resembling the experimental ones.
 | ||
| Scheme 28 Continuous-flow asymmetric epoxidation of chalcone with H2O2, catalyzed by poly-L-leucine.256 | ||
The oxidation of 1-methylcyclohexene catalyzed by Candida antarctica lipase B (Novozym® 435) with H2O2 in a continuous-flow packed-bed reactor was investigated by Wiles and Watts et al.257 They employed a borosilicate glass tube (3 mm ID, 2.5 mL) loaded with Novozym® 435. Ethyl acetate was used as the solvent and could be enzymatically hydrolyzed to form acetic acid and subsequently oxidized to peracetic acid, which was the final oxidant for 1-methylcyclohexene oxidation.258,259 Full conversion for this substrate was observed at 70 °C with a residence time of 2.6 min (99% yield, Scheme 29). The flow allowed one to use larger amounts of H2O2 (4× higher) before deactivation of the enzyme was noticed. The catalytic performance of the enzyme was consistent even after 24 h of use.
 | ||
| Scheme 29 Lipase-mediated epoxidation of 1-methylcyclohexene with H2O2 in a packed-bed microreactor.257 | ||
Organic peroxides are strong oxidizers, which are widely used in the chemical industry. However, they are difficult to handle due to easy decomposition and risk for explosion hazards. Ebrahimi et al. studied the sulfuric acid catalyzed synthesis of performic and peracetic acids from H2O2 in a PTFE capillary (1.0 or 1.6 mm ID, 0.59–10 mL).260 The formation of performic acid could reach equilibrium within 4 min at 40 °C, while the synthesis of peracetic acid needed 10 min at 70 °C. Furthermore, they designed a plate reactor, which allowed them to work on an industrial production scale. The reactor was composed of alternating heat transfer and reaction channels. This unit has potential to be applicable for on-site or on-demand production of performic and peracetic acids due to the high production capacity up to 100 t/a of performic acid and up to 170 t/a of peracetic acid. Based on the results from flow experiments, they developed several kinetic models.261 A laminar flow model with velocity distribution in the radial direction had the best accuracy according to the Markov chain Monte Carlo analysis.
Although sulfuric acid is an efficient catalyst for the synthesis of percarboxylic acids, the homogeneous catalyst causes corrosion of the reaction equipment and needs to be removed after reaction.262 Ebrahimi et al. further investigated the formation of performic acid in the presence of a cation exchange resin loaded in a packed-bed micro-structured reactor.263 Dowex 50Wx8 was selected as the optimal catalyst due to excellent reusability. The results showed that the reaction could reach equilibrium in 2 min at 40 °C, while it needs at least 30 min for the corresponding batch reaction. However, Dowex 50Wx8 had lower activity than H2SO4, indicating that the active sites of Dowex 50Wx8 were partially deactivated.
Sulfoxides and sulfones are important moieties to tailor the properties of pharmaceuticals, pesticides and polymer stabilizers. Noguchi et al. synthesized sulfoxides from sulfides by using 30% hydrogen peroxide and no catalyst in a stainless steel capillary microreactor (1 mm ID, 730 μL, Scheme 30).264 The oxidation of thioanisole showed that 97% yield of sulfoxide could be reached within a residence time of 2.12 s in microreactor. In a conventional batch reactor, it required 3 h in batch to reach full conversion with only 82% yield and 15% of the over-oxidized sulfone.
 | ||
| Scheme 30 Synthesis of sulfoxides from thioanisole with a 30% solution of H2O2 in a stainless steel capillary microreactor.264 | ||
Doherty and Hardacre, et al. have developed a new catalyst [PO4{WO(O2)2}4]@PIILP based on the polymer immobilized ionic liquid phase (PIILP) concept, which facilitates catalyst recovery and allows one to minimize the amount of ionic liquid.265 The catalyst can be loaded in a packed-bed microreactor and the catalyst bed is further diluted with silica. A high yield of sulfoxide (92%) was obtained in methanol with 4 min of residence time, while the corresponding sulfone could be obtained with 96% yield in acetonitrile in a 15 min residence time (Scheme 31). The catalyst remained active for 8 hours under continuous-flow conditions (TON = 46428). Amberlite IR-120H as a solid supported catalyst for the sulfide oxidation in flow has been reported as well.266
 | ||
| Scheme 31 The synthesis of sulfoxide and sulfones from thioanisole and H2O2 in a microreactor using a peroxometalate-based polymer immobilized ionic liquid phase catalyst.265 | ||
Adipic acid is a basic building block in the chemical industry, which is mainly used as the starting material for the synthesis of nylon 6,6.267 Hessel et al. reported the continuous-flow synthesis of adipic acid via the oxidation of cyclohexene with 50% aqueous H2O2.268 The biphasic mixture was introduced into a packed-bed microreactor with glass spheres, which allowed one to intensify the mixing efficiency. [CH3(n-C8H17)3N]HSO4 and Na2WO4·2H2O were used as a phase transfer catalyst and an oxidation catalyst, respectively. At 110 °C, a 50% isolated yield for adipic acid was obtained within 20 min of residence time (Scheme 32). The yield could be further increased by using a multi-stage temperature ramping approach.269 This strategy allowed one to avoid hot spot formation throughout the reactor and extensive degradation of H2O2. With a three reactor system and a temperature profile of 70–100–110 °C, respectively, a total isolated yield of 66% for adipic acid could be obtained within 20 min of residence time. Kappe et al. developed another protocol which did not require a phase transfer catalyst and employed 25% aqueous H2O2 in the presence of tungstic acid as a catalyst.270 The isolated yield of adipic acid reached 63% at 140 °C with just 20 min of residence time.
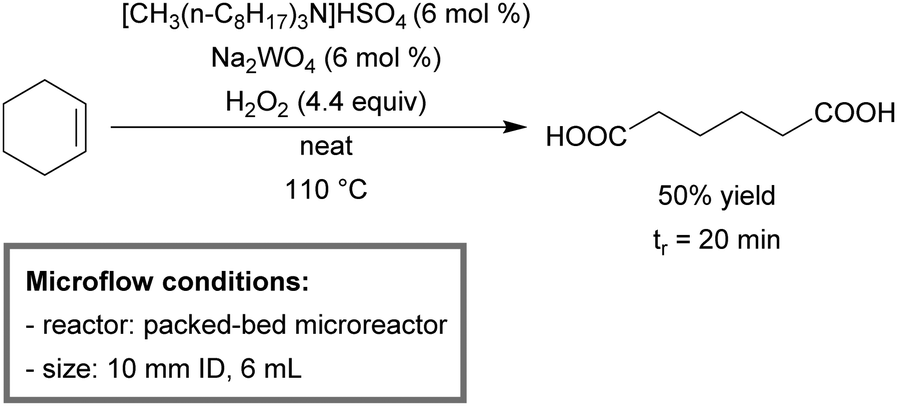 | ||
| Scheme 32 Continuous synthesis of adipic acid from cyclohexene with H2O2 in a packed-bed microreactor.268 | ||
The preparation of vitamin K3 (2-methyl-1,4-naphthoquinone) was carried out in a stainless steel capillary microreactor (1.0 mm ID, 8 mL).271 A high concentrated H2O2 solution (60%) was safely used for the oxidation process of 2-methylnaphthalene at 70 °C, but led to low selectivities. Alternatively, by using 26% aqueous peracetic acid, the selectivity could be improved at 80 °C (50% yield of vitamin K3).
Jensen et al. have developed a continuous-flow protocol for the direct oxidative amidation of aromatic aldehydes using H2O2 as the oxidant.272 In a silicon–Pyrex spiral microreactor (230 μL), a wide variety of aldehydes and amines were explored and the optimal reaction conditions for each aldehyde were rapidly selected due to the flexibility of microreactor assembly. Moreover, the use of elevated pressures allowed one to safely heat solvents above their boiling point, broadening the operational window significantly. The yield of the corresponding amides varied from 79–92% with 20–40 min of residence time at 80–110 °C (Scheme 33). Notably, the authors found that racemization was effectively suppressed when chiral amino acid derivatives were converted to their corresponding amides.
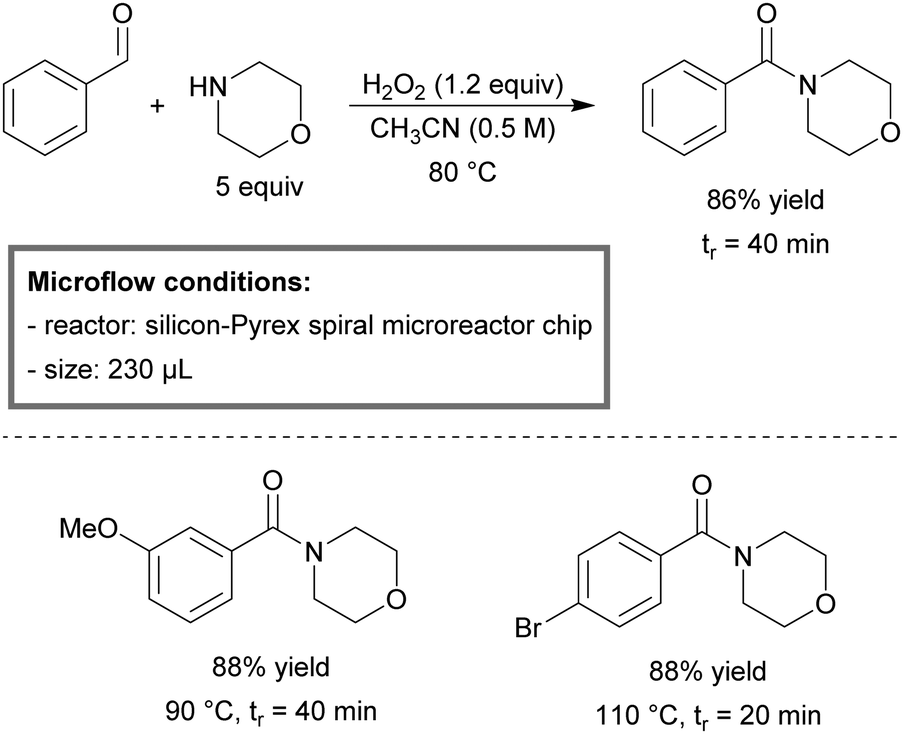 | ||
| Scheme 33 The direct oxidative amidation of aromatic aldehydes with H2O2 as the oxidant in a silicon–Pyrex microreactor.272 | ||
Park et al. have investigated the continuous-flow diacetoxylation of alkenes using peracetic acid (AcO2H) as the oxidant and triflic acid (TfOH) as the catalyst.273 AcO2H was prepared in situ by combining acetic anhydride (Ac2O) and H2O2 prior to combining it with the alkene substrate. A variety of alkenes were subsequently converted in flow within 10 min of residence time (Scheme 34).
