
Metallosupramolecular receptors for fullerene binding and release
Cristina
García-Simón
,
Miquel
Costas
* and
Xavi
Ribas
*
Grup de Química Bioinspirada, Supramolecular i Catàlisi (QBIS-CAT), Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus Montilivi, Girona E17071, Catalonia, Spain
First published on 12th October 2015
Abstract
Fullerene extracts are easily available from fullerene soot, but finding an efficient strategy to obtain them in pure form remains elusive, especially for higher fullerenes (Cx, x > 70). The properties of the latter remain unclear and their potential application to multiple research fields has not been developed mainly due to their purification difficulties. In this Tutorial Review we cover the use of molecular receptors for the separation of fullerenes by means of host–guest interactions. This strategy allows gaining selectivity, no specialized equipment is required and, ideally, recyclable systems can be designed. We focus on the metallosupramolecular receptors using the metal–ligand coordination approach, which offers a controlled and versatile strategy to design fullerene hosts, and the latest strategies to release the fullerene guest will be described. The field is probably in its beginnings but it is rapidly evolving and we are confident that this tutorial review will help researchers to rapidly gain a general overview of the main works and concepts that are leading this promising strategy and that may lead towards a useful methodology to purify fullerenes.
 Cristina García-Simón | Cristina García-Simón received her bachelor's in Chemistry from the University of Girona (UdG) in 2010. Thereafter she joined the QBIS-CAT group. In 2015, she received her PhD and currently she is working as a post-doc in the same group. Her interests are based on supramolecular nanocapsules, fullerene chemistry and supramolecular catalysis. |
 Miquel Costas | Miquel Costas (1971, Vigo) obtained his PhD in Chemistry from the University of Girona (UdG) in 1999, and moved to the group of Prof. Que, Jr, at the University of Minnesota for post-doctoral studies. He returned to Spain with a Ramon y Cajal Fellowship in 2002. He was appointed Associate professor in 2003. Since 2006 he has been a group leader at the QBIS-CAT research group in UdG. He got the ICREA-Acadèmia Award in 2009 and 2014, an ERC StG in 2009 and the 2014-RSEQ excellence in research award. His research interests involve synthetic bioinorganic chemistry, bioinspired oxidation catalysts and supramolecular chemistry. |
 Xavi Ribas | Xavi Ribas (1974, Santa Coloma de Farners) obtained his PhD in Chemistry from the University of Girona (UdG) in 2001, and he carried out a 3-year postdoc at ICMAB-CSIC in 2002–2004. He was promoted to Associate professor in Chemistry at UdG in 2006. He received an ICREA-Acadèmia Award in 2010 and an ERC-StG project in 2011. His research focuses on in-depth mechanistic understanding of the organometallic chemistry of copper, silver and gold. He is interested in coinage metal-catalyzed C–C and C–heteroatom cross-coupling reactions and C–H activation with directing groups. He is also interested in the involvement of Fe and Mn high oxidation states in alkane and alkene oxidations. The development of nanocages for catalysis and host–guest reactions is also pursued. |
Key learning points(a) Supramolecular hosts for fullerene encapsulation(b) Strategies for enhancing fullerene binding in supramolecular hosts (c) Strategies for fullerene release from supramolecular hosts (d) Fullerene purification strategies using supramolecular hosts |
1. Introduction
1.1 Relevance of fullerene separation
Fullerenes are the third most stable allotrope of carbon and in contrast to graphite or diamond, which are infinite extended arrays of sp2- and sp3-hybridized carbon atoms, respectively, fullerenes are spherical discrete molecules which contain a defined number of Csp2 atoms. Consequently fullerenes are soluble in various organic solvents, an essential requirement for chemical manipulations. These spherical molecules were first observed experimentally by Kroto and Smalley in 1985.1 The finding of fullerenes opened a gateway towards the discovery of other carbon nanoforms such as carbon nanotubes, graphene, nano-onions, peapods, etc.Fullerenes and their derivatives present a wide range of applications highlighting their use in materials science (polymers, thin films, liquid crystals, etc.),2 as electroactive materials in solar cells,3 as superconducting materials4 and for biological applications.5 However, all these applications are limited in origin by the current tedious and expensive methodologies for selective purification of fullerenes.
The crude product generated usually by arc discharge or laser vaporization on carbon sources contains mixtures of fullerenes of different sizes, as well as other carbon allotropes such as carbon nanotubes and amorphous forms of carbon. The first examples reported for the purification of fullerenes were based on controlled sublimation of the carbon soot.6 Other common methods to separate fullerenes from soot consist of extraction with organic solvents or purification by crystallization. Currently, chromatographic techniques are predominantly used for the purification of fullerenes. Alumina, graphite, activated carbon, polystyrene gel or γ-cyclodextrins have been used as stationary phases.7 In spite of the fact that efficient columns (COSMOSIL© columns) are available for the HPLC separation of fullerenes, most of the methods mentioned here require enormous amounts of solvent and can cause fullerene decomposition, irreversible absorption of fullerenes within the stationary phase, or give moderate yields. Furthermore, these methods are often tedious, and energy- and time-consuming, and in some cases, it is difficult to obtain specific fullerenes with high selectivity in a pure form. Therefore, designing an improved fullerene purification methodology is a challenging and desired goal.
1.2 Molecular hosts to selectively entrap fullerenes
The use of molecular receptors for fullerene separation in solution has emerged as an attractive alternative since it allows potential selectivity, do not require specific equipment and ideal recyclable hosts can be designed. Additionally, the encapsulation promotes the solubilisation and chemical modification of fullerenes. It also facilitates the selective extraction of higher fullerenes and their chiral resolution.8In this tutorial review some examples of relevant fullerene receptors will be described. Attention has been focused towards supramolecular receptors based on coordination bonds. Nevertheless, some examples of covalent and other supramolecular receptors will also be discussed in the following sections.
In order to design a molecular receptor for the selective separation of fullerenes, the most important factor to be taken into account is the complementarity between the host and the fullerene. On one hand, structural complementarity needs to be considered (i.e. size and shape of the receptor), and on the other hand, the electronic complementarity also plays a crucial role. For this reason, the majority of molecular hosts for fullerenes described so far were based on extended π-systems (i.e. π-extended tetrathiafulvalenes or porphyrins) which tend to act as π-donor moieties, favouring the interaction with fullerenes that behave as π-acceptor moieties.9 Another important factor to consider when designing a fullerene receptor is the structural and electronic tunability of the container. A tuneable and versatile receptor may pave the way for achieving good selectivity towards a specific fullerene within a mixture of fullerenes of different sizes. A host molecule can also be tuned in order to selectively encapsulate a particular higher fullerene or an endohedral fullerene. Besides, other factors such as solvophobic effects may also influence the fullerene–receptor interaction, and they also need to be considered.
1.2.2.1 Fully organic covalent hosts. Ringsdorf and Wennerström reported the first examples of covalent molecular receptors for fullerenes in 1992. They complexed fullerenes using aza-crown compounds and γ-cyclodextrins.10,11 Since then, a number of covalent fully organic receptors have been reported in the literature. For example, arene based receptors such as cyclodextrins (CD),12 calixarenes13 and cyclotriveratrylenes (CTVs)14 have proven to be efficient receptors for fullerenes, especially when two or more of these units are associated, through the formation of cage-like receptors (vide infra). There are also arene-based fullerene receptors such as cycloparaphenylenes or carbon nanotubes (CNTs), which can give rise to peapod structures.15 Another relevant example was reported by Martín who used π-extended derivatives of tetrathiafulvalene (exTTF) to wrap around different fullerenes.8 Multiple examples of fully organic fullerene receptors have been reported so far, and as such an extensive account is beyond the purpose of this review paper. Herein we will focus mainly on fully organic receptors having a 3D cage-like structure.
The suitability of dimeric cyclotriveratrylene (CTV)-based cage-like structures for fullerene separation was exemplified by Chiu and co-workers who synthesized a cage formed by two covalently linked CTV units (1) (Fig. 1a). Zigzagging alkyl chains were chosen as spacers in order to minimize the energy cost of structural reorganization during the guest binding event. Notably, receptor 1 is able to encapsulate C60 and C70 fullerenes at room temperature.16 The cage favours the formation of the hemicarceplex containing C70 in the presence of C60 and therefore, the system could be applied in the selective encapsulation of C70 from fullerene extracts. To a solution of 1 in tetrachloroethane (TCE), the fullerene extract was added and the mixture was heated to 313 K for 16 h, thereafter host–guest adducts with C70 fullerene were exclusively formed.
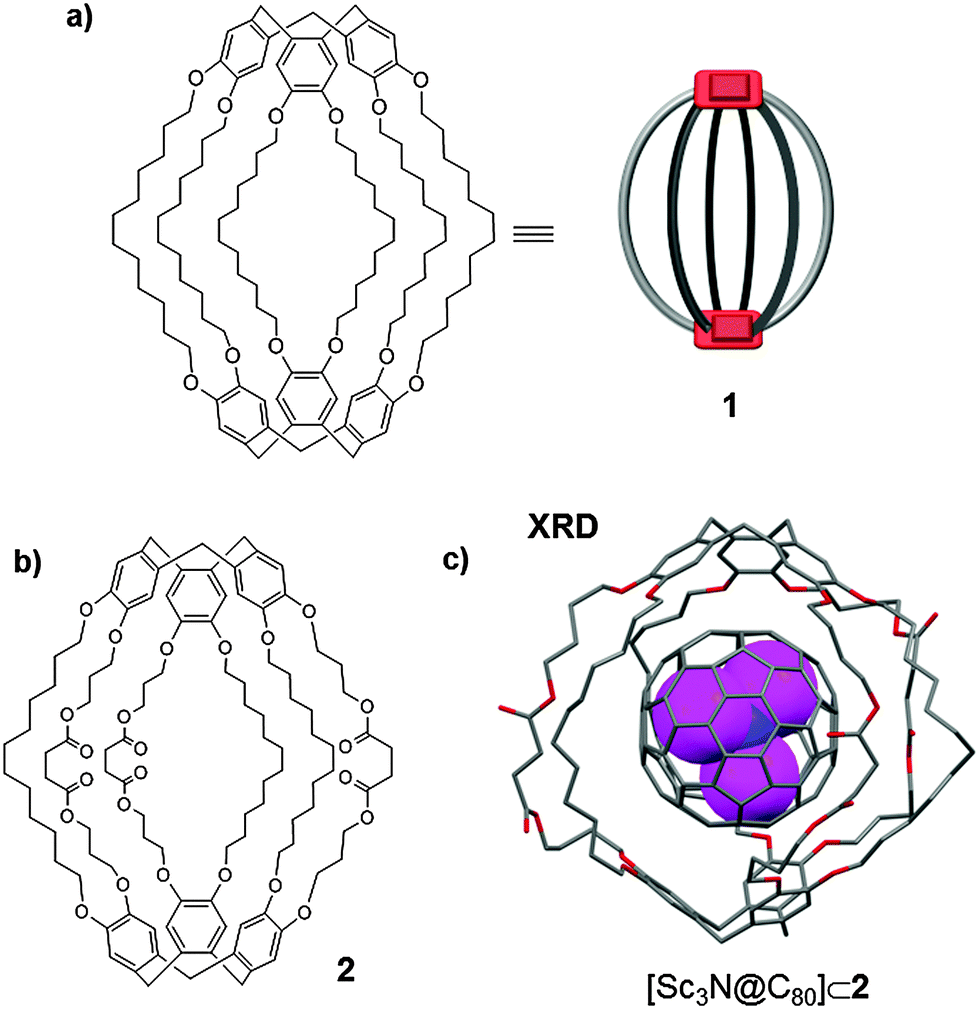 | ||
| Fig. 1 Chemical structure of CTV-based molecular cages 1 and 2 (a and b).16 (c) A representation of the solid state structure of the [Sc3N@C80]⊂2 host–guest adduct (hydrogen atoms are omitted for clarity).17 | ||
Very recently it was demonstrated that covalent cage 1 encapsulates Sc3N@C80 metallofullerene under solvent-free conditions. Sc3N@C80 exhibits two structural isomers D5h and Ih.17 The lack of solubility of these higher fullerenes makes their isolation and characterization challenging tasks. Moreover, the lack of hydrogen atoms in their structure prevents their study by 1H-NMR. Consequently, 13C-NMR, a less sensitive technique, is the most widely used technique for directly identifying the fullerene isomers. When Sc3N@C80 was sequestered inside cage 1, the 1H-NMR spectra displayed two sets of signals that were attributed to [Sc3N@D5h-C80]⊂1 and [Sc3N@Ih-C80]⊂1 adducts. Despite the fact that the solubility of the endofullerene in TCE was increased by approximately 50 times upon encapsulation, it was still too poor to permit its further characterization by 13C-NMR. The authors therefore decided to synthesise the more soluble cage 2 which contained three succinic diester linkages (Fig. 1b). The enlarged cage 2 was able to encapsulate Sc3N@C80 and a crystal structure was obtained for the [Sc3N@C80]⊂2 adduct (Fig. 1c), which confirmed the Russian doll (Matryoshka)-like multilayer structure. Remarkably, when Sc3N@C80 was encapsulated within cage 2, its solubility in TCE was 200 times greater than that of the free endofullerene, allowing its characterization by 1H-NMR and 13C-NMR. The 13C-NMR spectrum corresponding to [Sc3N@C80]⊂2 allowed for identification of the hemicarceplexes of both Sc3N@D5h-C80 and Sc3N@Ih-C80 isomers, which appear in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio. Moreover, the 1H-NMR spectrum also displayed two sets of signals of the aromatic C–H proton nuclei of the CTV units in the same 1:4 ratio.
:4 ratio. Moreover, the 1H-NMR spectrum also displayed two sets of signals of the aromatic C–H proton nuclei of the CTV units in the same 1:4 ratio.
Most of the fully organic covalent hosts for fullerenes reported in the literature are constructed via irreversible chemical reactions, which generally involve tedious and often low yielding complex synthetic procedures. Interestingly, recent advances in dynamic covalent chemistry (DCC), which takes advantage of reversible reactions such as alkyne methathesis/condensation, offer appropriate pathways to high-yielding synthesis of covalent organic capsule-like receptors. Strikingly in 2011, Zhang developed a shape persistent capsule containing non-metalated porphyrin units making use of DCC.18 A rectangular prismatic cage (4) was synthesised via alkyne metathesis of two tetrasubstituted porphyrins (3). Interestingly, the cage forms 1:1 complexes with C60 and C70 fullerenes, when the cage is mixed with fullerenes in toluene at room temperature (Fig. 2). It is well known that there is a favoured donor–acceptor interaction between porphyrins and fullerenes (vide infra). The rigidity of host 4 along with its combination of conjugated systems results in a cage with 3 orders of magnitude higher affinity towards C70 (Ka = 1.5 × 108 M−1) compared to C60 (Ka = 1.4 × 105 M−1). The significant difference in the affinity of the cage towards C70 over C60 facilitated the selective separation of C70 fullerene from a mixture containing both fullerenes.