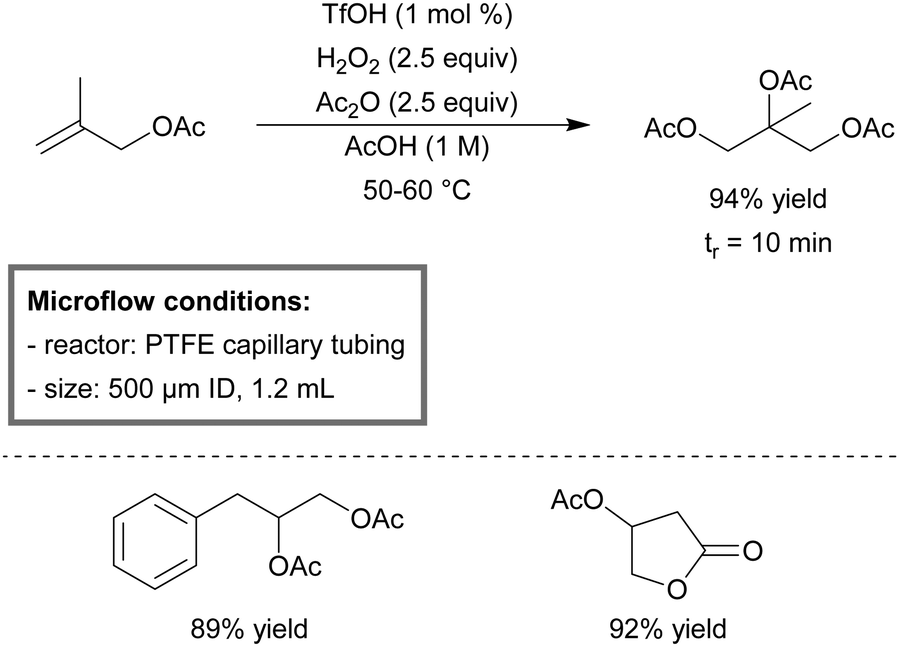 | ||
| Scheme 34 Trifluoromethanesulfonic acid-catalyzed diacetoxylation of alkenes with peracetic acid generated in situ from acetic anhydride and H2O2 in a microreactor.273 | ||
A continuous-flow protocol for the two-step hydroboration/oxidation of olefins was developed by Souto et al.274 In the first step, the olefin was hydroborated by BH3·THF in a PFA capillary microreactor (1 mm ID, 2 mL). Next, the reaction stream was merged with a basic H2O2 solution to oxidize the boron intermediate to the corresponding alcohol. The authors also used an inline extraction device to facilitate the work up and to minimize manual labor. The maximum productivity reached 120 mmol h−1 at a residence time of 70 s.
Other examples using H2O2 in flow involve the oxidation of methyl ethyl ketone,275 phenol276 and cyclohexene to the corresponding trans-1,2-cyclohexanediol.277
5. Ozonolysis in flow
The use of ozone (O3) in organic synthetic chemistry has been recognized for decades as it enables the oxidative cleavage of unsaturated molecules.278,279 In general, oxidation processes with O3 are considered environmentally friendly due to its atom efficiency. However, the exothermic nature of the reaction and production of the explosion hazardous ozonides require special safety considerations when using this reagent on a large scale. These concerns provide opportunities for microreactors to use ozone in a safe and reliable fashion.Kappe et al. used a commercially available reactor system (O-Cube™) to perform ozonolysis at atmospheric pressure and at temperatures between −25 °C and room temperature.280 The setup consists of a precooling section, which allows one to cool the substrate solution. Subsequently, the liquid stream is merged with the ozone gas flow, which is generated in situ. The segmented gas–liquid flow is then entering a thermostatic reactor coil (PTFE, 1 mm ID, 4 mL) and is quenched (oxidative or reductive cleavage) upon exiting the reactor coil. The in situ quench allows one to minimize the amount of explosive ozonides, making this process environmentally benign. A wide variety of substrates were processed in the reactor system, thus preparing aldehydes, ketones (Scheme 35), nitro compounds, sulfones and sulfoxides.
 | ||
| Scheme 35 Ozonolysis of β-pinene in a O-Cube™ microreactor.280 | ||
Ley et al. used a semipermeable capillary microreactor (Teflon AF-2400, 0.6 mm ID, 250 μL) which enables ozone transport through the reactor walls to the liquid reaction stream.281 Next, a variety of substrates were subjected to the ozonolysis conditions using methanol as the solvent. All reached full conversion within 1 h of residence time (Scheme 36).
 | ||
| Scheme 36 The ozonolysis of alkenes in a Teflon AF-2400 semipermeable capillary microreactor.281 | ||
Another practical ozonolysis procedure in flow was developed by Roydhouse and Gavriilidis et al.282,283 The authors used a commercially available Vapourtec flow system which was equipped with a cooled flow cell (1 mm ID, 2 mL). The use of the cooled flow cell was required to accommodate the highly exothermic reaction in a safe and reliable fashion. A wide variety of different substrates were oxidized by ozone, including heterocycles such as furan and quinolones. As an example, 2-(3-methoxyoxetan-3-yl)furan could by successfully converted into the corresponding oxetane acid (79%, Scheme 37), which represents a relevant pharmaceutical compound.
 | ||
| Scheme 37 Ozonolysis of 2-(3-methoxyoxetan-3-yl)furan to its oxetane acid in a Vapourtec cooled reaction module.283 | ||
Sun et al. have reported on the ozone degradation of pentachlorophenol in a T-junction microchannel reactor (400 μm × 500 μm, 14 μL).284 The authors have performed a systematic study on the ozone mass transfer characteristics in different gas–liquid flow regimes. An increase of the liquid side volumetric mass transfer coefficient (kLa) was observed with increasing superficial gas and liquid velocities. This increase was mainly caused by the increasing gas–liquid interfacial area. Also, it was found that an increase in pentachlorophenol and ozone concentration resulted in a higher ozone mass flux. The reaction between ozone and the substrate was instantaneous, which was confirmed by the absence of ozone in the bulk solution.
6. Miscellaneous oxidation processes in flow
Herein, other than aerobic, hydrogen peroxide or ozone mediated oxidation processes in continuous-flow microreactors are discussed. This chapter includes, metal oxide and anaerobic enzymatic oxidation reactions, Swern oxidation and electrochemical oxidation reactions. Furthermore, oxidations enabled by TEMPO, HOX, hypervalent iodine, and oxone will be discussed.6.1 Metal oxide oxidation processes in continuous flow
Kobayashi et al. developed a highly active, immobilized ruthenium catalyst for the non-aerobic flow oxidation of alcohols.285 Using a self-designed novel immobilization method, Kobayashi et al. could successfully immobilize RuCl2(PPh3)3 to an epoxide-containing copolymer. The solid catalyst (PI-Ru) was pre-oxidized and placed into a micro-column (2 mm ID, 214 μL). A reaction stream, containing the substrate and NMO as the co-oxidant, was sent over the catalytic bed. Notably, the immobilized PI-Ru catalyst showed higher activity as compared to its original non-immobilized counterpart. In the case of benzyl alcohol, 92% yield of the corresponding aldehyde was obtained during an 8 hour continuous operation (Scheme 38). No leaching of the catalyst was detected. | ||
| Scheme 38 Ruthenium catalyzed oxidation of benzyl alcohol to benzaldehyde in the flow system.285 | ||
In terms of sustainable chemistry, Kirschning et al. demonstrated the application of inductive heating for the continuous-flow oxidation of several functional organic compounds (i.e., alcohols, amines and alkenes).286 A packed-bed microreactor (4 mL) was filled with a mixture of superparamagnetic nanoparticles and solid oxides (CrO2 and NiO2). Operating under continuous flow, efficient heating was achieved inside the packed bed by exposing the highly conducting nanoparticles to a rapidly changing magnetic field (25 kHz). Full conversion was achieved for a single pass run.
Ley et al. demonstrated the use of microreactors for the efficient transformation of alcohols and aldehydes into carboxylic acids and nitro alkanes to their corresponding carbonyl compounds.287 Homogeneous permanganate (KMnO4) was used as a cheap oxidant. In order to avoid possible blockage of the capillary reactor (due to MnO2 solid formation), ultrasound was applied. Oxidized products could be isolated easily without the need for chromatography, providing high yields (58–98%) with excellent purity (>95%).
The same group reported the use of a pre-packed column of polymer supported perruthenate (PSP) for the oxidation reaction of commercially available 3,4-dimethoxybenzyl alcohol to its corresponding aldehyde.288 This transformation was then implemented in an integrated flow process for the multi-step synthesis of oxomaritidine, a bioactive molecule.
Recently, Ley reported a practical flow procedure for the in situ generation of unstable diazo compounds.289 A hydrazone solution was passed through a pre-oxidized manganese oxide (MnO2) supported micro-packed bed, generating diazo compounds as oxidized intermediates. Consecutively, these reactive species were quenched by aryl boronic acids, yielding the cross-coupling product in moderate to excellent yields (48–96%). The same procedure has been utilized for the cyclopropanation of flow-generated diazo compounds.290
OsO4 was immobilized in a PDMS microreactor by Kim et al.291 The microchannel was coated with a layer of polyvinylsilazane (PVSZ) polymer to increase its chemical resistance. Furthermore, the poly(4-vinylpyridine) (P4VP) polymer was attached to the PVSZ layer in a nanobrush-like manner. Finally, the repeating pyridine units on the P4VP polymer could be used for the immobilization of the OsO4 catalyst (Fig. 9). The microreactor was used for the room-temperature dihydroxylation and oxidative cleavage reaction of alkenes in the presence of NMI or NaIO4, respectively. All substrates could be converted quantitatively within 10 minutes of contact time, obtaining the respective products in good to excellent yields (67–95%).
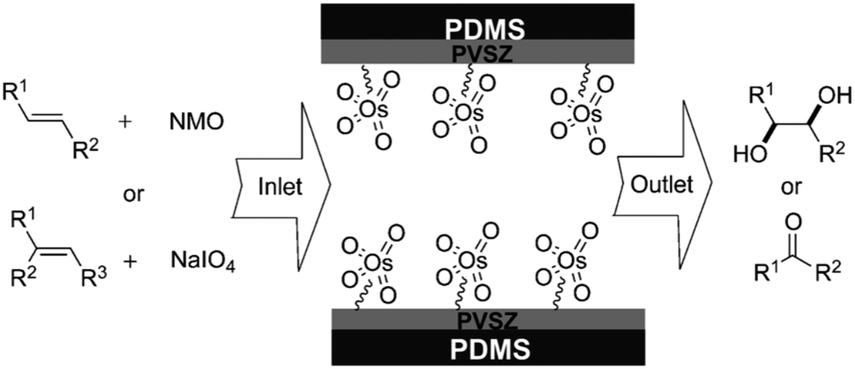 | ||
| Fig. 9 Continuous-flow dihydroxylation and oxidative cleavage of alkenes in an OsO4 immobilized nanobrush microreactor. Reproduced from ref. 291 with permission from John Wiley and Sons. | ||
Silica-supported Jones' reagents (CrO3/H2SO4) have been used in continuous-flow by Watts et al. for the oxidation of primary alcohols to their aldehydes or carboxylic acids, depending on the flow rates employed.292
6.2 Anaerobic enzymatic oxidation processes in continuous flow
Zelić et al. reported multiple papers on the enzymatic oxidation of n-hexanol to the green noted food additive, n-hexanal, in microreactors.293–295 Recently, they reported an alcohol dehydrogenase (ADH) catalyzed n-hexanol oxidation reaction in flow with an integrated NADH coenzyme regeneration system.296 The regeneration of NADH was conducted in a second microchip by the addition of acetaldehyde to the existing reaction stream. Finally, the enzyme and the coenzyme were reused immediately by the use of a recirculation loop to the first microreactor. The microfluidic setup could run up to three days without the addition of fresh NADH.The oxidation of phenols by the use of an immobilized biocatalyst in a microfluidic reactor was investigated by Parvulescu et al.297 Horseradish peroxidase (HRP) was deposited on a gold layered silicon wafer microreactor. Different layer thicknesses from the nano- to micro-scale were developed and the catalytic performance was measured. A maximum conversion of phenol (35%) was acquired. The immobilized enzyme remained active for multiple reaction cycles, proving that enzyme immobilized microchip devices can be utilized for potential biosensor applications.
In the field of biotechnology, a lot of efforts have been made to develop biochips capable of detecting low quantities of important chemical compounds (e.g., ethanol and glucose) with the emphasis on the applications in pharmaceutical, food, clinical and environmental analysis.298 Much research have been reported on the use of immobilized oxidase enzymes, such as horseradish peroxidase,299 alcohol oxidase,300 soybean peroxidase301 and glucose oxidase302 in microfluidic devices for flow injection analysis (FIA) purposes.