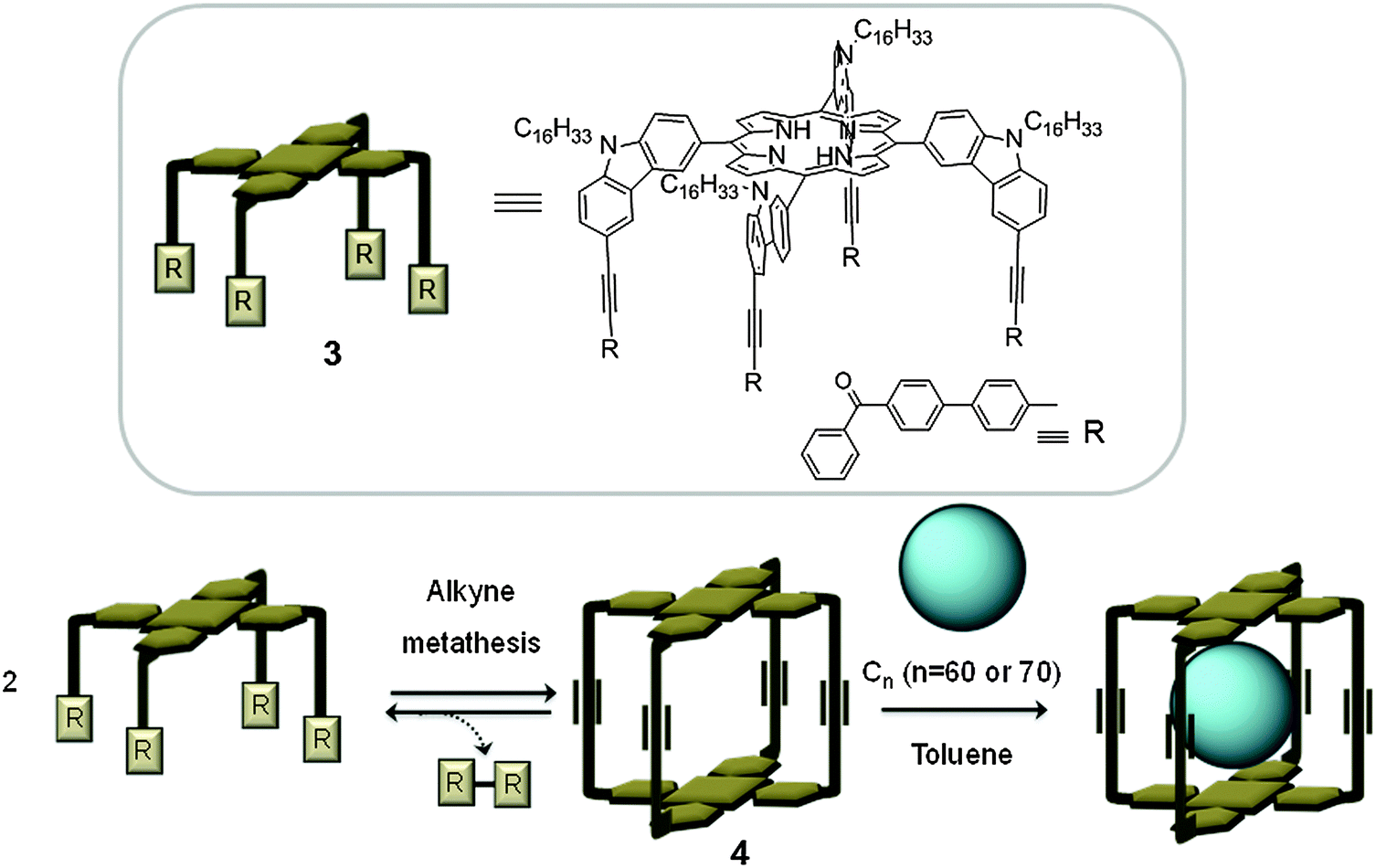 | ||
| Fig. 2 Top: Tetrasubstituted porphyrin (3), used for the preparation of the covalent capsule 4 reported by Zhang. Bottom: A schematic representation of the construction of capsule 4 by alkyne metathesis, and subsequent encapsulation of the fullerene.18 | ||
1.2.2.2 Metal containing covalent hosts. Metalloporphyrins contain a large π-conjugated system, thus they tend to act as π-donor moieties. Conversely, fullerenes behave as π-acceptors and this electronic complementarity favours the interaction between these two molecules.19 Nevertheless, this interaction is weak in solution and is difficult to visualize spectroscopically. For this reason, several examples of fullerene hosts containing two or more metalloporphyrin units have been reported in the literature, making attempts to enhance the strength of this fullerene–host interaction.8,20–23
One of the most well-known examples of fullerene receptors bearing metalloporphyrins was reported by Aida in 1999. The authors reported the synthesis of a covalently linked cyclic dimer of zinc(II) porphyrins (5) (Fig. 3).24 This cyclic host formed a highly stable 1:1 inclusion complex with C60 in benzene at room temperature (Ka = 6.7 × 105 M−1). Remarkably, in analogous experiments using C70 fullerene, an association constant 30 times larger than that of C60 (Ka(C70) ∼ 107 M−1) was obtained. It appears that C70 adopts a side-on conformation with respect to the two metalloporphyrin moieties in order to maximize the π-stacking interactions. With the intention to obtain more stable host–guest adducts, the authors modified the receptor structure by changing: (i) the porphyrin substituents, R, which could modify the π-basicity of the porphyrin or its conformation; (ii) the linker parts (Z) in order to gain flexibility or rigidity (aliphatic or acetylenic chains); and (iii) the metal ions of the porphyrin moieties. It was observed that hydrogenated separators (Z) gave more flexible structures (5a more flexible than 5c), and facilitated the host–guest interaction. Furthermore, when the porphyrin contains a more concave conformation (i.e. β-pyrrole-substituted porphyrin, host 5b) the host–guest contact is improved. Finally it was observed that the metal ions which have the highest association constants are those belonging to group 9 of the periodic table. Strikingly, the rhodium(III) porphyrin dimer showed 100 times higher affinity towards C60 and C70 than its zinc(II) analogue.25 Furthermore, in the case of the iridium(II) porphyrin cyclic dimer the association constant for C60 and C70 in benzene was too large to be evaluated accurately (Ka > 109 M−1). Studies in the solid state (XRD) and solution (NMR), at low temperature, indicated that dynamic organometallic bonds between the iridium(II) metal ions from the porphyrins and the π-electron-rich 6:6 junctions from the fullerenes were formed. This metal–fullerene interaction caused an ellipsoidal deformation of C60 and an end-on orientation of C70 relative to the porphyrin units.26 Finally, the authors successfully used the different binding ability of the hosts towards the fullerenes to perform the selective extraction of higher fullerenes (≥C76) from fullerene mixtures.27
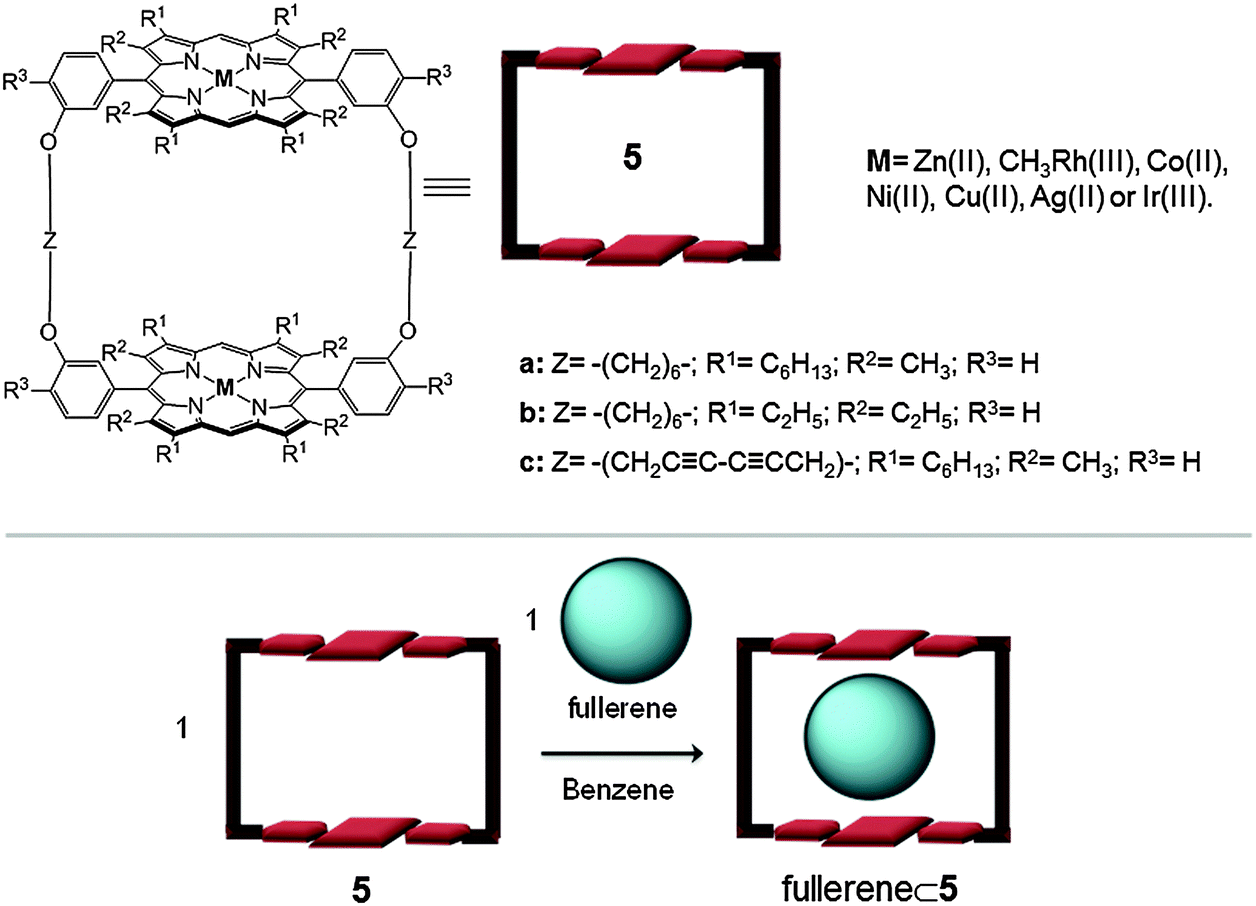 | ||
| Fig. 3 Top: Molecular structure and formulae of the metalloporphyrin dimer 5 reported by Aida.20 Bottom: schematic representation of fullerene encapsulation within receptor 5. | ||
The same authors later reported in 2011 the synthesis of a new cyclic receptor based on copper(II) or zinc(II) metalloporphyrins (6cyclo), which could be transformed into the cubic-cage (6cage) analogue by intramolecular ring-closing olefin metathesis (Fig. 4).28 Both structures were able to encapsulate La@C82 endofullerene in toluene at 25 °C (Ka ∼ 106–107 M−1). The inclusion experiments using the cyclic receptors based on copper(II) paramagnetic porphyrins showed ferromagnetic coupling with the paramagnetic La@C82 fullerene. However, a ferrimagnetic coupling was observed when the receptor was transformed into its cage analogue. This change in spin coupling was rationalized by the change in the geometry of the host–guest complex.
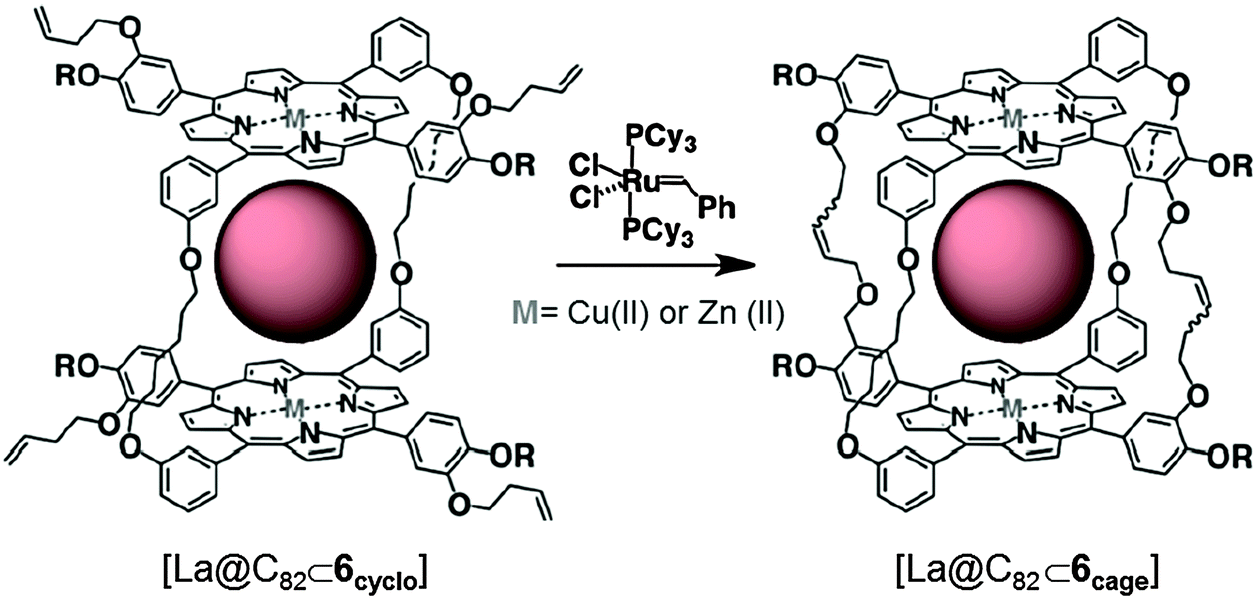 | ||
| Fig. 4 Representation of the molecular structures of [La@C82]⊂6cyclo and [La@C82]⊂6cage, and their transformation by ring-close metathesis. Adapted with permission from ref. 28. Copyright (2011) American Chemical Society. | ||
A different example of cyclic porphyrin dimers was investigated by Ballester. Cyclic covalent receptors were synthesized by dimerization of two non-β-pyrrole-substituted monoporphyrins, bearing four meso-aryl substituents (7), to give 8 (Fig. 5).21 Subsequently, hydrogenation of 8 afforded the flexible cyclic dimer 9. As Aida had observed previously in their receptors, the relative rigidity of the spacer units affected the shape and flexibility of the hosts and controlled their affinity towards different fullerene guests. Both receptors 8 and 9 form 1:1 host–guest adducts with C60 and C70 fullerenes at room temperature using toluene (TL) or dichloromethane (DCM) as solvents. The more rigid receptor 8, which bears fully unsaturated spacers (six carbon acetylenic chains), presented a relatively weak interaction with C70 (Ka(DCM) ∼ 104 M−1 and Ka(TL) ∼ 104 M−1) and an unobservable interaction with C60 (Ka < 103 M−1). On the other hand, the more flexible host 9, containing fully saturated six-carbon linkers, forms more stable complexes with C60 (Ka(DCM) ∼ 104 M−1 and Ka(TL) ∼ 104 M−1) and C70 (Ka(DCM) ∼ 105 M−1 and Ka(TL) ∼ 104 M−1) due to better adaptability. The host–guest adducts with both 8 and 9 receptors, were also characterized in the solid state. In collaboration with Echegoyen the authors discovered that both dimers were also able to form inclusion complexes with Sc3N@C80 endofullerene (Ka ∼ 105). For the first time, solid state structures were obtained for an endohedral fullerene bound to molecular bis-porphyrin receptors.29
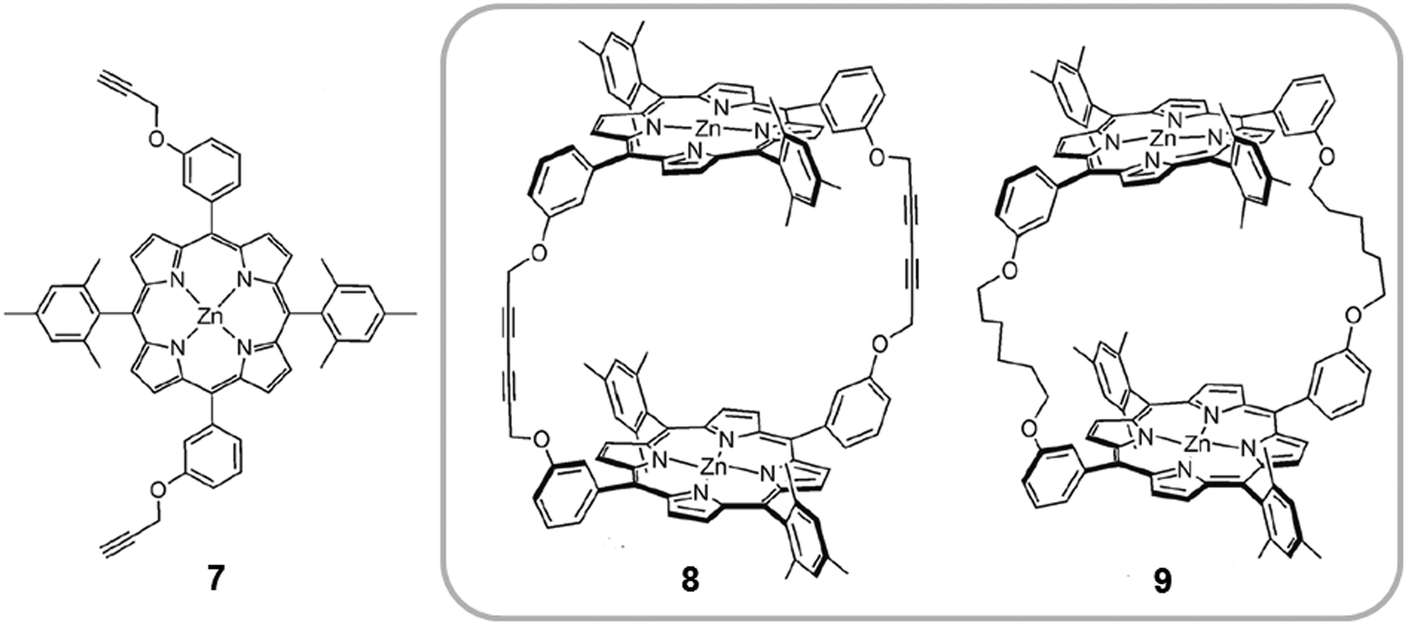 | ||
| Fig. 5 Molecular structure of the substituted zinc(II) porphyrin 7 used by Ballester to prepare covalent dimers 8 and 9. Reproduced in part from ref. 21 with permission of John Wiley and Sons. | ||
The association constant between fullerenes and porphyrins dramatically increased when using porphyrin dimers (vide supra) instead of a single porphyrin. In response, different research groups worked on the development of multiple-porphyrin systems with the aim of achieving even higher association constants.22 An interesting example was reported by Anderson who developed a cyclic porphyrin trimer that showed high affinity for fullerenes (Fig. 6a). The rigid cyclic porphyrin trimer (10) was synthesized by Sonogashira coupling of 3,4-diiodophthalimide and the alkyne-terminated linear porphyrin trimer. The linear porphyrin trimer was synthesized following an elaborated synthetic protocol.22 The trimeric receptor 10 formed 1:1 host–guest adducts with C60 at room temperature in different organic solvents (i.e. toluene). The association constant for C60⊂10 in toluene was (1.6 ± 0.7) × 106 M−1. On the other hand, the inclusion compound with C70 was two orders of magnitude more stable than that with C60 (Ka(C70) = (1.6 ± 0.7) × 108 M−1). The receptor also formed 1:1 adducts with higher fullerene C86 and La@C82 endofullerene, however, adducts are too stable for the equilibrium constant to be calculated by UV-Vis or fluorescence titrations (Ka > 109 M−1). The value obtained for the association constant between the cyclic trimer and C60 in toluene is slightly higher than the ones obtained for zinc(II)–porphyrin dimers, indicating that the presence of the third porphyrin moiety has a positive effect on the affinity towards C60. The effect of the third porphyrin is more relevant for C70 and higher fullerenes. The association constant of C70⊂10 is much higher than those reported for other zinc(II) porphyrin hosts and the association constants for C86⊂10 and La@C82⊂10 are still higher, therefore the larger size of the fullerene enables them to better interact with the three walls of the receptor.
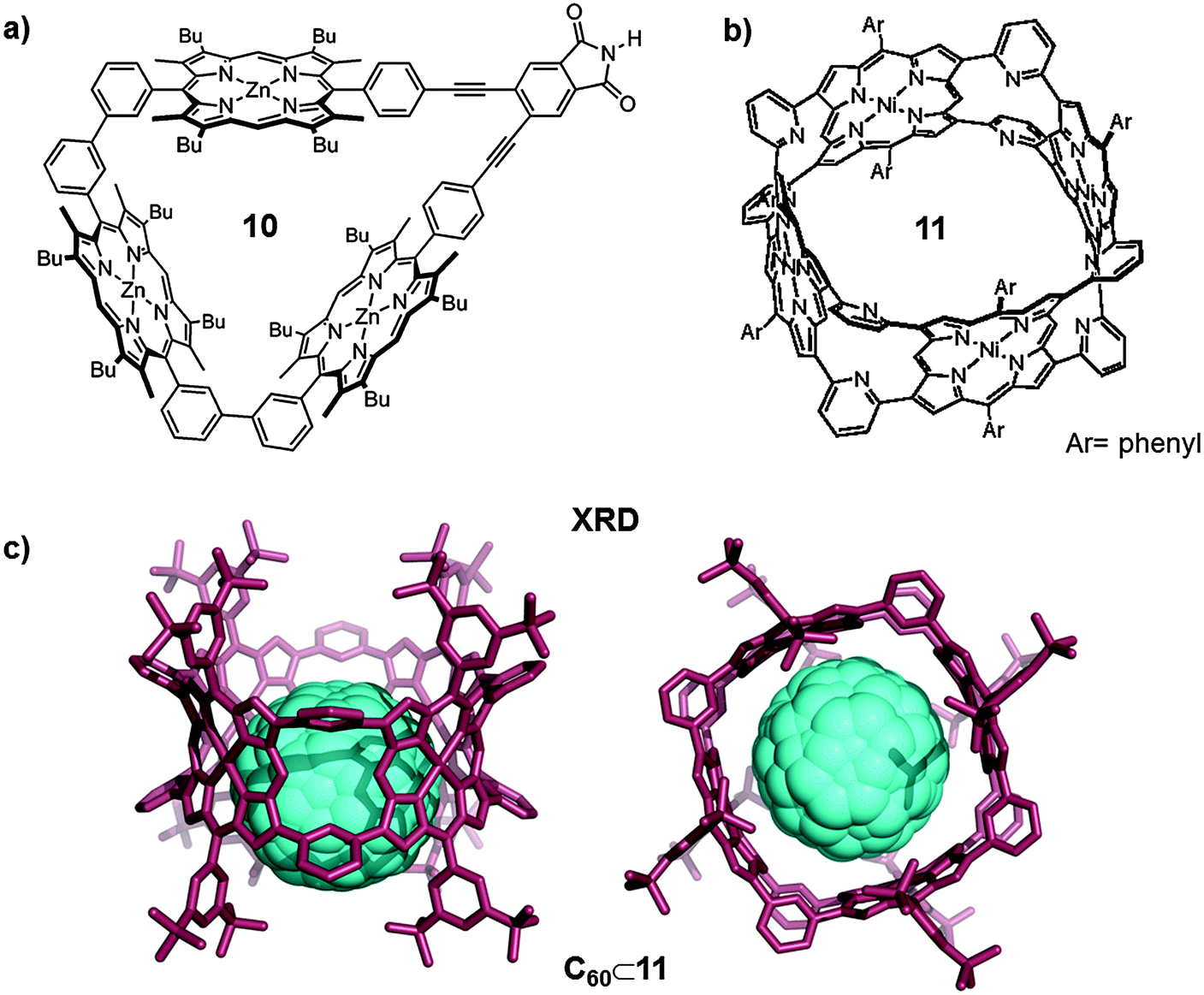 | ||
| Fig. 6 (a) Molecular structure of the cyclic porphyrin trimer 10 synthesized by Anderson. Adapted with permission from ref. 22. Copyright (2012) American Chemical Society. (b) Structure of nanobarrel 11. Adapted with permission from ref. 23. Copyright (2010) American Chemical Society. (c) Lateral and top views of the X-ray crystal structure of C60⊂11 (hydrogen atoms have been omitted for clarity). | ||
With these results in hand Osuka, Shionokubo and Aratani further increased the number of porphyrin units in order to improve the fullerene–porphyrin interaction. The authors synthesized the nanobarrel 11 bearing four porphyrin units (Fig. 6b), following a concise synthetic route. The addition of C60 to a solution of 11 in toluene, at room temperature, gave 1:1 inclusion adducts.23 Unexpectedly, the association constant for C60⊂11 calculated from UV-vis titrations in toluene was (5.3 ± 0.1) × 105 M−1, which is slightly smaller than those reported for cyclic porphyrin dimers and Anderson's trimer. The lower value for the association constant was rationalized by the high rigidity of this covalent receptor, which in this case is detrimental. Nevertheless, the host–guest adduct structure was unambiguously confirmed by X-ray diffraction studies (Fig. 6c).
Weak non-covalent interactions such as hydrogen bonds, van der Waals forces or π–π interactions have been used to hold the molecular building blocks together in order to form sophisticated supramolecular receptors. Moreover, some interactions that possess a significant covalent bond component (e.g. coordination metal–ligand bonds) are also widely used. In this review article special attention will be given to fullerene receptors based on metal–ligand coordination bonds.
1.2.3.1 Hydrogen bonded hosts. The suitability of dimeric CTV cage-like structures for fullerene separation was also exemplified by Mendoza who prepared a nanocapsule (13) by self-assembly, utilizing hydrogen bonding between two CTV (12) units that were modified with three high-affinity hydrogen-bonding units (1,1′-carbonyldiimidazole) (Fig. 7). This nanocapsule presents affinity towards C60 (Ka(C60) = 1.82 × 103 M−1) and C70 (Ka(C70) = 3.89 × 104 M−1).30 The higher affinity towards C70 was rationalized by an improved hydrogen-bonding interaction between the CTV units, giving rise to a more stable host–guest adduct. In contrast to the covalent CTV-based cage reported by Chiu and co-workers, which presents a more rigid structure, nanocapsule 13 presents a very flexible structure that allows for the encapsulation of higher fullerenes up to C84. When the capsule is reacted with fullerene extract (∼2% higher fullerenes), host–guest adducts with Cn (60, 70, 76, 78, 82 and 84) were observed by HPLC.31 Taking advantage of the different binding affinities and by changing the ratios of the nanocapsule/fullerene extract, the authors were able to selectively extract C70 and C84 fullerenes.
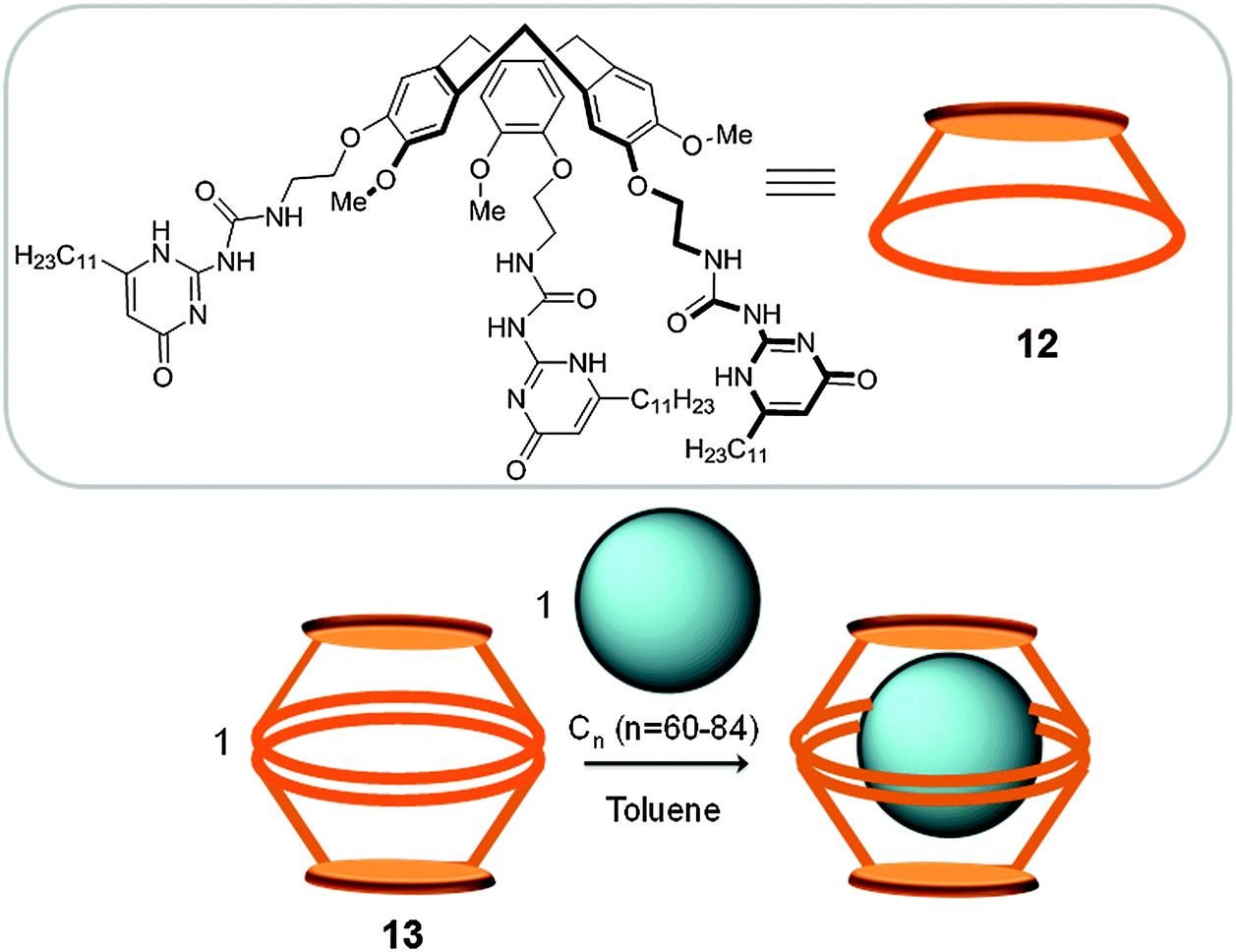 | ||
| Fig. 7 Top: CTV unit (12) used for the preparation of the supramolecular capsule 13 reported by Mendoza. Bottom: The reaction scheme for the encapsulation and release of fullerenes using 13.30 | ||
1.2.3.2 Fullerene hosts assembled through coordination bonds. When the complexity of the target supramolecular receptors increases, the assembly of small molecules into larger aggregates may turn into an increasingly impeding process. This is mostly a result of lack of control during the self-assembly process. As a response to these limitations, coordination-driven self-assembly has emerged as a powerful tool to regain control over supramolecular synthesis. It facilitates the formation of large complex molecules, ranging from ∼300 Å3 to more than 2000 Å3.32
The strength of coordination bonds is between that of a weak traditional supramolecular interaction and that of a strong covalent bond. Metal–ligand interactions offer a high degree of directionality as a result of predictable metal–ion coordination geometries. In addition, the diversity of transition metal complexes and ligands available as building blocks makes this strategy very versatile, allowing the generation of multiple supramolecular structures.