6.3 Swern-type oxidation processes in continuous flow
The Swern–Moffatt oxidation is known as a mild and selective method for the oxidation of primary and secondary alcohols to their corresponding aldehydes and ketones. The oxidation procedure is remarkable in terms of selectivity.303 However, in conventional set-ups, Swern–Moffatt oxidation reactions are conducted at low temperatures (<−50 °C) in order to suppress possible side reactions (e.g., Pummerer rearrangement).To make this transformation more appealing for industrial applications, Yoshida et al. reported the development of a room-temperature Swern-oxidation, enabled by the use of a micro-scale flow system.304 Three stainless steel tube microreactors were built in series (1 mm ID, 1 mL), each having a multilamination-type micromixer (40 μm channel width) at the inlet. In the first reactor, activation of DMSO was achieved by the addition of trifluoroacetic anhydride (TFAA) at room-temperature. It was found that by shortening the residence time up to 10 milliseconds in the first reactor, Pummerer rearrangement could be avoided. In the second reactor, the alcohol was added, forming the reactive intermediate with the activated DMSO. Finally, the reaction mixture was treated with triethylamine (Et3N) in a third microreactor, obtaining the corresponding carbonyl compound within 8.31 seconds of overall residence time. In the case of cyclohexanol, a 3 hour run resulted in a consistent yield of 91% cyclohexanone (Scheme 39).
 | ||
| Scheme 39 Room-temperature Swern oxidation of cyclohexanol in continuous flow.304 | ||
Other high-temperature continuous-flow Swern oxidations have been reported. McConnell et al. developed a semi-continuous process, with oxalyl chloride as the DMSO activator.305 The reaction took place in an in-line mixer (3.5 mL) with residence times of 0.1–1.4 seconds, before discharging the reaction mixture in a Et3N containing reactor vessel.
With the help of a continuous-flow microreactor setup, Kemperman et al. were capable of transforming testosterone into 4-androstene-3,17-dione (95% yield) in milliseconds.306 With the reactor running at 0 °C, the system was reliable for at least 1.5 hours of runtime, resulting in a production rate of 64 g h−1.
Rutjes et al. developed an automated microreactor platform for the optimization of the Swern oxidation procedure.103 Five parameters (temperature, stoichiometries of both TFAA and DMSO, substrate concentration and residence time) were screened systematically by the use of a 140 nL microreactor, generating large amounts of chemical information. Multidimensional contour plots were developed with the aid of the program MATLAB in order to find optimal reaction conditions. For the model reaction of benzyl alcohol to benzaldehyde, 70 °C (approximately 150 °C higher than conventional batch Swern oxidation reactions) was found to be the optimal temperature, yielding 96% of benzaldehyde within 32 milliseconds of residence time.
6.4 Electrochemical oxidation processes in continuous flow
Electrochemical microreactors have the advantage of providing a homogeneous electrical field, which avoids the formation of hot spots and, in some cases, the necessity to use electrolytes.307,308 Typically, more efficient reactions can be carried out in micro-flow due to the small diffusion distance to the electrodes.An interesting example of an electrochemical oxidation was given by Roth et al.309 They used a commercially available electrochemical flow reactor (Asia Flux) to facilitate electrochemical oxidations to prepare metabolites from commercial drug structures, thus mimicking the single electron transfer mechanism of cytochrome P450. Diclofenac, tolbutamide, primidone, albendazole and chlorpromazine were selected to demonstrate the potential of this device to enable different oxidation chemistries (i.e., aliphatic oxidation, aromatic hydroxylation, S oxidation, N oxidation and dehydrogenation). By using a continuous-flow protocol, the authors were able to prepare large quantities (10–100 mg) of metabolites, which are required for full characterization and toxicity studies (Scheme 40).
 | ||
| Scheme 40 Oxidation of chlorpromazine in a electrochemical reactor.309 | ||
Due to the fast mass transfer characteristics, microreactors are ideal for the preparation of unstable reactive intermediates and to use them in a subsequent trapping reaction. Atobe et al. have demonstrated this principle by preparing reactive ortho benzoquinones and subsequently consume them in a Michael addition with thiophenols.310,311 In a batch electrolytic cell, a maximum yield of 32% could be obtained. Black precipitation due to decomposition of ortho quinone was observed prior to addition of the thiol. However, in flow, the unstable ortho benzoquinone could be prepared rapidly and immediately quenched by 4-isopropyl-thiophenol providing the target compound in excellent yield (88% yield, Scheme 41).
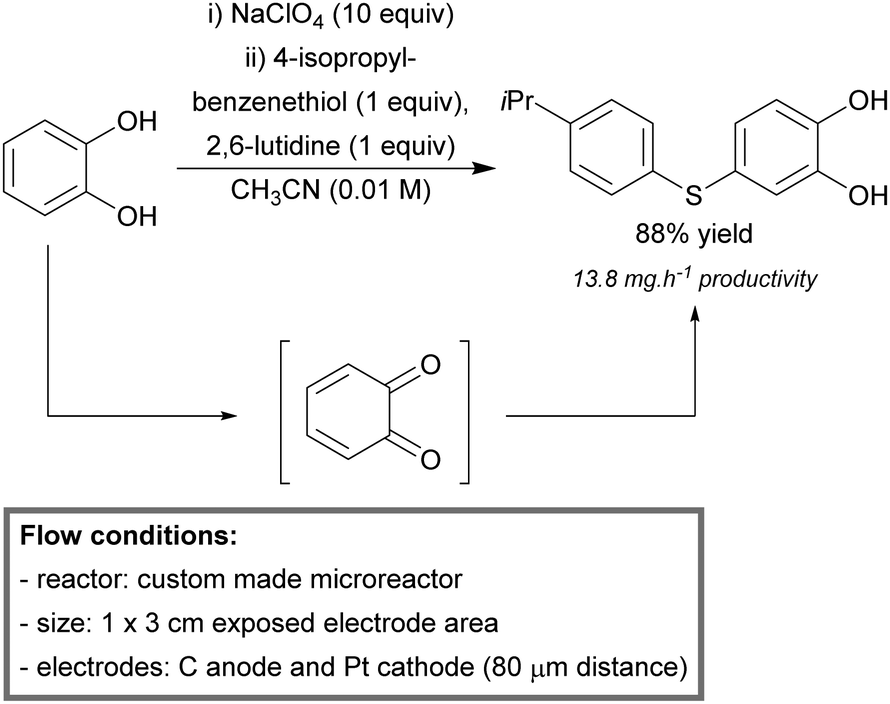 | ||
| Scheme 41 Oxidation of catechol in a electrochemical reactor to ortho benzoquinone and subsequent Michael addition.310,311 | ||
Similar setups, both commercially available and custom made ones, have been used by others to enable oxidations of furan,312 chlorophenols,313 and cyclic amines (Shono oxidation).314–316
6.5 TEMPO, HOX, hypervalent iodine and oxone mediated oxidation processes in continuous flow
Oxidation reactions which do not require transition metal catalysts are appealing since there is no need for removal and regeneration of precious metals.3172,2,6,6-Tetramethylpiperidine-1-oxyl (TEMPO) and other stable nitroxyl radicals are considered as well suited substitutes for metal oxide in oxidation reactions of primary and secondary alcohols. When used in catalytic amounts, the nitroxide radical (TEMPO) is readily oxidized into the highly electrophile oxoammonium species with the aid of a terminal oxidant (e.g., HOCl, BAIB, O2). It is this oxoammonium intermediate, which then serves as the active oxidizing agent in the alcohol oxidation reaction.
In order to overcome possible hot spot generation in the reaction mixture, Hampton et al. demonstrated the use of a spinning tube-in-tube microreactor (STT, 1.43 mL).318 The organic (TEMPO/tetrabutylammonium bromide/primary alcohol) and aqueous (sodium hypochlorite/sodium bicarbonate) solutions were merged and rapidly mixed inside the STT at rotor speeds of 4000–6000 RPM, preventing heat build-up. This demonstrates its heat transfer capacity, and the oxidation of benzyl alcohol was examined under neat conditions, with a high concentration of bleach (13% solution). Benzaldehyde could be isolated in excellent yields (91–95%) at a short residence time of 27 seconds, while no temperature increase was observed at the reactor output.
Another way of promoting excellent mixing between biphasic systems was demonstrated by McQuade et al.319 The TEMPO catalyst was immobilized on a commercially available resin (AMBERZINE® Oxirane) and was packed in polymer tubing (1.2 mL). A liquid–liquid slug flow was formed between the aqueous bleach phase and the organic alcohol phase before entering the packed bed. When passed through the catalytic bed, complete emulsification occurred, demonstrating the excellent mixing behaviour of the microreactor (Fig. 10). Within 5 minutes, a variety of substrates could be converted quantitatively, resulting in good to excellent yields (71–95%). Moreover, the TEMPO resins were completely recyclable (used for over 100 trials), showing no loss in activity.
 | ||
| Fig. 10 (A) The organic (colored solution) and aqueous phase (colorless solution) forming plugs at the Y-junction. (B) The phases mix upon reaching the packed bed, leading to a coalescence of drops at the outlet of the microchannel. Reproduced from ref. 319 with permission from Creative Commons. | ||
Further intensification was demonstrated by Hermans et al.320 A packed-bed microreactor was operated under three-phase conditions. The reactor was filled with silica immobilized TEMPO. Before entering the reactor, a liquid solution, containing the appropriate alcohol and catalytic amounts of HNO3, was merged with gaseous molecular oxygen. In this example molecular oxygen was used as the terminal oxidant in order to reactivate the TEMPO catalyst. This system proved to be much milder as compared to their previous reported research.321 The procedure was applicable for a broad substrate scope. Acid sensitive alcohols (e.g., prenol and isoprenol) could be smoothly oxidized to their corresponding carbonyls within two minutes of residence time. Furthermore, 5-hydroxymethylfurfural (HMF) could be selectively oxidized to its aldehyde or carboxylic acid depending on the residence time applied (2–6 minutes). To further extend the scope, the pharmaceutical intermediate, pyruvic acid, was synthesized successfully in high yield (96%, 15 seconds) from lactic acid, without decarboxylation (Scheme 42).
 | ||
| Scheme 42 TEMPO mediated synthesis of pyruvic acid from lactic acid in a packed-bed microreactor.320 | ||
The use of hypervalent iodine(III), the terminal oxidant in TEMPO catalyzed oxidations of alcohols in micro-flow, has been reported by Wirth et al.322
Kirschning et al. reported the use of a functionalized polymer with oxidizing bromate(I) anions in a packed-bed microreactor.323 The immobilized anions ensured the reactivation of the TEMPO catalyst during the oxidation of a steroid compound to its ketone.
In search of new catalytic systems for the oxidation of alcohols, Jamison et al. discovered that secondary alcohols could be readily oxidized in micro-flow, in the presence of only a stoichiometric amount of sodium hypochlorite (12.6% solution) and catalytic amounts of tetrabutylammonium bromide salt (TBAB), without the need for any additional supported catalyst.324 A liquid–liquid two-phase continuous-flow microfluidic setup (PFA tubing, 500 μm ID, 750 μL) was developed and optimized, and was capable of oxidizing secondary alcohols to ketones, aldehydes directly to their methyl ester and benzylic alcohols to aldehydes or methyl esters for over 60 substrates in total. Ethyl acetate could be used as the green organic solvent and 10 equivalents of methanol were used in the case of ester formation.
In the context of multi-step continuous-flow synthesis, Seeberger et al. developed the first non-iterative chemical assembly system.325 The system existed out of five different flow modules, each representing an essential transformation (e.g., oxidation, olefination, and hydrogenation), could be used interchangeably to give access to a variety of APIs. One module allowed the oxidation of primary and secondary alcohols in a biphasic manner, and the catalyst by TEMPO and bleach. With this proof-of-principle, the group was able to synthesize five APIs (i.e., Rolipram, Lyrica, Phenibut, Baclofen, and Gabapentin) in good overall yields (49–75%).