Over the years different supramolecular synthetic approaches have been developed based on coordination bonds. The most widely used strategies are the directional bonding (DBA), molecular panelling (MPA) and symmetry interaction (SIA) approaches. In addition, weak link (WLA) and bimetallic building block (DBBA) approaches have also led to complex coordination supramolecules. The corresponding literature has been extensively gathered in excellent reviews (see ref. 32) and will not be exhaustively discussed in this article.
The predictable directionality and geometry of metal–ligand coordination bonds have been used to synthesize the vast majority of the reported coordination supramolecular structures for fullerene recognition (2D and 3D), which usually are highly symmetric and resemble Archimedean or Platonic solids in shape. However, there are also structures with lower symmetry that have also been used as hosts for fullerene separation (i.e. heteroleptic species) and will also be described hereafter.33
Finally, apart from the metal–ligand interaction itself, there are other factors to consider which play a crucial role in the development of coordination receptors, such as the counter-anions in charged species, labilization agents, guest templates or solvent interactions. In many cases these subtle secondary interactions play a key role in the encapsulation of fullerenes.
2. 2D coordination receptors for fullerene recognition
The examples of coordination supramolecular polygons capable of interacting with fullerenes remain scarce, most likely due to the limited fullerene–receptor surface interaction (Ka up to 105 M−1). Nevertheless, some elegant examples of bi-dimensional coordination structures capable of hosting fullerenes have been reported in the literature.34–362.1 Aromatic heterocycles for fullerene binding
In 2012 Sallé reported on the synthesis of molecular triangles (15) and squares (16), obtained from metal–ligand directed self-assembly of cis-blocked square planar Pt(II) complexes and BPTTF-based electron-rich ligands (14, BPTTF: bis(pyrrolo)tetrathiafulvalene) (Fig. 8).34 When an equimolar mixture of ligand 14 and (dppp)Pt(CF3SO3)2 was mixed in dichloromethane both species 15 and 16 were generated, in a 2:1 ratio. The two species were easily separated by filtration and solvent evaporation. Whilst the square 16 is too large to accommodate electron-deficient C60 (cavity size ∼20 Å), UV-vis experiments indicated that the triangle 15 encapsulates C60 with an association constant of ∼104 M−1. The rigid and pre-organized structure of 15, with an inner cavity size of ∼13 Å, and the cooperative electron-donating ability of the three moderate π-extended BPTTF units, favors the interaction between the receptor and the substrate.
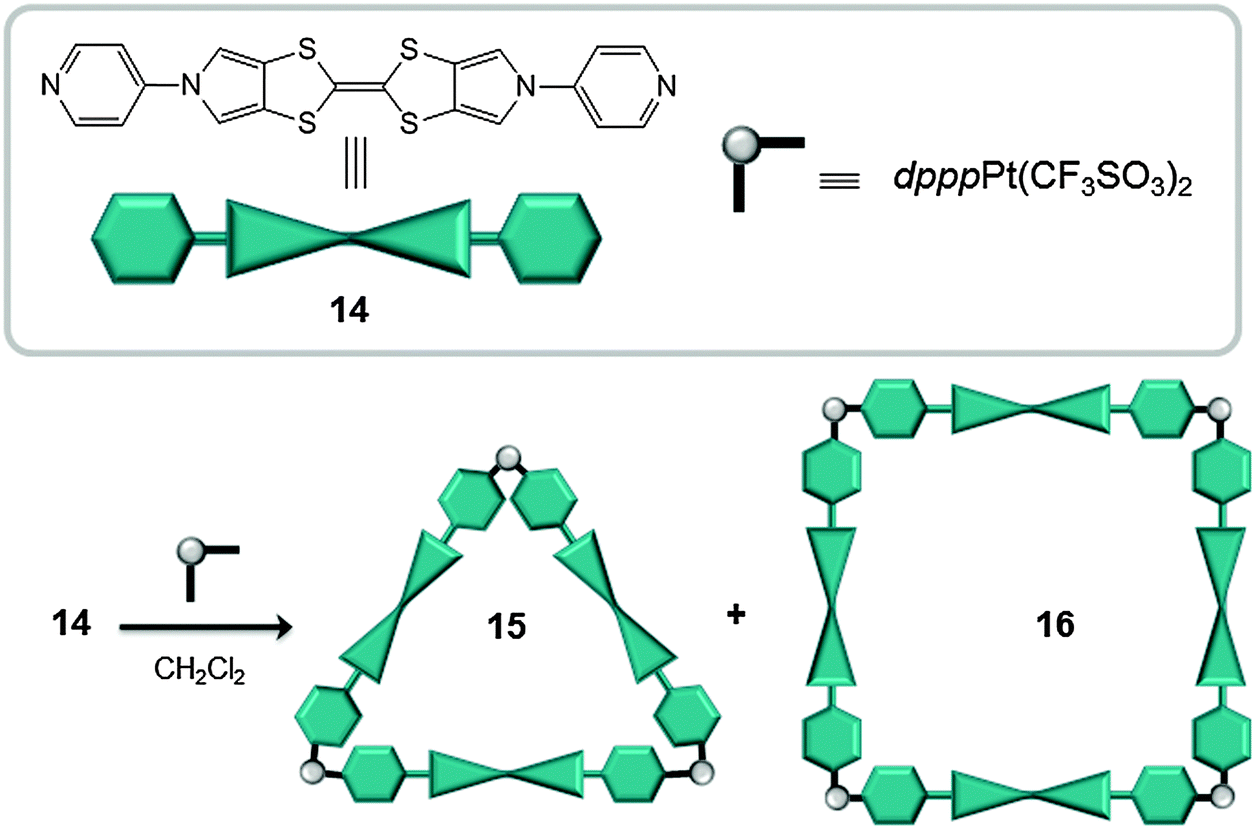 | ||
| Fig. 8 Chemical formula and schematic representation of ligand 14 (top) used for the self-assembly of coordination polygons 15 and 16 (bottom).34 | ||
In parallel studies Chi, Stang and Mukherjee worked on the synthesis of a 2D-metallamacrocycle that was also able to bind C60. The 2D coordination receptor was obtained from [2+2] self-assembly of a carbazole-based 90° dipyridyl donor 3,6-di(4-pyridylethynyl)carbazole ligand (17) and a cis-capped (dppf)Pd(CF3SO3)2 complex (Fig. 9).35 The host 18 was prepared under mild conditions when an equimolar mixture of ligand 17 and cis-(dppf)Pd(CF3SO3)2 was stirred for 5 h in nitromethane at 50 °C. Crystals suitable for single X-ray diffraction were obtained and the resulting structure showed a bowl-shaped conformation which is optimum for the interaction with the convex surface of fullerene (Fig. 9b). During fluorescence titration of the C60⊂18 adduct, the intensity of the band corresponding to 18 (λmax = 372) depleted rapidly upon addition of C60 (Fig. 9c). This quenching effect was rationalized by the formation of the C60⊂18 inclusion compound. Analysis of the fluorescence titration data gave a value of the association constant of ∼105 M−1, which is indicative of the formation of a stable host–guest adduct.
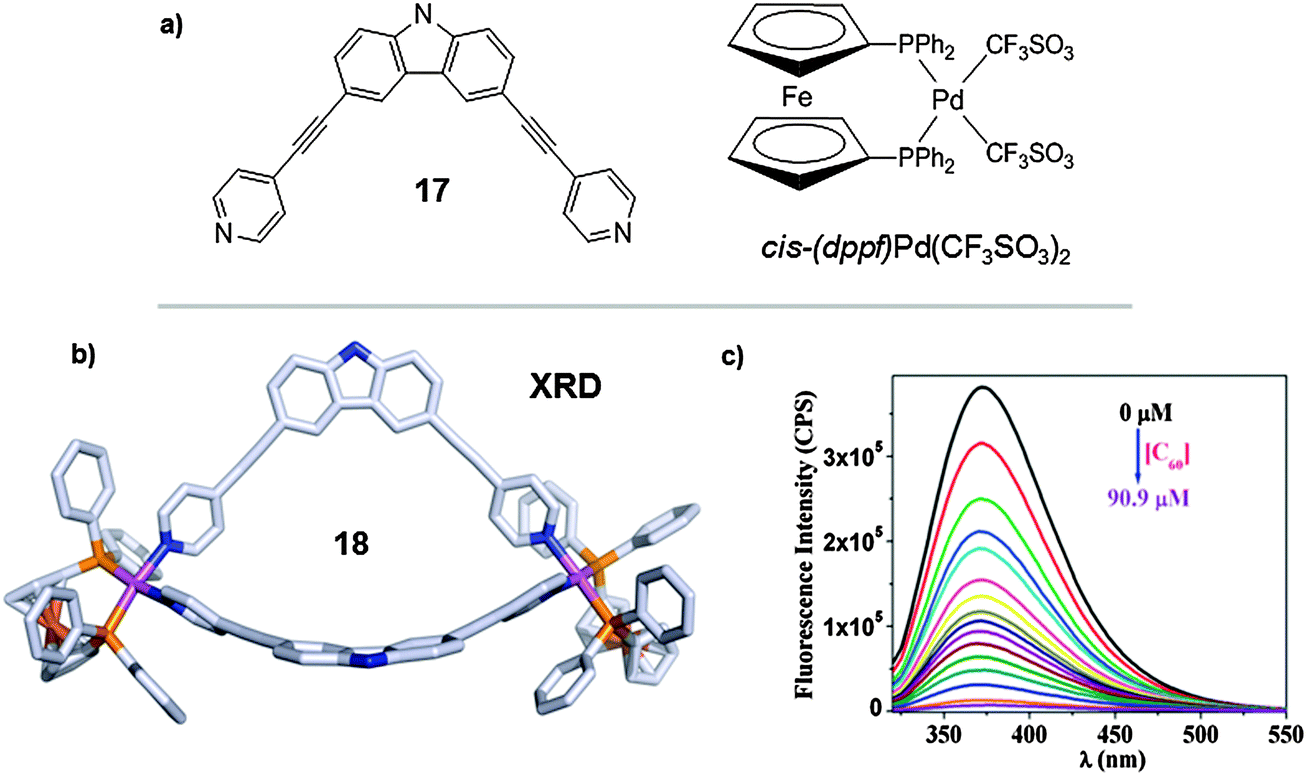 | ||
| Fig. 9 (a) Chemical formulae of compounds 17 and the cis-capped Pd(II) complex, cis-(dppf)Pd(CF3SO3)2. (b) Self-assembled coordination bowl 18. (c) Quenching of the emission intensity of macrocycle 18 upon the gradual emission of C60 fullerene. Adapted with permission from ref. 35. Copyright (2012) American Chemical Society. | ||
2.2 π-Extended systems
Recently, Yoshizawa and co-workers used a pyridine curved ligand 19, bearing large aromatic panels (two anthracene molecules linked by m-phenylene) and silver(I) as metal for the coordination-driven self-assembly of the tube-like receptor 20.36 The tubular host 20 was exclusively formed upon reaction of ligand 19 and AgNO3, in the presence of C60 in acetonitrile for 1 hour at room temperature in the dark, whereby the colour of the solution changed from yellow to red-purple (Fig. 10). C60 plays the role of a template molecule and as a result 1:1 host–guest adducts were formed. Silver(I) anions adopted a linear geometry forming complexes with two ligands (M2L2). Thanks to its open structure, receptor 20 was also able to encapsulate different C60-malonate and C60-indene derivatives, although no Ka was reported.
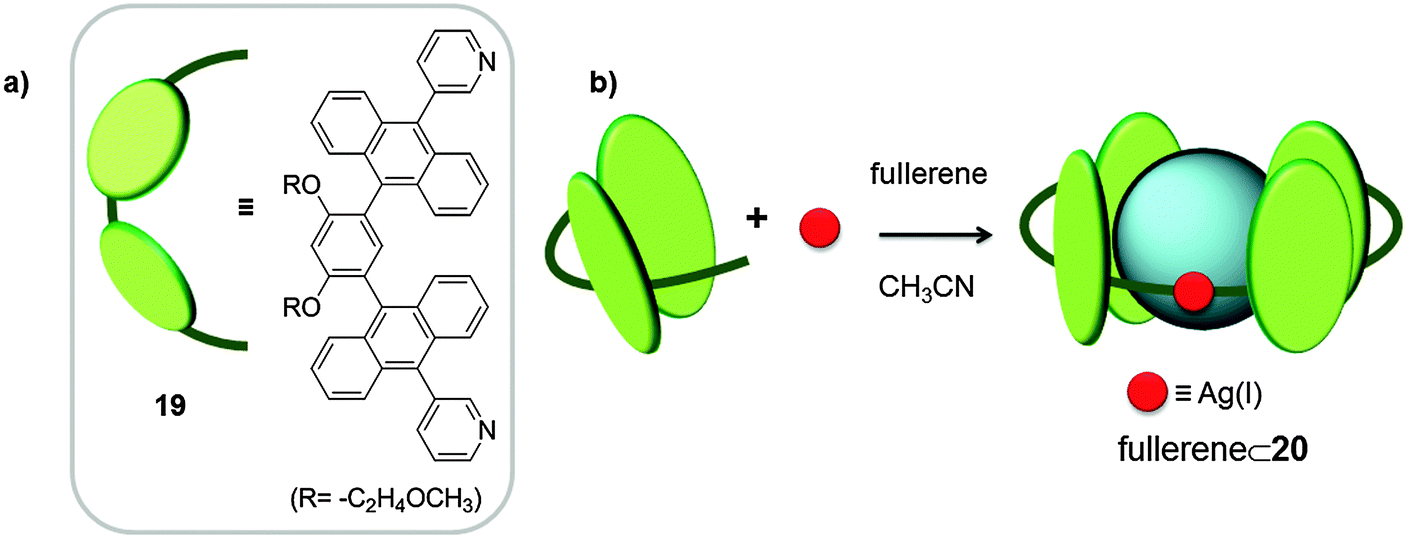 | ||
| Fig. 10 Chemical formula and schematic representation of ligand 19 (a) used for the self-assembly of tubular receptor 20 (b).36 | ||
3. 3D metallocages to entrap fullerenes
In contrast to bi-dimensional metalloreceptors, three-dimensional hosts offer a more confined environment to accommodate and isolate fullerenes. Besides, more ligand units can be incorporated in the structure further extending the receptor–fullerene surface contact, which translates into much higher association constants (Ka up to 108). As previously mentioned, the metal–ligand interaction allows for the construction of fullerene receptors in a straightforward manner, and it permits the fine-tuning of the receptor geometry or solubility. Additionally, coordination 3D structures present a more flexible and adaptable structure than the covalent ones, facilitating the interaction with the fullerene substrate. For all these reasons, attention of several research groups has been directed towards the use of the coordination-driven self-assembly approach to build sophisticated 3D cage-like receptors capable of catching fullerenes and herein, some examples will be highlighted.33,37–483.1 Coordination cage-like receptors containing π-extended moieties
Different π-extended systems such as calixarenes and resorcinarenes have also been used as molecular building blocks in the preparation of coordination cages for the isolation of fullerenes.37 As for the case of CTV units, these species can also establish a donor–acceptor interaction with fullerenes.One of the first examples of a coordination 3D cage-like receptor for fullerenes was reported in 1999 by Shinkai.37 The authors targeted the synthesis of a dimeric calix[n]arene cage for fullerene inclusion, by self-assembly through coordination bonds. Calixar[3]arenes units substituted with 3 pyridine moieties were reacted with [PdII(Ph2PCH2CH2PPh2)](CF3SO3)2 in a 2:3 ratio, to form the molecular cage 21 (Fig. 11a). The nanocapsule forms 1:1 host–guest adducts with C60 at 25 °C in 1,2-dichloroethane (Ka ∼ 50 M−1). Afterwards it was discovered that the Ka for C60 remarkably increased when lithium cations were bound to the ester lower rims of capsule 21, giving a new complex Li–21.49 In the Li–21 capsule the phenyl groups are more flattened than those in 21, in such a way that the three ethoxycarbonylmethoxy groups can interact with the bound lithium cation, improving the interaction between the fullerene and the calixarene walls (Ka ∼ 2 × 103 M−1 in 1,2-dichloroethane at 25 °C). Interestingly, when the bigger sodium cation interacts with the ester pockets to give Na–21, the phenyl groups stand up, which tapers the cage structure and as a consequence the cage adopted an ellipsoidal shape which impeded the inclusion of the C60 fullerene (Ka ∼ 5 M−1 in 1,2-dichloroethane at 25 °C) (Fig. 11b). This modification of the cage shape and size induced by cation binding was revealed as a good strategy for the controlled binding and release of encapsulated C60.