The HOF·MeCN system can be formed readily by introducing fluorine gas into a solution of wet acetonitrile, and the complex is known to be a uniquely effective electrophilic oxidizing agent.326 However, having a half-life of approximately four hours at room temperature, the use of HOF·MeCN in batch reactions is still underdeveloped. Since the complex should be produced ‘on demand’, Sandford et al. considered that the implementation of microreactors could offer an effective way of generating and consuming HOF·MeCN in situ.327 A microreactor setup was built for the epoxidation of alkenes and proved to be highly efficient. In a first microreactor, diluted fluorine gas (10% in N2) was introduced into a wet acetonitrile stream. At the outlet of the reactor, freshly generated HOF·MeCN was merged immediately with the alkene solution, producing the corresponding epoxide in moderate to excellent yields (38–99%). Cooling of the microreactor was unnecessary. The same microreactor setup was used for the oxidation of aliphatic amines into their corresponding nitro derivatives.328
Due to its stability, cost-effectiveness and non-toxic nature, the oxidizing agent, Oxone® (2KHSO5·KHSO4·K2SO4), shows potential as an effective substitute for conventional oxidants.329 However, known for its fire and explosion hazards, the large-scale use of Oxone® is considered impractical. Uozumi et al. demonstrated that these potentially hazardous conditions could be overcome by the use of a microfluidic system.330 The microreactor was composed of PTFE tubing (1 mm ID, 40 μL) and was used for the oxidative cyclization of alkenols. With a residence time of 5 minutes and the temperature set at 80 °C, the reaction proceeded smoothly to afford the corresponding cyclic ethers in high conversion (70–99%).
7. Scale up potential
Increasing the production scale of liquid phase oxidation processes is an important, though challenging and time-consuming task for the process engineer. Crucial in the design process is that the hydrodynamics and transport properties are maintained on each scale (from lab to pilot to production).331 Furthermore, the safety risk, associated with oxidation chemistry, further aggravates the complexity of the scale-up problem.121As discussed in detail in this review, microreactor technology can overcome many challenges associated with oxidation chemistry. However, scale-up of such devices is rather challenging as the throughput is mostly limited to small quantities. Several strategies to obtain larger quantities with continuous-flow reactors can be distinguished: (1) longer operation times + increasing the throughput by using higher flow rates; (2) numbering-up by placing several devices in parallel; and (3) smart scale out by a dimension enlarging strategy.
The use of longer operation times is probably the most used strategy on a laboratory scale as it is effective and simple to carry out. This strategy allows one to optimize the reaction parameters on small amounts of material and then directly scale up without re-optimization of the reaction protocol and the device itself. Typically, up to few hundreds of grams can be prepared in this fashion, which is more than sufficient for the different stages in a drug discovery process.332 Increasing the flow rate in microreactors is also an easy way to increase the throughput. Hereto, the length of the microreactor can be extended while the residence time is kept constant. Special care should be dedicated towards the variation of hydrodynamics and transport properties. Higher flow rates typically result in improved mixing efficiency and higher heat transfer rates, which can boost the reaction rate. It is important to note that many reactions have already been substantially accelerated in a microreactor due to the excellent transport properties, which allows one to increase the throughput in a “natural” way.
Numbering-up of individual microreactors has been communicated in the past as a straightforward way of scaling a microreactor process. However, this statement has been overruled recently as it became obvious that even flow distribution over the different channels is difficult.333 Especially, the distribution of multiphase flows, which are common in oxidation chemistry, is very hard to achieve. Schouten et al. have developed an efficient barrier-based distributor which allows one to achieve a homogeneous distribution within ±10% in eight different channels for gas–liquid flows (Fig. 11a).334,335 This is an example of internal numbering up which is achieved by using a single micro-structured device, with different microchannels in parallel, in combination with a process control system. Flow distributors are required to regulate and equalize the flow distribution over the different channels. Small differences in pressure drop can lead to large variations and thus to a different performance in each reaction channel. Examples of this principle in oxidation chemistry have been developed, such as the multichannel microreactors,336 the falling film microreactor,337 and the multichannel microcapillary films180 (Fig. 11b–d).
 | ||
| Fig. 11 Examples of internal numbering-up: (a) barrier-based microchannel reactor with a gas–liquid distributor unit.335 (b) Gas–liquid multichannel microreactor for ozonolysis, top: picture of the reactor; bottom: details of the reaction channels and the pressure drop channels.336 (c) Falling-film microreactor for ozonolysis.337 (d) FEP multichannel microcapillary for singlet oxygen chemistry.180 Reprinted with permission from ref. 180 and 335–337. Copyright of American Chemical Society. | ||
External numbering-up is achieved by placing different microreactors and their individual process control units in parallel.338 This is a reliable strategy which ensures equal process conditions in each microreactor. In the case of failure of an individual device, other devices can operate autonomously. However, external numbering-up is very expensive as each microreactor requires its own process control (e.g., pumps and mass flow controllers).
Another strategy for the scale-up of oxidation chemistry involves increasing the characteristic dimensions. Such milli-scale reactors can be designed to have similar heat and mass transfer characteristics to microreactors by incorporating active mixers (smart scale out). Lonza has developed the FlowPlate™ concept, which allows one to scale flow chemistry from lab scale to pilot plant production (Fig. 12).339–343 The reactor consists of stainless steel plates containing tangential mixing elements. The plates are stacked between thermal conductive aluminium plates to ensure high heat transfer. Higher throughput is obtained by increasing the hydraulic diameter whilst keeping the heat and mass transfer characteristics constant. The lab plate is used as a development tool and produces a few grams, while A6, A5 to A4 plates are used to subsequently increase the reaction scale to up to 2500 kg for the A4 plate.
 | ||
| Fig. 12 The Lonza FlowPlate™ concept for a step-wise scale-up of continuous-flow reactions: (a) a single reactor plate with tangential mixing elements. (b) Laboratory scale reactor with a transparent view glass for optical inspection. (c) Lonza's plate stack reactors for step-wise scale-up. Reproduced from ref. 339 with permission from Elsevier. | ||
A similar concept has been developed by Corning and uses glass flow reactors with heart-shaped split-and-recombine mixers.88,343,344 Jensen et al. have used the Corning Low Flow (0.45 mL) and the Advanced Flow Reactor (8.9 mL volume) to enable scalability of the oxidation of alcohols and aldehydes using bleach (Fig. 13).345 Compared to a silicon microreactor chip (0.25 mL volume, 0.0064 g min−1), the productivity of the preparation acetophenone could be gradually increased in the Low Flow Reactor (0.37 g min−1) and the Advanced Flow Reactor (4.08 g min−1) by a factor of 58 and 640, respectively.
 | ||
| Fig. 13 Scalability of the oxidation of alcohols using bleach: (a) the silicon microreactor chip, (b) Corning low flow reactor plates, and (c) Corning advanced flow reactor plates. Reprinted with permission from ref. 345. Copyright (2014) American Chemical Society. | ||
8. Conclusions and outlook
The use of continuous-flow microreactor technology, as outlined in this review, has many benefits for liquid phase oxidation chemistry, including high heat and mass transfer rates, safe handling of explosive reaction mixtures, and potential to scale up efficiently. In the last decade, we have witnessed a remarkable expansion in the use of this technology for liquid phase oxidation chemistry. Commercial available flow devices exist for both lab and production scale making the technology broadly available. In addition, more and more chemists get acquainted with the use of flow chemistry in their own laboratories. Given these trends, we anticipate that more researchers will continue to be attracted by these advantages.However, progress in this field is still highly desired and will require a combined effort of chemists, engineers and materials scientists. Only now, due to the improved safety characteristics, researchers dare to use pure oxygen gas or other hazardous reagents in organic synthetic chemistry. In other words, we have only seen the tip of the iceberg and still many different opportunities lay ahead of us. An example of this is the preparation of short living species (e.g., peroxides), which can be subsequently used in a follow-up reaction. To exploit the full potential of these reagents, fundamentals of multiphase transport phenomena are required and need to be further developed in combination with the chemistry.
To make progress in continuous-flow oxidation chemistry, full support from the industry is crucial. Interest from the industry is fuelled by the increased safety, the higher reaction selectivity and the overall efficiency of the oxidation protocol. Such collaborative efforts are not only important from a funding perspective but industrial applications will facilitate the widespread use of this technology, reduce the overall cost, and stimulate innovations in real life examples.346,347 Examples which still require substantial collaborative research efforts are, e.g., (i) controlled scalability from lab to full production scale; (ii) efficient recycling of metal catalysts; (iii) safe use of non-diluted oxygen sources and their impact on the reaction kinetics.317
Acknowledgements
Financial support is provided by the Dutch Science Foundation (NWO) via an ECHO grant (Grant No. 713.013.001) and a VENI grant for T.N. (Grant No. 12464). We also acknowledge the European Union for a Marie Curie CIG grant for T.N. (Grant No. 333659), for a Marie Curie Intra-European Fellowship for Y.S. (Grant No. 622415), and an ERC Advanced Grant for V.H. (Grant No. 267443). T.N. and R.L. would like to thank generous funding from the European Union within the Photo4Future Innovative Training Network (Grant No. 641861). M.S. thanks Chinese Scholarship Council for financial support.Notes and references
- M. G. Clerici and O. A. Kholdeeva, Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications, John Wiley & Sons, Hoboken, 2013 Search PubMed.
- J. C. J. Bart and S. Cavallaro, Ind. Eng. Chem. Res., 2015, 54, 1–46 CrossRef CAS.
- J. C. J. Bart and S. Cavallaro, Ind. Eng. Chem. Res., 2015, 54, 567–576 CrossRef CAS.
- F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508–534 CrossRef CAS PubMed.
- Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- L. Boisvert and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 899–910 CrossRef CAS PubMed.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797–9804 CrossRef CAS PubMed.
- B. L. Ryland, S. D. McCann, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 12166–12173 CrossRef CAS PubMed.
- M. L. Scheuermann and K. I. Goldberg, Chem. – Eur. J., 2014, 20, 14556–14568 CrossRef CAS PubMed.
- M. N. Kashid and L. Kiwi-Minsker, Ind. Eng. Chem. Res., 2009, 48, 6465–6485 CrossRef CAS.
- M. P. Duduković, F. Larachi and P. L. Mills, Catal. Rev., 2002, 44, 123–246 CrossRef.
- M. N. Kashid, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2011, 66, 3876–3897 CrossRef CAS.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed.
- I. R. Baxendale, R. D. Braatz, A. J. Florence, B. K. Hodnett, K. F. Jensen, M. D. Johnson, P. Sharratt and J. Sherlock, J. Pharm. Sci., 2015, 104, 781–791 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- F. G. Finelli, L. S. M. Miranda and R. O. M. A. de Souza, Chem. Commun., 2015, 51, 3708–3722 RSC.
- I. M. Mándity, S. B. Ötvös and F. Fülöp, ChemistryOpen, 2015, 4, 212–223 CrossRef PubMed.
- R. Munirathinam, J. Huskens and W. Verboom, Adv. Synth. Catal., 2015, 357, 1093–1123 CrossRef CAS.
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC.
- L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC.
- K. F. Jensen, B. J. Reizman and S. G. Newman, Lab Chip, 2014, 14, 3206–3212 RSC.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 5750–5758 CrossRef CAS PubMed.
- V. Hessel, H. Lowe, A. Muller and G. Kolb, Chemical Micro Process Engineering: Processing and Plants, Wiley-VCH, Weinheim, 2005 Search PubMed.