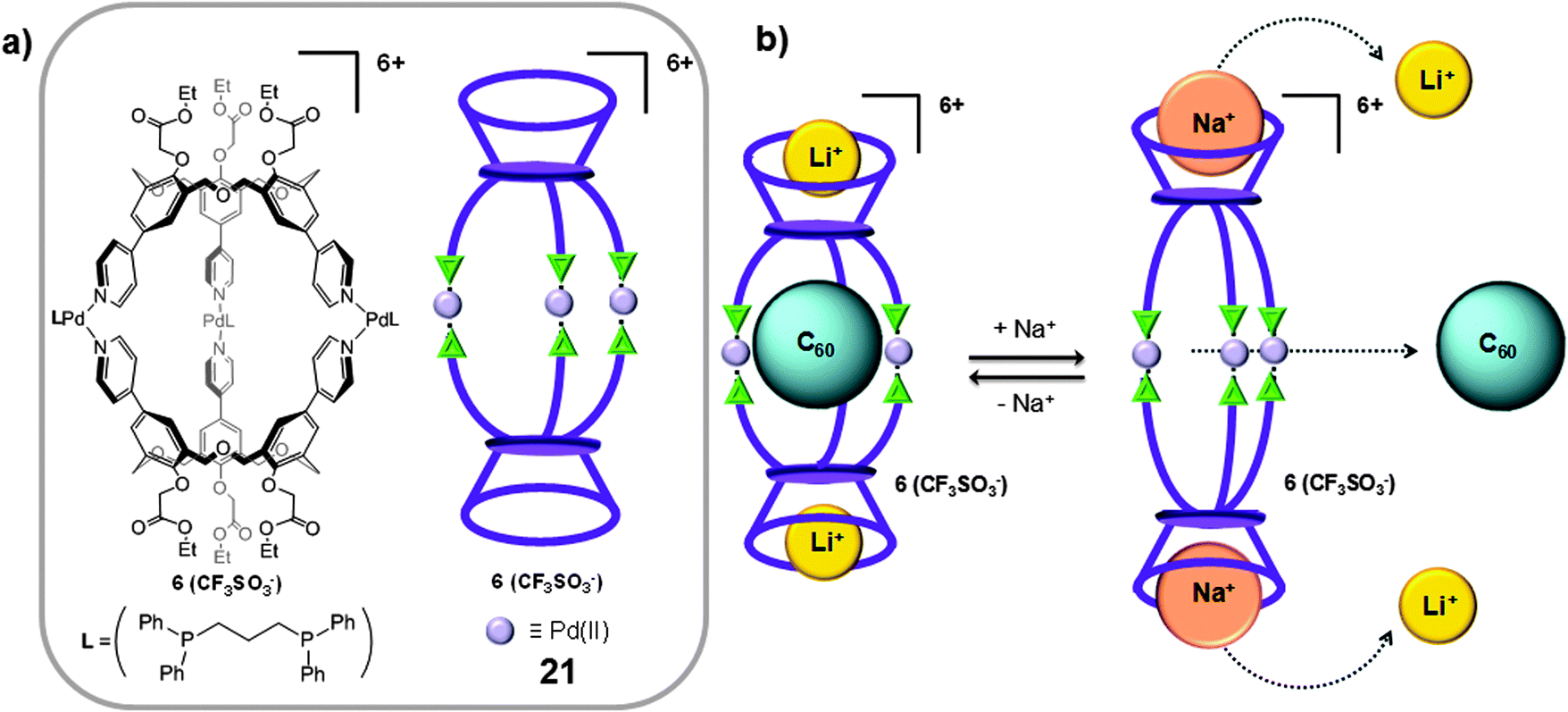 | ||
| Fig. 11 (a) Chemical structure and schematic representation of the coordination cage 21 reported by Shinkai and co-workers. (b) Flattening of the homocalix[3]arene units induced by lithium cations and release of C60 induced by the interaction with sodium cations. Adapted with permission from ref. 37. Copyright (1999) American Chemical Society. | ||
Taking advantage of the predictability and high directionality of the pyridine-N⋯Pd coordination bond, and using subphthalocyanine substituted with pyridines (22) as building blocks, Torres reported on the synthesis of a library of M3L2 metallic cages suitable for the encapsulation of fullerenes (Fig. 12). Subphthalocyanines (SubPcs) have a concave geometry comprising a 14 π-electron core with the ability to interact with fullerenes.50 The synthesis of metallocages with C3 symmetry (23a–e) was performed by mixing stoichiometric amounts of SubPc (22) and the corresponding palladium(II) or platinum(II) complex and stirring the solution in DCM under reflux. First a cage containing ethylenediamine as a ligand and palladium(II) as the metal was reported (23a). In solution the cage revealed the presence of two diastereoisomers which slowly exchange until reaching a thermodynamic equilibrium. Cages 23b–e were prepared by self-assembly of palladium(II) or platinum(II) metal ions and phosphine ligands, which in turn allowed their characterization by 31P NMR. The host–guest adduct between capsule 23a and C60 was formed by adding 5 equivalents of fullerene to a previously equilibrated solution of the host in acetone at room temperature. The formation of 1:1 (23a⊂C60) complex was confirmed by NMR and ESI-MS experiments. The encapsulation in acetone dramatically increases the solubility of the fullerene in this solvent, which makes the capsule of potential interest as a phase-transfer catalyst for C60. Host–guest experiments with cages 23b–e under the same conditions (acetone, r.t.) also gave 1:1 host guest adducts with C60, C70, [6,6]-phenyl-C61-butyric acid methyl ester ([60]PCBM) and [6,6]-phenyl-C71-butyric acid methyl ester ([70]PCBM) fullerenes. These experiments were also performed following a fullerene template approach.38 Initially, SubPc (22) was stirred with the fullerene for one hour and subsequently the metal complex was added. The association constants for [60]PCBM and [70]PCBM were calculated by monitoring the changes in the NMR spectra. Unfortunately, for the palladium(II) cages the equilibrium was too fast and no significant shifts were observed upon addition of the fullerenes. For the platinum(II) analogues (23d–e), the Ka for 23d was found to be 4.6 × 104 M−1 and 1.5 × 105 M−1 for [60]PCBM and [70]PCBM, respectively, and 3.2 × 102 M−1 and 2.2 × 103 M−1 for [60]PCBM and [70]PCBM for 23e. [70]PCBM forms more stable adducts with the capsules because its size is more appropriate for interaction with the cage.
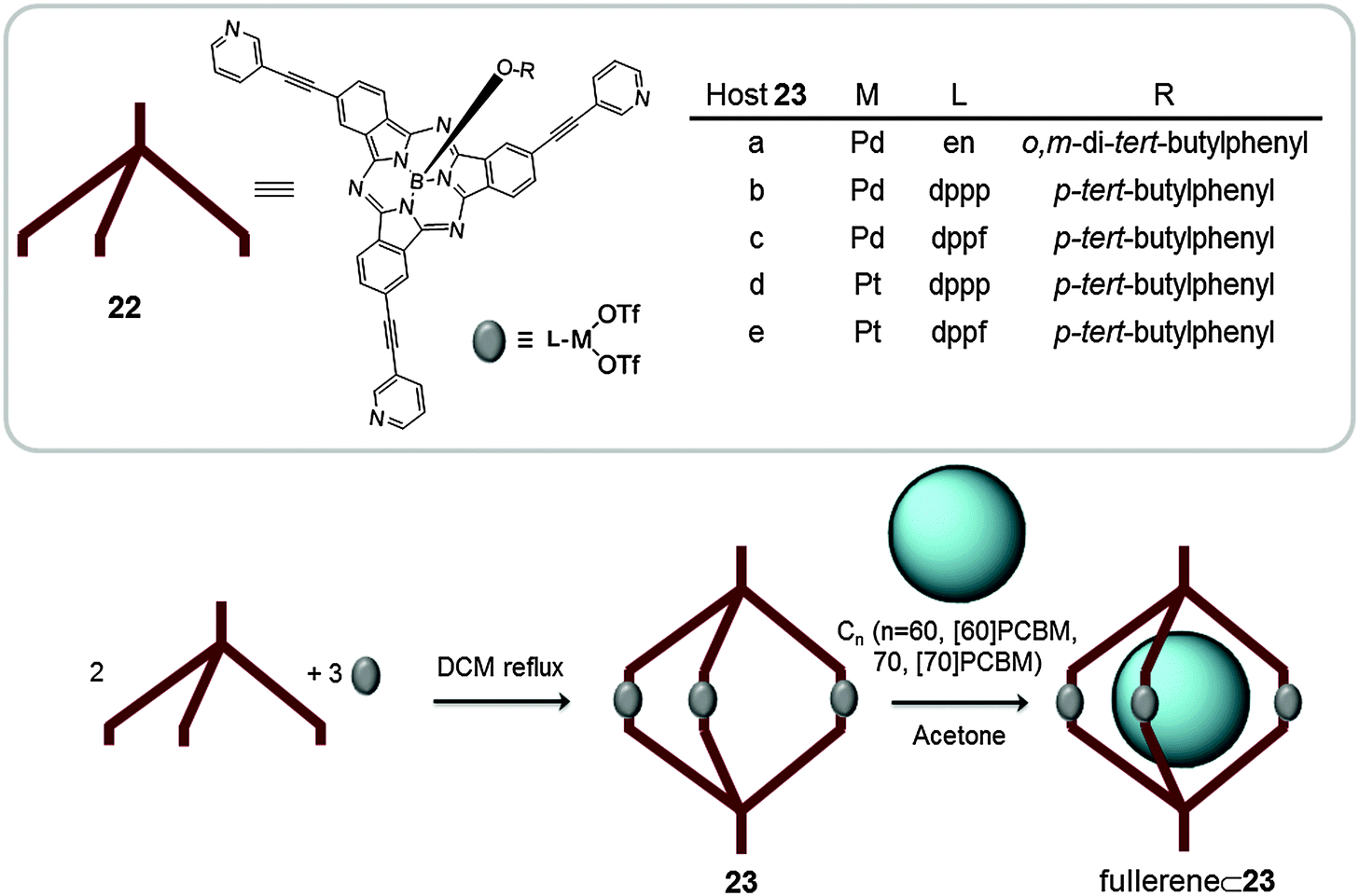 | ||
| Fig. 12 Representation of the self-assembly reaction between SubPc (22) and the corresponding metal complexes to give the coordination cage 23. Cages 23a–e encapsulate different fullerenes in acetone (en = ethylenediamine, dppp = 1,1′-bis(diphenylphosphino)propane and dppf = 1,1′-bis(biphenylphosphino)ferrocene).38 | ||
Yoshizawa and co-workers moved from 2D receptors (Fig. 10) to 3D receptors by changing the metal ions used for the self-assembly reaction (Fig. 13). Unlike silver (I) ions, palladium(II) adopted a distorted square planar geometry with four ligands (M2L4). The coordination 3D cage 24 was obtained by self-assembly of ligand 19 (see Fig. 10) and Pd(NO3)2 in DMSO at 100 °C.39 The large aromatic panels (two anthracene molecules linked by m-phenylene) of ligand 19 provided an aromatic shell that possesses an enclosed cavity which can facilitate stronger host–guest interactions in comparison with receptor 20 (Fig. 10 and 13a). Interestingly the cage cavity was large enough (∼1 nm in diameter) for the encapsulation of large guest molecules. The reaction between cage 24 and an excess of C60 in acetonitrile at 80 °C for 3 h, gave 1:1 host–guest complexes. The palladium(II) based cage irreversibly binds C60 through π-stacking interactions (Fig. 13b). Remarkably, cage 24 showed selective molecular recognition abilities and could only encapsulate C60 of complementary shape and size, from a mixture of C60 and C70.
 | ||
| Fig. 13 (a) X-Ray crystal structure of the Pd(II)-based molecular capsule 24. (b) Schematic representation of the C60⊂24 host–guest adduct.39 (c) Mercury(II)-based cage 25 and tube 26 reported by Yoshizawa.40 | ||
Afterwards, the same authors reported on the synthesis of a new cage 25 analogous to 24 in which mercury(II) metal ions were used as a nexus.40 This M2L4 capsule easily interconverts to a tube-like structure 26 (analogous to 20) in response to modulation of the metal-to-ligand ratio (Fig. 10 and 13c). When ligand 19 and Hg(CF3SO3)2 were mixed in a 2:1 ratio in acetonitrile at room temperature for 5 min, nanocapsule 25 was obtained. On the other hand, if the curved ligand 19 and Hg(CF3SO3)2 were mixed in a 1:1 molar ratio under similar conditions, 2D receptor 26 was obtained. Both structures were fully characterized by means of NMR, ESI-MS and XRD analyses. Fast interconversion between the cage-like structure 25 and the tube 26 was achieved by changing the metal-to-ligand ratio, by the addition of further equivalents of the ligand or the metal ion. When 2 equivalents of the mercury(II) salt were added to 1 equivalent of cage 25 (M2L4), the cage changed its conformation to give two units of tube 26 (M2L2) in 15 min at room temperature in acetonitrile. Additionally, tube 26 was quickly transformed into cage 25 by adding 2 equivalents of ligand 19 for each equivalent of tube. In spite of the fact that both receptors 25 and 26 possess large inner cavities with similar diameters (∼1 nm) they present different affinities towards fullerenes. When a colourless solution of capsule 25 in acetonitrile was mixed in a 1:3 ratio with fullerene C60 at room temperature for 15 min, the solution turned blue-violet owing to the formation of the C60⊂25 host–guest adduct. Cage 25 was also able to encapsulate C70, however as it was observed for cage 24, a better fit of C60 fullerene inside the cavity resulted in higher association constants than for C70. Unexpectedly, tube 26 showed no affinity towards C60 or C70 and the authors reasoned that in the case of the analogous tube-like structure 20, the fullerene⊂receptor interaction was possible due to the presence of Ag–π interactions, whereas those interactions were absent for the Hg(II) analogue.
Very recently, the same authors tuned the length of ligand 19 by replacing the phenylene spacer for a naphthalene spacer (Fig. 14);41 the elongated ligand 27 formed a new elliptical cage (28) when it was reacted with [PdCl2(DMSO)2] and AgNO3 in a 4:2:5 ratio in DMSO, at 100 °C for 2 h. The Pd⋯Pd distance was enlarged from 13.36 Å in cage 24 to 16.1 Å in cage 28, while the widths of the capsules's cavities were quite similar (∼10.3 Å). When cage 28 was suspended in acetonitrile together with C70, a stable 1:1 host–guest adduct was obtained. The anisotropic expansion of the cage enhanced its host capability, and the elliptical shape of the cavity which is complementary to the elliptical form of C70 optimized the interaction between the two species.
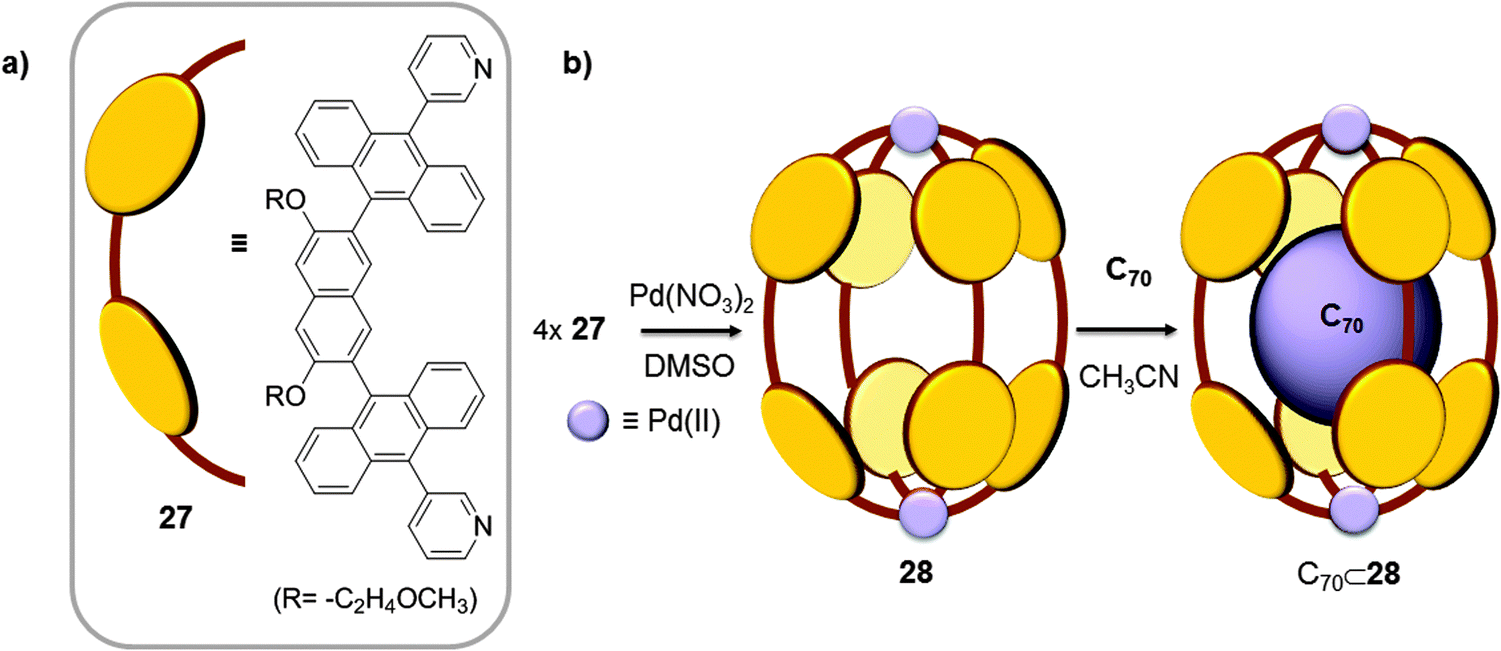 | ||
| Fig. 14 Chemical formula and schematic representation of ligand 27 (a) used for the self-assembly of cage 28 (b), reported by Yoshizawa which displays selectivity towards C60 fullerene.41 | ||
First row transition metals, which are attractive from environmental and economic points of view, have also been used as a nexus between pyridine ligands to give cage-like receptors for fullerenes. Würthner used FeX2 salts (X = BF4−, CF3SO3−) to link perylene bisimide (PBI) dyes, containing 2,2′-bipyridine groups (29), in order to prepare a M4L6 tetrahedral structure 30 (Fig. 15), which is one of the largest M4L6 tetrahedra ever reported.42 As a result of the known electronic interaction between C60 and the PBI units and considering the large volume of the cage cavity (∼950–2150 Å3), nanocapsule 30 was reacted with an excess of C60 (1:10 ratio) in a CH3CN/CHCl3 mixture (9/1) and the reaction crude was heated to 70 °C overnight. Afterwards the solvents were removed and the remaining mixture was dissolved in CH3CN and filtered to separate the excess of C60 that is insoluble in CH3CN. The HRMS spectrum displayed peaks corresponding to 30, C60⊂30 and 2·C60⊂30 host–guest adducts (Fig. 15b and c). Molecular force field (MMFF) geometry optimization of singly and doubly occupied host–guest complexes indicated that the fullerenes are preferentially located closer to the corners of the tetrahedral cage.42
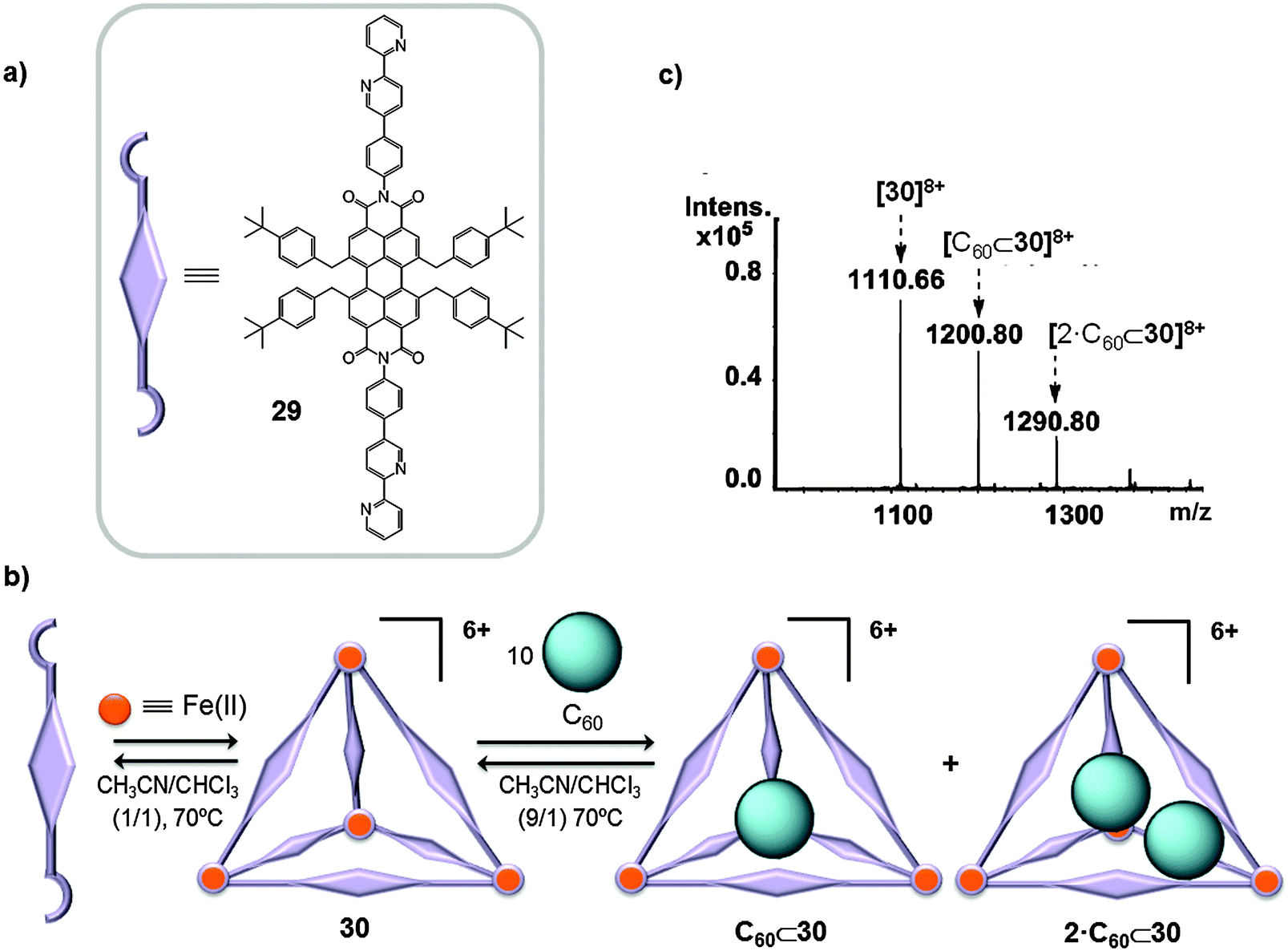 | ||
| Fig. 15 (a) PBI-based ligand 29 used in the self-assembly of the giant tetrahedral host 30 (b) which was able to accommodate one or two molecules of C60 fullerene. (c) ESI-TOF mass spectrum of 30, C60⊂30 and 2·C60⊂30 (CH3CN). Adapted with permission from ref. 42. Copyright (2013) American Chemical Society. | ||
Recently, Nitschke reported on the synthesis of a Fe(II)-based M6L6 tetragonal cage by self-assembly of diamine 1,6-pyrene-based ligands (6 equiv.) with 2-formylpyridine (12 equiv.) and iron(II)triflimide (4 equiv.). It was found that the resulting cage product consisted of a mixture of three diastereoisomers.43 Interestingly, both fullerenes C60 and C70 were observed to form 1:1 host–guest adducts with the tetrahedral cage, moreover the cage was able to adapt its structure when binding the fullerene, favouring the formation of the diastereoisomers best able to encapsulate the fullerene in order to maximize the cage–fullerene interaction.
A different strategy was used by Fujita and co-workers who, in the context of metal–organic frameworks (MOFs), developed crystalline sponges for fullerene encapsulation. Previously, the same authors had reported on the synthesis of multiple positively charged nano-vessels by self-assembly of rigid tridentate pyridine ligands through coordination with palladium(II) and platinum(II) (Fig. 16a). The corresponding M6L4 octahedral flasks were proven to be able to create an isolated micro-environment with properties different from those in the bulk solution and allow small organic molecules to be confined. The authors reported in 2010 on the synthesis of a crystalline coordination network, 32, generated from an infinite array of octahedral M6L4 cages (M = Co(NCS)2), 32a, in which each metal corner is shared between two adjacent octahedra (Fig. 16b and c).44 The cage framework 32a resembles the octahedral cages previously reported with palladium(II) and platinum(II) ions (31, Fig. 16a). The crystalline structure of the MOF revealed the presence of other types of cavities besides the M6L4 octahedral cage motifs. The interstitial void spaces between 32a moieties (63% of the total lattice volume) define alternating M12L8 (32b) and M12L24 (32c) cuboctahedral molecular cages, that are analogous to the palladium(II)-based discrete nanocapsules previously reported. Thus, a crystalline infinite network 32 provides three distinct molecular cage environments (32a–c).
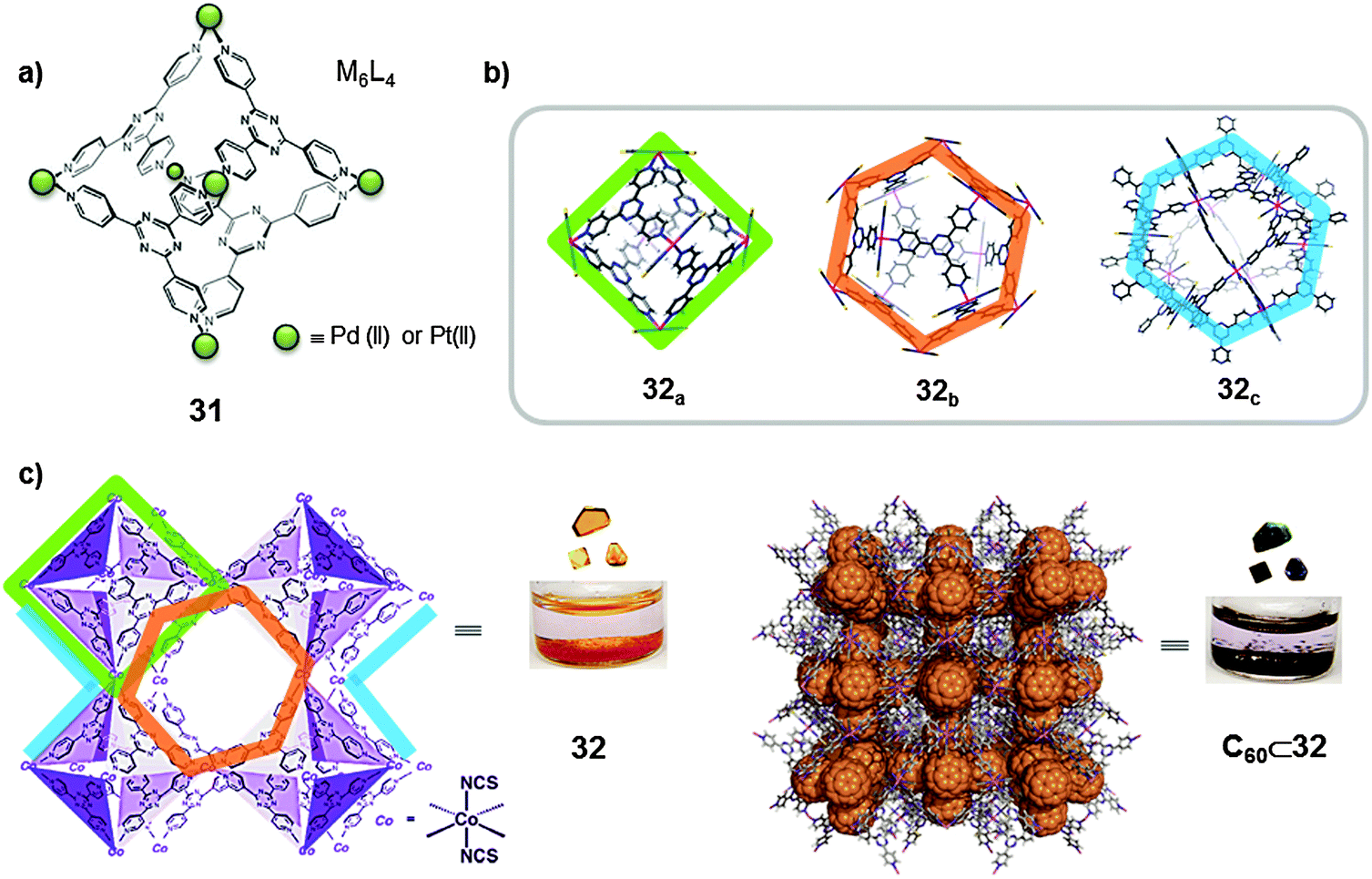 | ||
| Fig. 16 (a) Octahedral coordination Pd(II)- or Pt(II)-based capsules (31) reported by Fujita. (b) Representation of the crystal structures of the three distinct polyhedral molecular cages in the infinite crystalline network 32. (c) Inclusion of C60 into the crystalline sponge 32, and photographs of complex 32 before (yellow) and after (black) inclusion of C60. Adapted by permission from Macmillan Publishers Ltd: [Nature Chemistry] (ref. 44), copyright (2010). | ||
The octahedral cage 32a has a narrow window which allows the binding of small organic guests, i.e. tetrathiafulvalene (TTF), through solid–liquid host–guest recognition. On the other hand, the larger cuboctahedra 32b–c are of ideal size for the encapsulation of large aromatic guests such as fullerenes. To perform the encapsulation of fullerenes, a crystalline sample of MOF 32 was soaked in C60 fullerene containing toluene at 60 °C for a week. During this time the yellow crystals of 32 turned black (Fig. 16c). X-ray data were obtained for the inclusion compound and it was found that 35 wt% C60 fullerene was accommodated. An analogous experiment was performed using C70 and 34 wt% C70 was accommodated. The half-lives of the inclusion complexes C60⊂32 and C70⊂32 in toluene were found to be 15 and 25 days respectively. Cages 32b–c displayed preferential encapsulation towards C70 than for C60 (C60/C70, 8/2 ratio). The cuboctahedral cages were also used to encapsulate fullerene soot (containing ∼7% fullerenes), where interestingly the C70 content was enriched from C60/C70 = 90/10 to C60/C70 = 76/24. Moreover, the cuboctahedral cages also showed a preferential encapsulation of higher fullerenes from fullerene soot (C76, C78, C82 and C84). The content of higher fullerenes relative to C60 increased ∼3 times upon inclusion.