- V. Hessel, A. Renken, J. C. Schouten and J. Yoshida, Micro Process Engineering: A Comprehensive Handbook, Wiley-VCH, Weinheim, 2009 Search PubMed.
- C. Wiles and P. Watts, Micro Reaction Technology in Organic Synthesis, CRC Press, Boca Raton, FL, 2011 Search PubMed.
- T. Wirth, Microreactors in Organic Chemistry and Catalysis, Wiley-VCH, Weinheim, 2013 Search PubMed.
- J.-I. Yoshida, Basics of Flow Microreactors Synthesis, Springer, Japan, Kyoto, 2015 Search PubMed.
- A. Pohar and I. Plazl, Chem. Biochem. Eng. Q., 2009, 23, 537–544 CAS.
- K. Boodhoo and A. Harvey, Process Intensification Technologies for Green Chemistry: Engineering Solutions for Sustainable Chemical Processing, John Wiley & Sons, Chichester, 2013 Search PubMed.
- A. Gorak and A. I. Stankiewicz, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 431–451 CrossRef CAS PubMed.
- V. Kumar and K. D. P. Nigam, Green Process. Synth., 2012, 1, 79–107 CAS.
- G. Kolb, Chem. Eng. Process. Process Intensif., 2013, 65, 1–44 CrossRef CAS.
- G. Kolb, J. Schürer, D. Tiemann, M. Wichert, R. Zapf, V. Hessel and H. Löwe, J. Power Sources, 2007, 171, 198–204 CrossRef CAS.
- G. Kolb, V. Cominos, C. Hofmann, H. Pennemann, J. Schürer, D. Tiemann, M. Wichert, R. Zapf, V. Hessel and H. Löwe, Chem. Eng. Res. Des., 2005, 83, 626–633 CrossRef CAS.
- T. Noël and V. Hessel, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2014, pp. 1–35, DOI:10.1002/14356007.o16_o02.
- Z. Hou, N. Theyssen, A. Brinkmann and W. Leitner, Angew. Chem., Int. Ed., 2005, 44, 1346–1349 CrossRef CAS PubMed.
- E. Garcia-Verdugo, J. Fraga-Dubreuil, P. A. Hamley, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2005, 7, 294–300 RSC.
- E. Garcia-Verdugo, E. Venardou, W. B. Thomas, K. Whiston, W. Partenheimer, P. A. Hamley and M. Poliakoff, Adv. Synth. Catal., 2004, 346, 307–316 CrossRef CAS.
- J. Fraga-Dubreuil, E. Garcia-Verdugo, P. A. Hamley, E. M. Vaquero, L. M. Dudd, I. Pearson, D. Housley, W. Partenheimer, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2007, 9, 1238–1245 RSC.
- R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322–5325 CrossRef CAS PubMed.
- A. O. Chapman, G. R. Akien, N. J. Arrowsmith, P. Licence and M. Poliakoff, Green Chem., 2010, 12, 310–315 RSC.
- X. Han, R. A. Bourne, M. Poliakoff and M. W. George, Chem. Sci., 2011, 2, 1059–1067 RSC.
- J. F. B. Hall, R. A. Bourne, X. Han, J. H. Earley, M. Poliakoff and M. W. George, Green Chem., 2013, 15, 177–180 RSC.
- U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. Da Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1–17 CrossRef CAS.
- J. H. J. Kluytmans, A. P. Markusse, B. F. M. Kuster, G. B. Marin and J. C. Schouten, Catal. Today, 2000, 57, 143–155 CrossRef CAS.
- P. L. Mills and R. V. Chaudhari, Catal. Today, 1999, 48, 17–29 CrossRef CAS.
- G. Langhendries, D. E. De Vos, B. F. Sels, I. Vankelecom, P. A. Jacobs and G. V. Baron, Clean Technol. Environ. Policy, 1998, 1, 21–29 CrossRef.
- P. L. Mills and R. V. Chaudhari, Catal. Today, 1997, 37, 367–404 CrossRef CAS.
- H. Yin, C. Sheng, Z. Chen, S. Yuan, H. Li and J. Mei, Chem. Eng. Technol., 2014, 37, 1797–1804 CrossRef CAS.
- F. Wang, J. Xu, X. Li, J. Gao, L. Zhou and R. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987–1992 CrossRef CAS.
- H. Ge, G. W. Chen, Q. Yuan and H. Q. Li, Catal. Today, 2005, 110, 171–178 CrossRef CAS.
- V. Tukač, J. Vokál and J. Hanika, J. Chem. Technol. Biotechnol., 2001, 76, 506–510 CrossRef.
- R. Pohorecki, J. Bałdyga, W. Moniuk, A. Krzysztoforski and Z. Wójcik, Chem. Eng. Sci., 1992, 47, 2559–2564 CrossRef CAS.
- T. Chou and F. Lin, Can. J. Chem., 1983, 61, 1295–1300 CrossRef CAS.
- H. Nakajima, Mass Transfer – Advanced Aspects, InTech, 2011 Search PubMed.
- G. R. Astbury, Org. Process Res. Dev., 2002, 6, 893–895 CrossRef CAS.
- J.-R. Chen, Process Saf. Prog., 2004, 23, 72–81 CrossRef CAS.
- T. A. Kletz, J. Hazard. Mater., 1975, 1, 165–170 CrossRef.
- V. Tukač, J. Hanika and V. A. Chyba, Catal. Today, 2003, 79–80, 427–431 CrossRef.
- A. Pintar, G. Berčič and J. Levec, Chem. Eng. Sci., 1997, 52, 4143–4153 CrossRef CAS.
- V. Hessel, P. Angeli, A. Gavriilidis and H. Löwe, Ind. Eng. Chem. Res., 2005, 44, 9750–9769 CrossRef CAS.
- Y. Su, Y. Zhao, G. Chen and Q. Yuan, Chem. Eng. Sci., 2010, 65, 3947–3956 CrossRef CAS.
- Y. Zhao, G. Chen and Q. Yuan, AIChE J., 2007, 53, 3042–3053 CrossRef CAS.
- A. L. Dessimoz, L. Cavin, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2008, 63, 4035–4044 CrossRef CAS.
- J. H. Xu, J. Tan, S. W. Li and G. S. Luo, Chem. Eng. J., 2008, 141, 242–249 CrossRef CAS.
- C. Yao, Z. Dong, Y. Zhao and G. Chen, Chem. Eng. Sci., 2014, 112, 15–24 CrossRef CAS.
- K. S. Elvira, X. Casadevall i Solvas, R. C. R. Wootton and A. J. de Mello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 CrossRef CAS PubMed.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS PubMed.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- Y. Su, G. Chen, Y. Zhao and Q. Yuan, AIChE J., 2009, 55, 1948–1958 CrossRef CAS.
- J. Yue, L. Luo, Y. Gonthier, G. Chen and Q. Yuan, Chem. Eng. Sci., 2009, 64, 3697–3708 CrossRef CAS.
- T. Cubaud, M. Sauzade and R. Sun, Biomicrofluidics, 2012, 6 DOI:10.1063/1.3693591.
- A. Leclerc, M. Alamé, D. Schweich, P. Pouteau, C. Delattre and C. de Bellefon, Lab Chip, 2008, 8, 814–817 RSC.
- S. Waelchli and P. Rudolf von Rohr, Int. J. Multiphase Flow, 2006, 32, 791–806 CrossRef CAS.
- K. A. Triplett, S. M. Ghiaasiaan, S. I. Abdel-Khalik and D. L. Sadowski, Int. J. Multiphase Flow, 1999, 25, 377–394 CrossRef CAS.
- Y. Zhao, G. Chen, C. Ye and Q. Yuan, Chem. Eng. Sci., 2013, 87, 122–132 CrossRef CAS.
- J. Yue, L. Luo, Y. Gonthier, G. Chen and Q. Yuan, Chem. Eng. Sci., 2008, 63, 4189–4202 CrossRef CAS.
- C. X. Zhao and A. P. J. Middelberg, Chem. Eng. Sci., 2011, 66, 1394–1411 CrossRef CAS.
- A. Woitalka, S. Kuhn and K. F. Jensen, Chem. Eng. Sci., 2014, 116, 1–8 CrossRef CAS.
- G. Berčič and A. Pintar, Chem. Eng. Sci., 1997, 52, 3709–3719 CrossRef.
- J. M. van Baten and R. Krishna, Chem. Eng. Sci., 2004, 59, 2535–2545 CrossRef CAS.
- P. Sobieszuk, R. Pohorecki, P. Cygański and J. Grzelka, Chem. Eng. Sci., 2011, 66, 6048–6056 CrossRef CAS.
- M. T. Kreutzer, F. Kapteijn, J. A. Moulijn and J. J. Heiszwolf, Chem. Eng. Sci., 2005, 60, 5895–5916 CrossRef CAS.
- C. Yao, Z. Dong, Y. Zhao and G. Chen, Chem. Eng. Sci., 2015, 123, 137–145 CrossRef CAS.
- W. E. Carroll, J. Organomet. Chem., 1981, 205, 14–15 CrossRef.
- J. K. Kochi, Organometallic Mechanisms and Catalysis, Academic Press, New York, 1978 Search PubMed.
- C. Masters, Homogeneous Transition-metal Catalysis: A Gentle Art, Springer, London, New York, 1981 Search PubMed.
- P. M. Henry, Palladium Catalyzed Oxidation of Hydrocarbons, Springer, 1980, vol. 2 Search PubMed.
- L. Kucka, J. Richter, E. Y. Kenig and A. Górak, Sep. Purif. Technol., 2003, 31, 163–175 CrossRef CAS.
- J. Fischer, T. Lange, R. Boehling, a. Rehfinger and E. Klemm, Chem. Eng. Sci., 2010, 65, 4866–4872 CrossRef CAS.
- Y. Su, V. Hessel and T. Noël, AIChE J., 2015, 61, 2215–2227 CrossRef CAS.
- A. K. Suresh, M. M. Sharma and T. Sridhar, Ind. Eng. Chem. Res., 2000, 39, 3958–3997 CrossRef CAS.
- J. I. Yoshida, A. Nagaki and T. Yamada, Chem. – Eur. J., 2008, 14, 7450–7459 CrossRef CAS PubMed.
- P. J. Nieuwland, K. Koch, N. Van Harskamp, R. Wehrens, J. C. M. Van Hest and F. P. J. T. Rutjes, Chem. – Asian J., 2010, 5, 799–805 CrossRef CAS PubMed.
- T. Schwalbe, V. Autze, M. Hohmann and W. Stirner, Org. Process Res. Dev., 2004, 8, 440–454 CrossRef CAS.
- V. Hessel and T. Noël, Ullmanns Encycl. Ind. Chem., Wiley-VCH, 2012, pp. 2–54, DOI:10.1002/14356007.b16_b37.pub2.
- R. Dey, T. Das and S. Chakraborty, Heat Transfer Eng., 2012, 33, 425–446 CrossRef CAS.
- P. Rosa, T. G. Karayiannis and M. W. Collins, Appl. Therm. Eng., 2009, 29, 3447–3468 CrossRef CAS.
- N. Kockmann, M. Gottsponer, B. Zimmermann and D. M. Roberge, Chem. – Eur. J., 2008, 14, 7470–7477 CrossRef CAS PubMed.
- T. Illg, V. Hessel, P. Löb and J. C. Schouten, Green Chem., 2012, 14, 1420–1433 RSC.
- J. Haber, M. N. Kashid, A. Renken and L. Kiwi-Minsker, Ind. Eng. Chem. Res., 2012, 51, 1474–1489 CrossRef CAS.
- F. Incropera, A. Lavine and D. DeWitt, Fundamentals of heat and mass transfer, John Wiley & Sons, Hoboken, 2011 Search PubMed.