In the same year, Fujita also reported the synthesis of sphere-like molecular nanocapsules that are able to isolate fullerenes. The nanosphere 34 was synthesised by self-assembly of ligand 33 treated with Pd(NO3)2 in DMSO for 4 h at 70 °C, resulting in a M12L24 cage (Fig. 17). The sphere 34 contains 24 large aromatic coronene molecules concentrated inside, which behave as a “nanodroplet” able to host C60. Confined C60⊂34 is 30 times more soluble in DMSO than in toluene.45
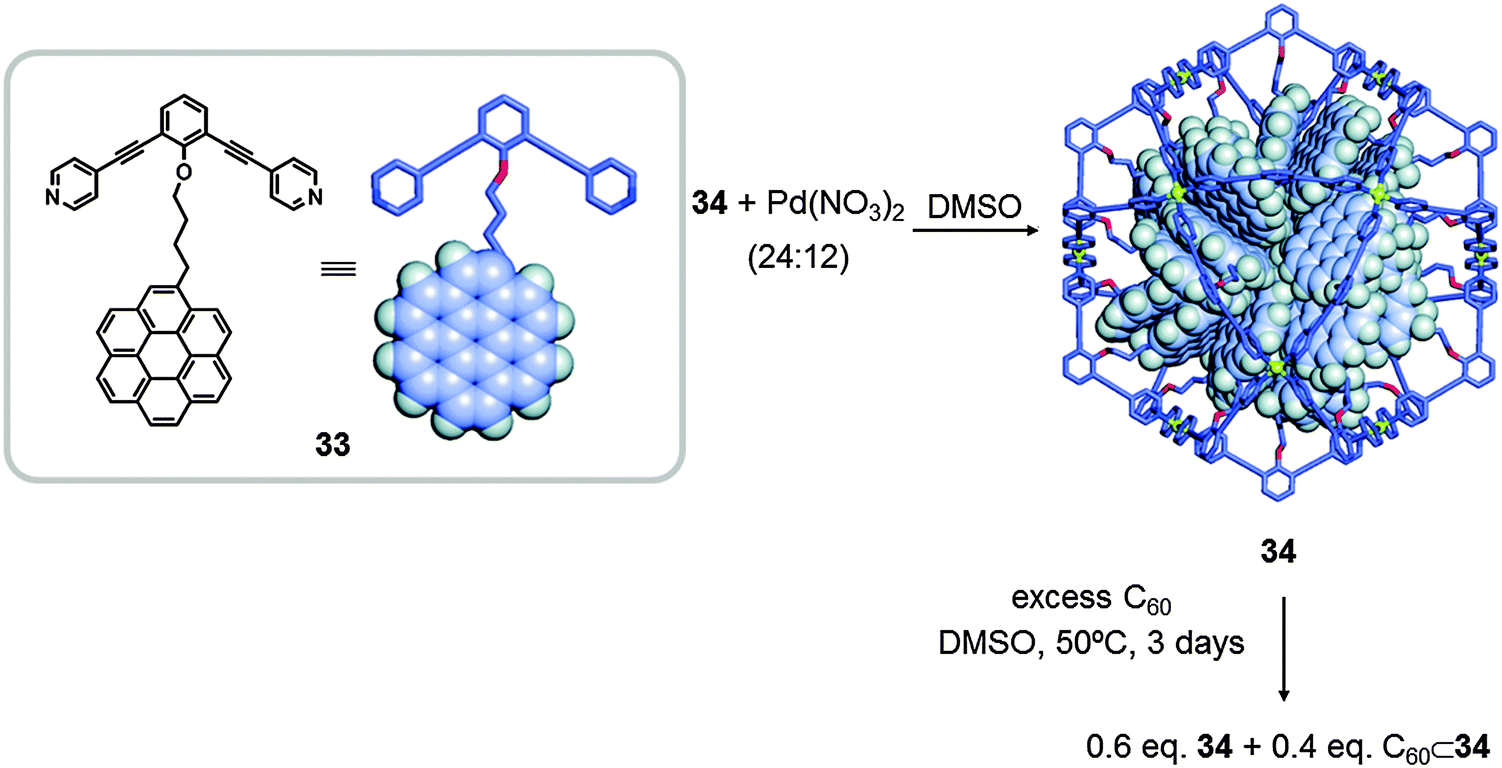 | ||
| Fig. 17 Self-assembly of M12L24 coordination sphere 34, using Pd(NO)3 and the pendant coronene molecule 34 as molecular building blocks. Adapted with permission from ref. 45. Copyright (2010) American Chemical Society. | ||
3.2 Metalloporphyrin-based coordination capsules
It has been shown that when covalent-multiporphyrin hosts are used to entrap fullerenes in solution, the association constants dramatically increase compared with the acyclic analogues. However, the covalent synthesis of these receptors is tedious, synthetically complex and often low yielding. Moreover, highly rigid structures are usually formed which in some cases can be detrimental to the host–guest interaction.23 On the other hand, dimeric porphyrin hosts linked by hydrogen-bonds have also been reported in the literature, but the Ka values observed are usually smaller than those of their covalent analogues.51 With these precedents, metal–ligand coordination receptors bearing porphyrins appear to be a promising alternative.Along these lines, Schmittel reported in 2008 the synthesis of a heteroleptic structure (M6L2L′3) prepared by self-assembly through copper(I) ions (Fig. 18). The reaction of [Cu(CH3CN)4]PF6 with ligands 35 (L) and 36 (L′) gave nanocapsule 37 which contains three zinc(II)-porphyrin units, along with some undesired oligomeric aggregates. A subsequent addition of C60 to the product mixture provided an extra driving force which shifted the reaction equilibrium towards complete formation of the desired 3D nanocapsule, which contained the fullerene within its cavity (C60⊂37).33 In this example, fullerene C60 is not just a guest molecule but it also acts as a template leading to a more stable host–guest complex avoiding the formation of undesired oligomers. Interestingly, the encapsulation of C60 into nanocapsule 37 leads to atypical proton shifts and splitting in the 1H-NMR spectrum. These changes were rationalized by a lateral distortion of the host, resembling an accordion, in order to maximize its interaction with the substrate molecule, leading to a constricted prism which strongly interacts with the fullerene (adaptive structure).
 | ||
| Fig. 18 (a) Ligands 35 and 36 used for (b) the self-assembly of capsule 37, which encapsulates C60 through its adaptive structure.33 | ||
A similar strategy was used by Shionoya who reported another example of a metallocage in which C60 fullerene acts as a template molecule, and helps to stabilize the desired product (Fig. 19).46 To perform the metal–ligand directed self-assembly reaction the authors selected zinc(II), a metal centre with a versatile coordination sphere. Despite the variety of geometries that zinc(II) can adopt, by using coordinating anions and solvent molecules, and an appropriate template molecule (C60), the authors were able to synthesize a discrete tetra-porphyrin structure ([M8L4X16]n+ X = solvent molecule or p-CH3C6H4SO3− (38)). Initially, the synthesis of cage 39 was attempted by mixing 4 equivalents of tetrakis-(bispyridyl)-porphyrin and 8 equivalents of Zn(p-CH3C6H4SO3)2, in a 1:1 mixture of CHCl3/MeOH. However, the major product of this reaction was a porphyrin trimer [M8(38)3X12]n+, which was in equilibrium with the desired porphyrin tetramer [M8(38)4X16]n+ (39). Finally, it was found that the addition of C60 was necessary to shift the equilibrium towards the formation of the C60⊂39 adduct (75%). Single crystal X-ray diffraction analysis confirmed that C60⊂39 was formed by four zinc(II) porphyrins wrapping around a single C60 molecule. The tetrameric barrel 39 was not formed when weakly-coordinating anions were used (i.e. Zn(CF3SO3)2). These results indicated that the p-CH3C6H4SO3− anions play a key role as capping ligands. Thus p-CH3C6H4SO3− molecules are able to modulate the coordination sphere of the zinc(II) anions and they control the self-assembly interaction between the metal ion and the ligands.
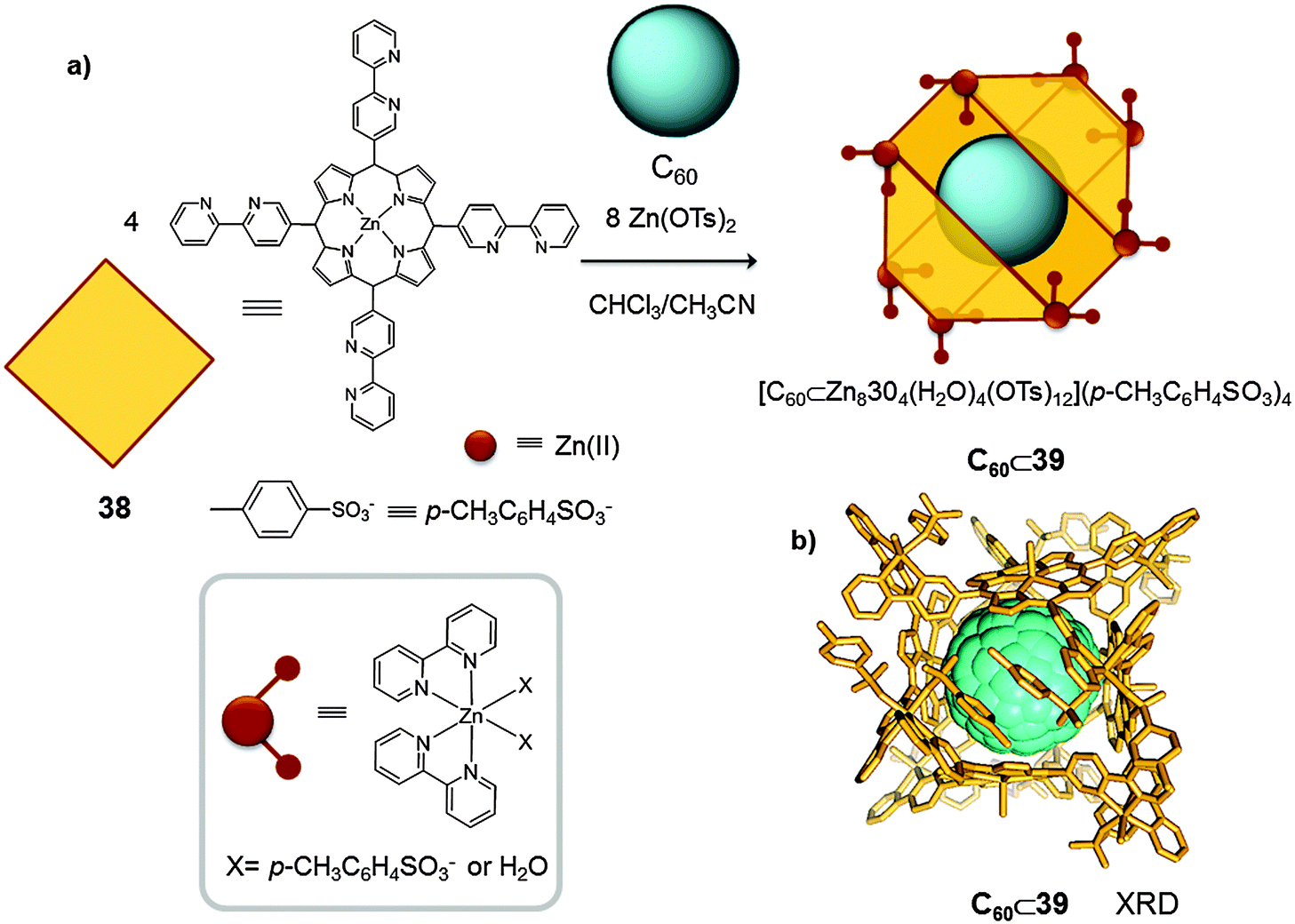 | ||
| Fig. 19 (a) Building blocks used for zinc(II) mediated self-assembly of the porphyrin barrel complex C60⊂39. (b) X-ray crystal structure of complex C60⊂39 (hydrogen atoms are omitted for clarity).46 | ||
Another example was reported by Nitschke in 2011 who synthesized a closed-face cubic metallo-cage containing six porphyrin units, which allowed for the isolation of the guest molecules from the bulk solution.47 In this work the self-assembly reaction was exclusively controlled by the metal ions (iron(II)) and the geometry of the selected pyridine-based ligands. Cage 40 was obtained by reacting nickel(II) tetrakis-(4-aminophenyl)porphyrin, 2-formylpyridine and Fe(CF3SO3)2 in dimethylformamide (DMF) (Fig. 20). The cage was obtained as a single product which was confirmed by NMR spectroscopy and ESI-MS. Crystals suitable for X-ray diffraction were obtained and the crystallographic data revealed the cage to have an octahedral geometry, containing six porphyrins on its faces and eight iron(II) centres located at the corners (M8L6). The volume of the inner cavity was 1340 Å3, large enough to host large aromatic guests such as fullerenes.
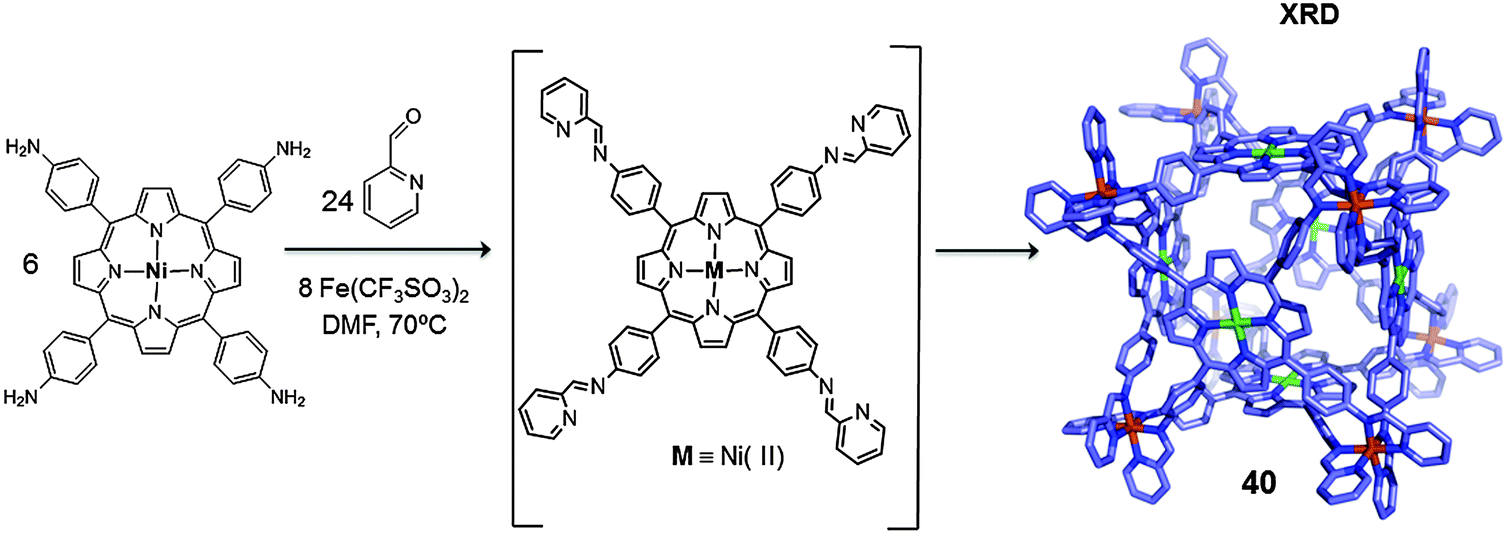 | ||
| Fig. 20 Schematic representation of the synthesis of cubic cage 40 through component self-assembly and its X-ray structure (hydrogens and anions are omitted for clarity).47 | ||
The potential of cage 40 to encapsulate fullerenes was first explored with C60. Cage 40 and C60 (5 eq.) were mixed in DMF and the reaction mixture was then heated at 70 °C for 5 days. The reaction resulted in a 35% conversion to the C60⊂40 adduct (Ka = 5.5 × 103 M−1). In a subsequent experiment 2 equivalents of C70 were added to a solution of the cage in DMF and full conversion to the C70⊂40 adduct was obtained after stirring the reaction mixture for 3 h at 70 °C. These experiments showed a higher affinity of 40 for C70 than for C60, which can be rationalized by the larger surface area of the former which facilitates a larger number of stabilizing π–π interactions (Fig. 20). To further investigate these findings, an experiment in which 2 equivalents of C70 were added to a C60⊂40 solution resulted in complete conversion towards C70⊂40 as confirmed by ESI-MS. Nanocapsule 40 was then mixed with fullerene soot (C60 53%, C70 1.54%, higher fullerenes 0.14%) in a 1:10 weight ratio, and after stirring the mixture for 12 h at 70 °C, neither 40 nor C60⊂40 compounds were observed in the ESI-MS spectrum. However, host–guest adducts with C70, C76, C78, C82 and C84 fullerenes were observed. Although mass spectra are unable to provide quantitative information, comparable mass ionization behaviour was found for 40 and its fullerene adducts. Therefore from the ESI-MS experiments it was concluded that cage 40 displayed a higher affinity towards higher fullerenes.
Very recently, García-Simón et al. reported the synthesis of a tetragonal prismatic coordination cage (42), built by metal-directed self-assembly of two tetra-carboxylated Zn(II) porphyrins and four dipalladium(II)-based macrocyclic synthons Pd–41 (Fig. 21).48 The coordination nanocapsule 42 was fully characterized by NMR, HRMS and UV-Vis spectroscopy. Moreover, crystallographic data for the cationic form of the capsule were obtained by synchrotron X-ray resolution (Fig. 22).
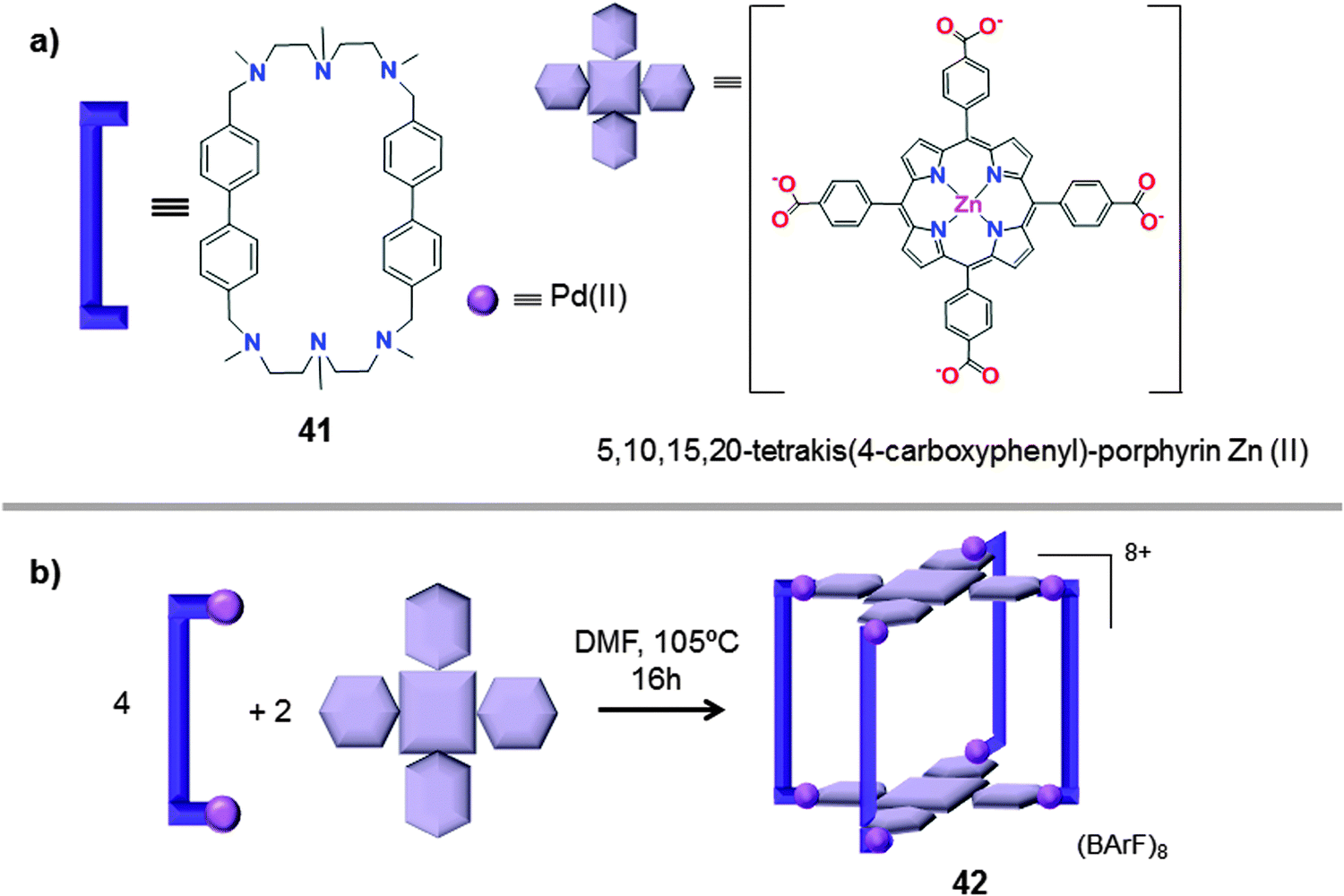 | ||
| Fig. 21 Building blocks (a) used for the self-assembly (b) of the coordination nanocapsule 42 (BArF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate).48 | ||
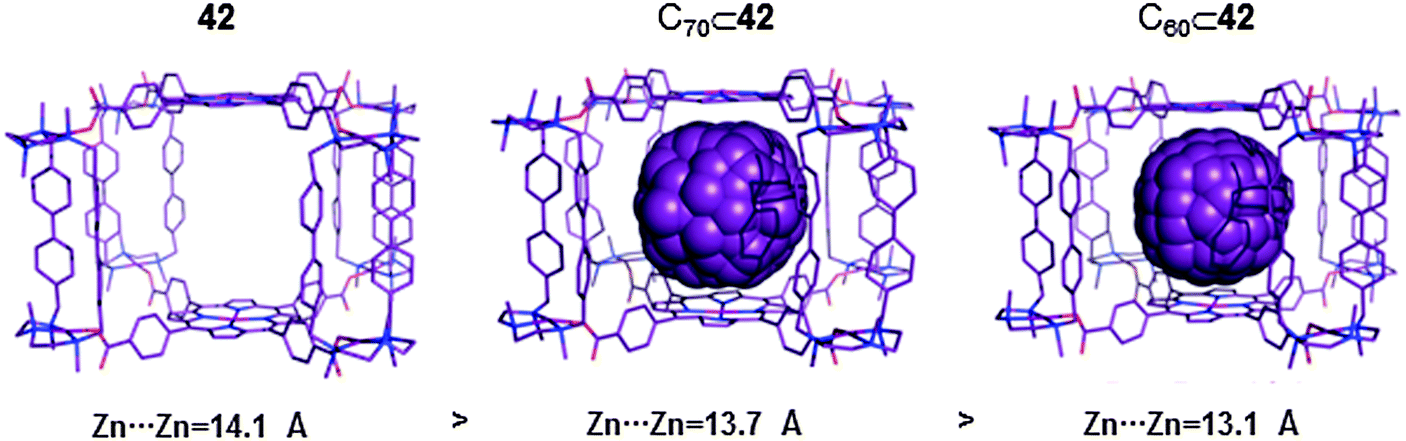 | ||
| Fig. 22 Crystallographic data for the cationic capsule 42 (left) and it corresponding fullerene inclusion compounds C70⊂42 (middle) and C60⊂42 (right). In all structures hydrogens have been omitted for clarity.48 | ||
The large void volume of the cage cavity (∼696 Å) prompted the authors to explore the possibility to encapsulate fullerene molecules. Delightfully, fast inclusion of C60 and C70 fullerenes occurred during the mixing of a solution of capsule 42 in acetonitrile and the corresponding fullerene in toluene in a 1:1 molar ratio. The data obtained from the UV-vis and fluorescence titration experiments indicated that the capsule has high affinity towards C60 and C70 fullerenes, Ka(C60) ∼ 3 × 10−7 M−1 and Ka(C70) ∼ 4 × 10−8 M−1. Structural data for C60⊂42 and C70⊂42 were also obtained by synchrotron radiation. Interestingly, a comparison of the crystallographic data for the empty capsule 42, and C60⊂42 and C70⊂42 adducts showed that the distance between the two Zn(II) porphyrins is maximized when guest molecules are absent. Indeed, the distance is reduced upon inclusion of C70 or C60 (Fig. 22). This experimental trend that was confirmed by state-of-the-art DFT calculations indicated that the cage has the ability to adapt its structure upon guest encapsulation (breathing ability). These experimental results encouraged the authors to explore the possibility of encapsulating a bulkier [60]PCBM derivative and higher fullerene C84. NMR, UV-Vis and HRMS experiments indicated that 1:1 host–guest adducts were obtained with both substrates. Fast inclusion was also observed when fullerene mixtures, fullerene extract (C60 70%, C70 28%, higher fullerenes 2%) or fullerene soot (C60 53%, C70 1.54%, higher fullerenes 0.14%) were used. Host–guest adducts with the more abundant C60 and C70 fullerenes, as well as inclusion compounds of higher fullerenes such us C76, C78 and C84 were observed. More interestingly, the encapsulation of the fullerenes can be performed by simply soaking a sample of the solid capsule in fullerene containing toluene. When the solid nanocapsule was suspended in a toluene solution of C60 or C70 and after stirring for 5–10 minutes at room temperature, full encapsulation was confirmed by HRMS. The same results were obtained for a solution of fullerene extract in toluene. This solid–liquid encapsulation strategy is similar to the one reported by Fujita in which a MOF crystal was used to catch fullerenes (Fig. 16). The solid–liquid encapsulation of fullerenes within capsule 42 supports the idea that the fullerenes enter the nanocapsule through one of the four lateral apertures of the capsule, which are large enough to allow easy mobility of the fullerenes through them.
4. Release of the sequestered fullerene molecules
As it has been mentioned in this review article, there are key factors to consider when designing a molecular receptor for binding fullerenes, such as the fullerene–receptor complementarity and the receptor tunability. However, from a practical point of view it is indispensable to design a system capable of liberating the sequestered fullerenes in an efficient manner, for the selective delivery or utilization of the captured fullerenes. The controllable binding–release of the fullerene would allow obtaining reusable receptors and recovery of the fullerene molecule. Therefore, reversibility is envisioned as the key to obtain sustainable purification of fullerene molecules by using molecular host platforms. Usually, the release of fullerene causes irreversible cage disassembly or the receptor is blocked by a high-affinity secondary guest.24 For these reasons the use of host–guest systems as a means of separating mixtures of fullerenes remains a challenging task. Nevertheless, there are few methods described in the literature in which the release of the fullerene from the receptor has been achieved, and the host platforms have been recovered, in some cases with excellent yields. Examples of controlled fullerene release will be discussed in this last section.4.1 Covalent systems
Covalent receptors generally have a more robust structure than receptors based on supramolecular interactions. Consequently, harsh methodologies such as high temperatures or acidic conditions can be applied in order to release the entrapped fullerene without causing host decomposition.By using CTV-based dimeric capsule 1 (Fig. 1), Chiu and co-workers were able to selectively encapsulate C70 fullerene from a mixture of fullerenes (Fig. 23a).16 Taking advantage of the higher affinity towards C70, the authors designed an experimental protocol to isolate C70 from the fullerene extract. The separation process consists of 3 main steps: (1) formation of the C70⊂1 complex. First the molecular host 1 and the fullerene extract were mixed in a TCE solution (in a 1/6-cage/extract weight ratio), the remaining solution was heated at 313 K for 16 h; afterwards, TCE was removed by evaporation, the remaining solid was re-dissolved in DCM and the resulting solution was filtered in order to remove the fullerenes in excess. (2) Isolation of the C70⊂1 adduct. The filtrate was concentrated and introduced into a short SiO2 column, and eluted with CS2 in order to further remove any uncomplexed or dissociated fullerene. Free host 1 and its corresponding C70⊂1 adduct were recovered from the chromatographic column using DCM/hexane and DCM/MeOH as mobile phases. (3) Release of C70. Finally, the filtrate from the SiO2 column was dried and the product was re-dissolved in toluene. The toluene solution was heated up to 303 K for 12 h. The host 1 precipitated from the solution as a white solid while the liberated C70 and the residual C70⊂1 complex remained in solution. Isolated C70 was separated from the residual host–guest complex by centrifugation. The purity of the isolated C70, determined through HPLC analysis, was ∼99%. After the first release cycle ∼85% of receptor 1 was recovered, and it was used in a second cycle without further purification. The dissociation of C70 from recycled 1, following the same experimental protocol, gave C70 with ∼92% purity. To our knowledge, the amount of receptor 1 recovered from the second release cycle was not reported.