- W. M. Deen, Analysis of Transport Phenomena, Oxford University Press, Oxford, 1998 Search PubMed.
- A. Odedra and P. H. Seeberger, Angew. Chem., Int. Ed., 2009, 48, 2699–2702 CrossRef CAS PubMed.
- A. C. Hordijk and J. J. De Groot, Thermochim. Acta, 1986, 101, 45–63 CrossRef CAS.
- C. W. Jones, Applications of Hydrogen Peroxide and Derrivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed.
- T. Illg, V. Hessel, P. Löb and J. C. Schouten, ChemSusChem, 2011, 4, 392–398 CrossRef CAS PubMed.
- M. T. Musser, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012, pp. 49–58 Search PubMed.
- M. L. Campbell, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012, pp. 134–182 Search PubMed.
- J. R. Chen, H. H. Yang and C. H. Wu, Org. Process Res. Dev., 2004, 8, 252–255 CrossRef CAS.
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015 DOI:10.1021/op500328f.
- R. A. Baker, W. E. Cox, P. A. Westine, P. S. Kulesz and J. J. Strehlow, Explosion Hazards and Evaluation, Elsevier Scientific Publishing Company, Amsterdam, Oxford, New York, 1983 Search PubMed.
- R. A. Strehlow, AIAA J., 1985, 23, 1139 CrossRef.
- M. Gödde, C. Liebner and H. Hieronymus, Chem. Ing. Tech., 2009, 81, 73–78 CrossRef.
- C. Liebner, J. Fischer, S. Heinrich, T. Lange, H. Hieronymus and E. Klemm, Process Saf. Environ. Prot., 2012, 90, 77–82 CrossRef CAS.
- S. Heinrich, F. Edeling, C. Liebner, H. Hieronymus, T. Lange and E. Klemm, Chem. Eng. Sci., 2012, 84, 540–543 CrossRef CAS.
- N. B. Siccama and K. R. Westerterp, Ind. Eng. Chem. Res., 1993, 32, 1304–1314 CrossRef CAS.
- N. B. Siccama and K. R. Westerterp, Ind. Eng. Chem. Res., 1995, 34, 1755–1768 CrossRef CAS.
- J. W. Bolk and K. R. Westerterp, AIChE J., 1999, 45, 124–144 CrossRef CAS.
- J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42 CrossRef CAS PubMed.
- J. Alagy, P. Trambouze and H. Van Landeghem, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 317–323 Search PubMed.
- J. Hao, H. Cheng, H. Wang, S. Cai and F. Zhao, J. Mol. Catal. A: Chem., 2007, 271, 42–45 CrossRef CAS.
- N. Theyssen, Z. Hou and W. Leitner, Chem. – Eur. J., 2006, 12, 3401–3409 CrossRef CAS PubMed.
- G. Veser, Chem. Eng. Sci., 2001, 56, 1265–1273 CrossRef CAS.
- R. Jevtic, P. A. Ramachandran and M. P. Dudukovic, Chem. Eng. Res. Des., 2010, 88, 255–262 CrossRef CAS.
- X. Liu and K. F. Jensen, Green Chem., 2013, 15, 1538–1541 RSC.
- U. Neuenschwander and K. F. Jensen, Ind. Eng. Chem. Res., 2014, 53, 601–608 CrossRef CAS.
- S. Kuhn, R. L. Hartman, M. Sultana, K. D. Nagy, S. Marre and K. F. Jensen, Langmuir, 2011, 27, 6519–6527 CrossRef CAS PubMed.
- D. V. Bavykin, A. A. Lapkin, S. T. Kolaczkowski and P. K. Plucinski, Appl. Catal., A, 2005, 288, 175–184 CrossRef CAS.
- P. K. Plucinski, D. V. Bavykin, S. T. Kolaczkowski and A. A. Lapkin, Ind. Eng. Chem. Res., 2005, 44, 9683–9690 CrossRef CAS.
- P. K. Plucinski, D. V. Bavykin, S. T. Kolaczkowski and A. A. Lapkin, Catal. Today, 2005, 105, 479–483 CrossRef CAS.
- E. Cao, W. B. Motherwell and A. Gavriilidis, Chem. Eng. Technol., 2006, 29, 1372–1375 CrossRef CAS.
- E. Cao, M. Sankar, S. Firth, K. F. Lam, D. Bethell, D. K. Knight, G. J. Hutchings, P. F. McMillan and A. Gavriilidis, Chem. Eng. J., 2011, 167, 734–743 CrossRef CAS.
- D. Obermayer, A. M. Balu, A. A. Romero, W. Goessler, R. Luque and C. O. Kappe, Green Chem., 2013, 15, 1530–1537 RSC.
- G. Wu, A. Constantinou, E. Cao, S. Kuhn, M. Morad, M. Sankar, D. Bethell, G. J. Hutchings and A. Gavriilidis, Ind. Eng. Chem. Res., 2015, 54, 4183–4189 CrossRef CAS.
- E. Cao, M. Sankar, E. Nowicka, Q. He, M. Morad, P. J. Miedziak, S. H. Taylor, D. W. Knight, D. Bethell, C. J. Kiely, A. Gavriilidis and G. J. Hutchings, Catal. Today, 2013, 203, 146–152 CrossRef CAS.
- M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed.
- T. Noël and V. Hessel, ChemSusChem, 2013, 6, 405–407 CrossRef PubMed.
- A. L. Tarasov, L. M. Kustov, A. A. Bogolyubov, A. S. Kiselyov and V. V. Semenov, Appl. Catal., A, 2009, 366, 227–231 CrossRef CAS.
- N. Wang, T. Matsumoto, M. Ueno, H. Miyamura and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 4744–4746 CrossRef CAS PubMed.
- K. Kaizuka, K.-Y. Lee, H. Miyamura and S. Kobayashi, J. Flow Chem., 2012, 2, 1–4 CrossRef CAS.
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157–2163 RSC.
- D. S. Mannel, S. S. Stahl and T. W. Root, Org. Process Res. Dev., 2014, 18, 1503–1508 CrossRef CAS PubMed.
- N. Asao, N. Hatakeyama, Menggenbateer, T. Minato, E. Ito, M. Hara, Y. Kim, Y. Yamamoto, M. Chen, W. Zhang and A. Inoue, Chem. Commun., 2012, 48, 4540–4542 RSC.
- V. Hessel, B. Cortese and M. H. J. M. de Croon, Chem. Eng. Sci., 2011, 66, 1426–1448 CrossRef CAS.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- S. R. Chaudhuri, J. Hartwig, L. Kupracz, T. Kodanek, J. Wegner and A. Kirschning, Adv. Synth. Catal., 2014, 356, 3530–3538 CrossRef CAS.
- T. Osako, K. Torii and Y. Uozumi, RSC Adv., 2015, 5, 2647–2654 RSC.
- J. H. Holland, Adaptation on Natural and Artificial Systems, MIT Press, Cambridge, MA, 1992 Search PubMed.
- R. Leardi, J. Chemom., 2001, 15, 559–569 CrossRef CAS.
- R. Leardi, J. Chromatogr. A, 2007, 1158, 226–233 CrossRef CAS PubMed.
- Y. Chu, W. Heyndrickx, G. Occhipinti, V. R. Jensen and B. K. Alsberg, J. Am. Chem. Soc., 2012, 134, 8885–8895 CrossRef CAS PubMed.
- J. E. Kreutz, A. Shukhaev, W. Du, S. Druskin, O. Daugulis and R. F. Ismagilov, J. Am. Chem. Soc., 2010, 132, 3128–3132 CrossRef CAS PubMed.
- L. Vanoye, M. Pablos, N. Smith, C. de Bellefon and A. Favre-Réguillon, RSC Adv., 2014, 4, 57159–57163 RSC.
- B. Pieber and C. O. Kappe, Green Chem., 2013, 15, 320–324 RSC.
- J. De Houwer, K. Abbaspour Tehrani and B. U. W. Maes, Angew. Chem., Int. Ed., 2012, 51, 2745–2748 CrossRef CAS PubMed.
- S. L. Bourne and S. V. Ley, Adv. Synth. Catal., 2013, 355, 1905–1910 CrossRef CAS.
- B. Gutmann, P. Elsner, D. Roberge and C. O. Kappe, ACS Catal., 2013, 3, 2669–2676 CrossRef CAS.
- B. Saha and J. H. Espenson, J. Mol. Catal. A: Chem., 2007, 271, 1–5 CrossRef CAS.
- W. Partenheimer, Adv. Synth. Catal., 2004, 346, 297–306 CrossRef CAS.
- J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247–1251 CrossRef CAS.
- X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180–1186 RSC.
- L. Vanoye, M. Pablos, C. de Bellefon and A. Favre-Réguillon, Adv. Synth. Catal., 2015, 357, 739–746 CrossRef CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- T. Noël, X. Wang and V. Hessel, Chim. Oggi, 2013, 31, 10–14 Search PubMed.
- Z. J. Garlets, J. D. Nguyen and C. R. J. Stephenson, Isr. J. Chem., 2014, 54, 351–360 CrossRef CAS PubMed.
- M. Oelgemoeller, Chem. Eng. Technol., 2012, 35, 1144–1152 CrossRef.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- M. Oelgemöller and O. Shvydkiv, Molecules, 2011, 16, 7522–7550 CrossRef PubMed.
- R. C. R. Wootton, R. Fortt and A. J. De Mello, Org. Process Res. Dev., 2002, 6, 187–189 CrossRef CAS.
- K. S. Elvira, R. C. R. Wootton, N. M. Reis, M. R. Mackley and A. J. deMello, ACS Sustainable Chem. Eng., 2013, 1, 209–213 CrossRef CAS.
- S. Meyer, D. Tietze, S. Rau, B. Schäfer and G. Kreisel, J. Photochem. Photobiol., A, 2007, 186, 248–253 CrossRef CAS.
- C. P. Park, R. A. Maurya, J. H. Lee and D.-P. Kim, Lab Chip, 2011, 11, 1941–1945 RSC.
- R. A. Maurya, C. P. Park and D. P. Kim, Beilstein J. Org. Chem., 2011, 7, 1158–1163 CrossRef CAS PubMed.
- K. Loponov, J. Lopes, M. Barłóg, E. V. Astrova, A. V. Malkov and A. Lapkin, Org. Process Res. Dev., 2014, 18, 1443–1454 CrossRef CAS.
- C. Y. Park, Y. J. Kim, H. J. Lim, J. H. Park, M. J. Kim, S. W. Seo and C. P. Park, RSC Adv., 2015, 5, 4233–4237 RSC.
- C. Y. Park, J. H. Park, H. J. Lim, G.-S. Hwang and C. P. Park, Bull. Korean Chem. Soc., 2014, 35, 983–984 CrossRef CAS.
- F. Lévesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRef PubMed.
- T. Etrych, M. Šírovaé, L. Starovoytova, B. Řiéhovaé and K. Ulbrich, Mol. Pharmaceutics, 2010, 7, 1015–1026 CrossRef CAS PubMed.
- M. D. Altman, A. Ali, G. S. K. K. Reddy, M. N. L. Nalam, S. G. Anjum, H. Cao, S. Chellappan, V. Kairys, M. X. Fernandes, M. K. Gilson, C. A. Schiffer, T. M. Rana and B. Tidor, J. Am. Chem. Soc., 2008, 130, 6099–6113 CrossRef CAS PubMed.
- F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706–1709 CrossRef PubMed.
- D. Kopetzki, F. Lévesque and P. H. Seeberger, Chem. – Eur. J., 2013, 19, 5450–5456 CrossRef CAS PubMed.
- D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557–561 CrossRef CAS PubMed.
- S. Vukelić, D. B. Ushakov, K. Gilmore, B. Koksch and P. H. Seeberger, Eur. J. Org. Chem., 2015, 3036–3039 CrossRef.
- D. B. Ushakov, M. B. Plutschack, K. Gilmore and P. H. Seeberger, Chem. – Eur. J., 2015, 21, 6528–6534 CrossRef CAS PubMed.
- Y. Nagasawa, K. Tanba, N. Tada, E. Yamaguchi and A. Itoh, Synlett, 2015, 412–415 CAS.
- A. Talla, B. Driessen, N. J. W. Straathof, L.-G. Milroy, L. Brunsveld, V. Hessel and T. Noël, Adv. Synth. Catal., 2015, 357, 2180–2186 CrossRef CAS.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- C. Richard, A. M. Martre and P. Boule, J. Photochem. Photobiol., A, 1992, 66, 225–234 CrossRef CAS.
- T. S. A. Heugebaert, C. V. Stevens and C. O. Kappe, ChemSusChem, 2015, 8, 1648–1651 CrossRef CAS PubMed.
- H. C. Aran, D. Salamon, T. Rijnaarts, G. Mul, M. Wessling and R. G. H. Lammertink, J. Photochem. Photobiol., A, 2011, 225, 36–41 CrossRef CAS.
- T. Carofiglio, P. Donnola, M. Maggini, M. Rossetto and E. Rossi, Adv. Synth. Catal., 2008, 350, 2815–2822 CrossRef CAS.
- E. K. Lumley, C. E. Dyer, N. Pamme and R. W. Boyle, Org. Lett., 2012, 14, 5724–5727 CrossRef CAS PubMed.
- R. Gorges, S. Meyer and G. Kreisel, J. Photochem. Photobiol., A, 2004, 167, 95–99 CrossRef CAS.
- H. Lindstrom, R. Wootton and A. Iles, AIChE J., 2007, 53, 695–702 CrossRef CAS.
- A. A. Lapkin, V. M. Boddu, G. N. Aliev, B. Goller, S. Polisski and D. Kovalev, Chem. Eng. J., 2008, 136, 331–336 CrossRef CAS.
- S. Lacombe, J. P. Soumillion, A. El Kadib, T. Pigot, S. Blanc, R. Brown, E. Oliveros, C. Cantau and P. Saint-Cricq, Langmuir, 2009, 25, 11168–11179 CrossRef CAS PubMed.
- S. Hejda, M. Drhova, J. Kristal, D. Buzek, P. Krystynik and P. Kluson, Chem. Eng. J., 2014, 255, 178–184 CrossRef CAS.
- J. Růžička and E. H. Hansen, Anal. Chim. Acta, 1986, 179, 1–58 CrossRef.
- I. Dencic, J. Meuldijk, M. Croon and V. Hessel, J. Flow Chem., 2011, 1, 13–23 CrossRef.
- I. Denčić, T. Noël, J. Meuldijk, M. De Croon and V. Hessel, Eng. Life Sci., 2013, 13, 326–343 CrossRef.
- V. Hessel, J. Tibhe, T. Noël and Q. Wang, Chem. Biochem. Eng. Q., 2014, 28, 167–188 CrossRef CAS.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS PubMed.
- T. B. Goriushkina, A. P. Soldatkin and S. V. Dzyadevych, J. Agric. Food Chem., 2009, 57, 6528–6535 CrossRef CAS PubMed.
- G. A. M. Mersal and U. Bilitewski, Electrophoresis, 2005, 26, 2303–2312 CrossRef CAS PubMed.
- J. Wang, M. P. Chatrathi and G. E. Collins, Anal. Chim. Acta, 2007, 585, 11–16 CrossRef CAS PubMed.
- S. Rissom, U. Schwarz-Linek, M. Vogel, V. I. Tishkov and U. Kragl, Tetrahedron: Asymmetry, 1997, 8, 2523–2526 CrossRef CAS.
- U. Schwarz-Linek, A. Krödel, F.-A. Ludwig, A. Schulze, S. Rissom, U. Kragl, V. I. Tishkov and M. Vogel, Synthesis, 2001, 947–951 CrossRef CAS.
- M. P. C. Marques, P. Fernandes, J. M. S. Cabral, P. Žnidaršič-Plazl and I. Plazl, Chem. Eng. J., 2010, 160, 708–714 CrossRef CAS.
- M. P. C. Marques, P. Fernandes, J. M. S. Cabral, P. Žnidaršič-Plazl and I. Plazl, Nat. Biotechnol., 2012, 29, 227–234 CAS.
- A. J. Tušek, M. Tišma, V. Bregović, A. Ptičar, Ž. Kurtanjek and B. Zelić, Biotechnol. Bioprocess Eng., 2013, 18, 686–696 CrossRef.
- S. Illner, C. Hofmann, P. Löb and U. Kragl, ChemCatChem, 2014, 6, 1748–1754 CrossRef CAS.
- B. Tomaszewski, R. C. Lloyd, A. J. Warr, K. Buehler and A. Schmid, ChemCatChem, 2014, 6, 2567–2576 CrossRef CAS.
- B. Tomaszewski, A. Schmid and K. Buehler, Org. Process Res. Dev., 2014, 18, 1516–1526 CrossRef CAS.
- E. M. Forsberg, J. R. A. Green and J. D. Brennan, Anal. Chem., 2011, 83, 5230–5236 CrossRef CAS PubMed.
- R. Chirillo, G. Caenaro, B. Pavan and A. Pin, Clin. Chem., 1979, 25, 1744–1748 CAS.
- L. Babich, A. F. Hartog, M. A. Van Der Horst and R. Wever, Chem. – Eur. J., 2012, 18, 6604–6609 CrossRef CAS PubMed.
- S. Wang, P. Su and Y. Yang, Anal. Biochem., 2012, 427, 139–143 CrossRef CAS PubMed.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- J. A. A. Ho, L. C. Wu, N. C. Fan, M. S. Lee, H. Y. Kuo and C. S. Yang, Talanta, 2007, 71, 391–396 CrossRef CAS PubMed.
- U. Schuchardt, W. A. Carvalho and E. V. Spinace, Synlett, 1993, 713–718 CrossRef CAS.
- C. Fräulin, G. Rinke and R. Dittmeyer, J. Flow Chem., 2013, 3, 87–91 CrossRef.
- M. Hamano, K. D. Nagy and K. F. Jensen, Chem. Commun., 2012, 48, 2086–2088 RSC.
- X.-M. Lv, L.-J. Kong, Q. Lin, X.-F. Liu, Y.-M. Zhou and Y. Jia, Synth. Commun., 2011, 41, 3215–3222 CrossRef CAS.
- L. Vanoye, A. Aloui, M. Pablos, R. Philippe, A. Percheron, A. Favre-Réguillon and C. de Bellefon, Org. Lett., 2013, 15, 5978–5981 CrossRef CAS PubMed.
- Z. He and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 3353–3357 CrossRef CAS PubMed.
- F. Diederich and A. de Meijere, Metal-Catalyzed Cross-coupling Reactions, Wiley-VCH, New York, 2004 Search PubMed.
- N. Miyaura, Cross-Coupling Reactions: A Practical Guide, Springer, 2002, vol. 219 Search PubMed.
- T. Noël and S. L. Buchwald, Chem. Soc. Rev., 2011, 40, 5010–5029 RSC.
- D. Cantillo and C. O. Kappe, ChemCatChem, 2014, 6, 3286–3305 CrossRef CAS.
- Y. Nishihara, Applied Cross-Coupling Reactions, Springer, 2013, vol. 80 Search PubMed.
- C. P. Park and D. P. Kim, J. Am. Chem. Soc., 2010, 132, 10102–10106 CrossRef CAS PubMed.
- D. M. Rudzinski and N. E. Leadbeater, Green Process. Synth., 2013, 2, 323–328 CAS.
- H. P. L. Gemoets, V. Hessel and T. Noël, Org. Lett., 2014, 16, 5800–5803 CrossRef CAS PubMed.
- T. P. Petersen, A. Polyzos, M. O'Brien, T. Ulven, I. R. Baxendale and S. V. Ley, ChemSusChem, 2012, 5, 274–277 CrossRef CAS PubMed.
- M. Brzozowski, J. A. Forni, G. Paul Savage and A. Polyzos, Chem. Commun., 2015, 51, 334–337 RSC.
- E. Dolusic, P. Larrieu, L. Moineaux, V. Stroobant, L. Pillote, D. Colau, L. Pochet, B. Van den Eynde, B. Masereel, J. Wouters and R. Frédérick, J. Med. Chem., 2011, 54, 5320–5334 CrossRef CAS PubMed.
- Y. S. S. Wan, J. L. H. Chau, A. Gavriilidis and K. L. Yeung, Chem. Commun., 2002, 878–879 RSC.
- Y. S. S. Wan, J. L. H. Chau, K. L. Yeung and A. Gavriilidis, J. Catal., 2004, 223, 241–249 CrossRef CAS.
- O. Fenollar, D. García, L. Sánchez, J. López and R. Balart, Eur. Polym. J., 2009, 45, 2674–2684 CrossRef CAS.
- B. Cortese, M. De Croon and V. Hessel, Ind. Eng. Chem. Res., 2012, 51, 1680–1689 CrossRef CAS.
- E. Santacesaria, A. Renken, V. Russo, R. Turco, R. Tesser and M. Di Serio, Ind. Eng. Chem. Res., 2012, 51, 8760–8767 CrossRef CAS.
- W. He, Z. Fang, D. Ji, K. Chen, Z. Wan, X. Li, H. Gan, S. Tang, K. Zhang and K. Guo, Org. Process Res. Dev., 2013, 17, 1137–1141 CrossRef CAS.
- A. Srinivasan, X. Wu, M. Y. Lee and J. S. Dordick, Biotechnol. Bioeng., 2003, 81, 563–569 CrossRef CAS PubMed.
- S. P. Kee and A. Gavriilidis, Org. Process Res. Dev., 2009, 13, 941–951 CrossRef CAS.
- C. Wiles, M. J. Hammond and P. Watts, Beilstein J. Org. Chem., 2009, 5 DOI:10.3762/bjoc.5.27.
- F. Björkling, S. E. Godtfredsen, O. Kirk, F. Björkling, S. E. Godtfredsen and O. Kirk, J. Chem. Soc., Chem. Commun., 1990, 1301–1303 RSC.
- F. Bjorkling, H. Frykman, S. E. Godtfredsen and O. Kirk, Tetrahedron, 1992, 48, 4587–4592 CrossRef CAS.
- F. Ebrahimi, E. Kolehmainen, P. Oinas, V. Hietapelto and I. Turunen, Chem. Eng. J., 2011, 167, 713–717 CrossRef CAS.
- F. Ebrahimi, E. Kolehmainen, A. Laari, H. Haario, D. Semenov and I. Turunen, Chem. Eng. Sci., 2012, 71, 531–538 CrossRef CAS.
- S. Leveneur, J. Wärnå, K. Eränen and T. Salmi, Chem. Eng. Sci., 2011, 66, 1038–1050 CrossRef CAS.
- F. Ebrahimi, E. Kolehmainen and I. Turunen, Chem. Eng. J., 2012, 179, 312–317 CrossRef CAS.
- T. Noguchi, Y. Hirai and M. Kirihara, Chem. Commun., 2008, 3040–3042 RSC.
- S. Doherty, J. G. Knight, M. A. Carroll, J. R. Ellison, S. J. Hobson, S. Stevens, C. Hardacre and P. Goodrich, Green Chem., 2015, 17, 1559–1571 RSC.
- R. Maggi, S. Chitsaz, S. Loebbecke, C. G. Piscopo, G. Sartori and M. Schwarzer, Green Chem., 2011, 13, 1121–1123 RSC.
- A. Castellan, S. Cavallaro and J. C. J. Bart, Catal. Today, 1991, 9, 237–254 CrossRef CAS.
- M. Shang, T. Noël, Q. Wang and V. Hessel, Chem. Eng. Technol., 2013, 36, 1001–1009 CrossRef CAS.
- M. Shang, T. Noël, Q. Wang, Y. Su, K. Miyabayashi, V. Hessel and S. Hasebe, Chem. Eng. J., 2015, 260, 454–462 CrossRef CAS.
- M. Damm, B. Gutmann and C. O. Kappe, ChemSusChem, 2013, 6, 978–982 CrossRef CAS PubMed.
- K. Yube and K. Mae, Chem. Eng. Technol., 2005, 28, 331–336 CrossRef CAS.
- X. Liu and K. F. Jensen, Green Chem., 2012, 14, 1471 RSC.
- J. H. Park, C. Y. Park, H. S. Song, Y. S. Huh, G. H. Kim and C. P. Park, Org. Lett., 2013, 15, 752–755 CrossRef CAS PubMed.
- J. Souto, R. Stockman and S. V. Ley, Org. Biomol. Chem., 2015, 13, 3871–3877 Search PubMed.
- W. Wu, G. Qian, X. G. Zhou and W. K. Yuan, Chem. Eng. Sci., 2007, 62, 5127–5132 CrossRef CAS.
- K. Yube, M. Furuta and K. Mae, Catal. Today, 2007, 125, 56–63 CrossRef CAS.
- A. Hartung, M. A. Keane and A. Kraft, J. Org. Chem., 2007, 72, 10235–10238 CrossRef CAS PubMed.
- P. Bailey, Chem. Rev., 1958, 58, 925–1010 CrossRef CAS.
- S. G. Van Ornum, R. M. Champeau and R. Pariza, Chem. Rev., 2006, 106, 2990–3001 CrossRef CAS PubMed.
- M. Irfan, T. N. Glasnov and C. O. Kappe, Org. Lett., 2011, 13, 984–987 CrossRef CAS PubMed.
- M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Lett., 2010, 12, 1596–1598 CrossRef PubMed.
- M. D. Roydhouse, A. Ghaini, A. Constantinou, A. Cantu-Perez, W. B. Motherwell and A. Gavriilidis, Org. Process Res. Dev., 2011, 15, 989–996 CrossRef CAS.
- M. D. Roydhouse, W. B. Motherwell, A. Constantinou, A. Gavriilidis, R. Wheeler, K. Down and I. Campbell, RSC Adv., 2013, 3, 5076 RSC.
- J. Ren, S. He, C. Ye, G. Chen and C. Sun, Chem. Eng. J., 2012, 210, 374–384 CrossRef CAS.
- S. Kobayashi, H. Miyamura, R. Akiyama and T. Ishida, J. Am. Chem. Soc., 2005, 127, 9251–9254 CrossRef CAS PubMed.
- J. Wegner, S. Ceylan, C. Friese and A. Kirschning, Eur. J. Org. Chem., 2010, 4372–4375 CAS.
- J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618–3621 CrossRef CAS PubMed.
- I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566–2568 RSC.
- D. N. Tran, C. Battilocchio, S.-B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120–1125 RSC.
- N. M. Roda, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 Search PubMed.
- K. C. Basavaraju, S. Sharma, R. A. Maurya and D. P. Kim, Angew. Chem., Int. Ed., 2013, 52, 6735–6738 CrossRef CAS PubMed.
- C. Wiles, P. Watts and S. J. Haswell, Tetrahedron Lett., 2006, 47, 5261–5264 CrossRef CAS.
- A. Šali, A. Tušek, Ž. Kurtanjek and B. Zelić, Biotechnol. Bioprocess Eng., 2011, 16, 495–504 CrossRef.
- A. Tušek, A. Šalić, Ž. Kurtanjek and B. Zelić, Eng. Life Sci., 2012, 12, 49–56 CrossRef.
- A. Šalić, K. Pindrić and B. Zelić, Appl. Biochem. Biotechnol., 2013, 171, 2273–2284 CrossRef PubMed.
- A. Šalić and B. Zelić, RSC Adv., 2014, 4, 41714–41721 RSC.
- M. Tudorache, D. Mahalu, C. Teodorescu, R. Stan, C. Bala and V. I. Parvulescu, J. Mol. Catal. B: Enzym., 2011, 69, 133–139 CrossRef CAS.
- P. J. Keay and Y. Wang, Trends Biotechnol., 1997, 15, 76–81 CrossRef CAS.
- A. M. Azevedo, V. Vojinović, J. M. S. Cabral, T. D. Gibson and L. P. Fonseca, J. Mol. Catal. B: Enzym., 2004, 28, 121–128 CrossRef CAS.
- A. M. Azevedo, J. M. S. Cabral, T. D. Gibson and L. P. Fonseca, J. Mol. Catal. B: Enzym., 2004, 28, 45–53 CrossRef CAS.
- M. Y. Lee, A. Srinivasan, B. Ku and J. S. Dordick, Biotechnol. Bioeng., 2003, 83, 20–28 CrossRef CAS PubMed.
- J. Drott, K. Lindström, L. Rosengren and T. Laurell, J. Micromech. Microeng., 1999, 7, 14–23 CrossRef.
- T. T. Tidwell, Org. React., 2004, 39, 297–555 Search PubMed.
- T. Kawaguchi, H. Miyata, K. Ataka, K. Mae and J. I. Yoshida, Angew. Chem., Int. Ed., 2005, 44, 2413–2416 CrossRef CAS PubMed.
- J. R. Mcconnell, J. E. Hitt, E. D. Daugs and T. A. Rey, Org. Process Res. Dev., 2008, 12, 940–945 CrossRef CAS.
- J. J. M. van der Linden, P. W. Hilberink, C. M. P. Kronenburg and G. J. Kemperman, Org. Process Res. Dev., 2008, 12, 911–920 CrossRef CAS.
- K. Watts, A. Baker and T. Wirth, J. Flow Chem., 2014, 4, 2–11 CrossRef.
- F. J. Del Campo, Electrochem. Commun., 2014, 45, 91–94 CrossRef CAS.
- R. Stalder and G. P. Roth, ACS Med. Chem. Lett., 2013, 4, 1119–1123 CrossRef CAS PubMed.
- T. Kashiwagi, F. Amemiya, T. Fuchigami and M. Atobe, Chem. Commun., 2012, 48, 2806–2808 RSC.
- F. Amemiya, T. Fuchigami and M. Atobe, J. Flow Chem., 2012, 3, 17–22 Search PubMed.
- K. Watts, W. Gattrell and T. Wirth, Beilstein J. Org. Chem., 2011, 7, 1108–1114 CrossRef CAS PubMed.
- O. Radovici, A. Banu and C. Pirvu, ECS Trans., 2009, 16, 1–9 CAS.
- R. A. Green, R. C. D. Brown and D. Pletcher, J. Flow Chem., 2015, 5, 31–36 CrossRef CAS.
- M. A. Kabeshov, B. Musio, P. R. D. Murray, D. L. Browne and S. V. Ley, Org. Lett., 2014, 16, 4618–4621 CrossRef CAS PubMed.
- J. Kuleshova, J. T. Hill-Cousins, P. R. Birkin, R. C. D. Brown, D. Pletcher and T. J. Underwood, Electrochim. Acta, 2011, 56, 4322–4326 CrossRef CAS.
- For continuous-flow precious metal recycling see: I. Vural Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2015, 17, 2012–2026 RSC.
- P. D. Hampton, M. D. Whealon, L. M. Roberts, A. A. Yaeger and R. Boydson, Org. Process Res. Dev., 2008, 12, 946–949 CrossRef CAS.
- A. Bogdan and D. T. McQuade, Beilstein J. Org. Chem., 2009, 5 DOI:10.3762/bjoc.5.17.
- C. Aellig, D. Scholz, S. Conrad and I. Hermans, Green Chem., 2013, 15, 1975–1980 RSC.
- C. Aellig, D. Scholz and I. Hermans, ChemSusChem, 2012, 5, 1732–1736 CrossRef CAS PubMed.
- N. Ambreen, R. Kumar and T. Wirth, Beilstein J. Org. Chem., 2013, 9, 1437–1442 CrossRef PubMed.
- A. Kirschning, C. Altwicker, G. Dräger, J. Harders, N. Hoffmann, U. Hoffmann, H. Schönfeld, W. Solodenko and U. Kunz, Angew. Chem., Int. Ed., 2001, 40, 3995–3998 CrossRef CAS PubMed.
- A. B. Leduc and T. F. Jamison, Org. Process Res. Dev., 2012, 16, 1082–1089 CrossRef CAS.
- D. Ghislieri, K. Gilmore and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 54, 678–682 Search PubMed.
- S. Rozen, Eur. J. Org. Chem., 2005, 2433–2447 CrossRef CAS.
- C. B. McPake, C. B. Murray and G. Sandford, Tetrahedron Lett., 2009, 50, 1674–1676 CrossRef CAS.
- C. B. Mcpake, C. B. Murray and G. Sandford, Chim. Oggi, 2010, 28, 28–29 Search PubMed.
- B. R. Travis, M. Sivakumar, G. O. Hollist and B. Borhan, Org. Lett., 2003, 5, 1031–1034 CrossRef CAS PubMed.
- Y. M. A. Yamada, K. Torii and Y. Uozumi, Beilstein J. Org. Chem., 2009, 5, 1–5 Search PubMed.
- M. Zanfir and A. Gavriilidis, Org. Process Res. Dev., 2007, 11, 966–971 CrossRef CAS.
- L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed.
- M. Saber, J. M. Commenge and L. Falk, Chem. Eng. Sci., 2010, 65, 372–379 CrossRef CAS.
- M. Al-Rawashdeh, F. Yu, T. A. Nijhuis, E. V. Rebrov, V. Hessel and J. C. Schouten, Chem. Eng. J., 2012, 207–208, 645–655 CrossRef CAS.
- M. Al-Rawashdeh, J. Zalucky, C. Müller, T. A. Nijhuis, V. Hessel and J. C. Schouten, Ind. Eng. Chem. Res., 2013, 52, 11516–11526 CrossRef CAS.
- Y. Wada, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2006, 45, 8036–8042 CrossRef CAS.
- S. Hubier, U. Bentrup, U. Budde, K. Lovis, T. Dietrich, A. Freitag, L. Küpper and K. Jahnisch, Org. Process Res. Dev., 2009, 13, 952–960 CrossRef.
- A. Yavorskyy, O. Shvydkiv, N. Hoffmann, K. Nolan and M. Oelgemöller, Org. Lett., 2012, 14, 4342–4345 CrossRef CAS PubMed.
- N. Kockmann, M. Gottsponer and D. M. Roberge, Chem. Eng. J., 2011, 167, 718–726 CrossRef CAS.
- D. M. Roberge, M. Gottsponer, M. Eyholzer and N. Kockmann, Chim. Oggi, 2009, 27, 8–11 CAS.
- N. Kockmann, Chem. Ing. Tech., 2012, 84, 646–659 CrossRef CAS.
- N. Kockmann and D. M. Roberge, Chem. Eng. Process. Process Intensif., 2011, 50, 1017–1026 CrossRef CAS.
- G. S. Calabrese and S. Pissavini, AIChE J., 2011, 57, 828–834 CrossRef CAS.
- M. J. Nieves-Remacha, A. A. Kulkarni and K. F. Jensen, Ind. Eng. Chem. Res., 2013, 52, 8996–9010 CrossRef CAS.
- Y. Zhang, S. C. Born and K. F. Jensen, Org. Process Res. Dev., 2014, 18, 1476–1481 CrossRef CAS.
- Q. Michaudel, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2015, 48, 712–721 CrossRef CAS PubMed.
- C. J. Welch, J. M. Hawkins and J. Tom, Org. Process Res. Dev., 2014, 18, 481–487 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |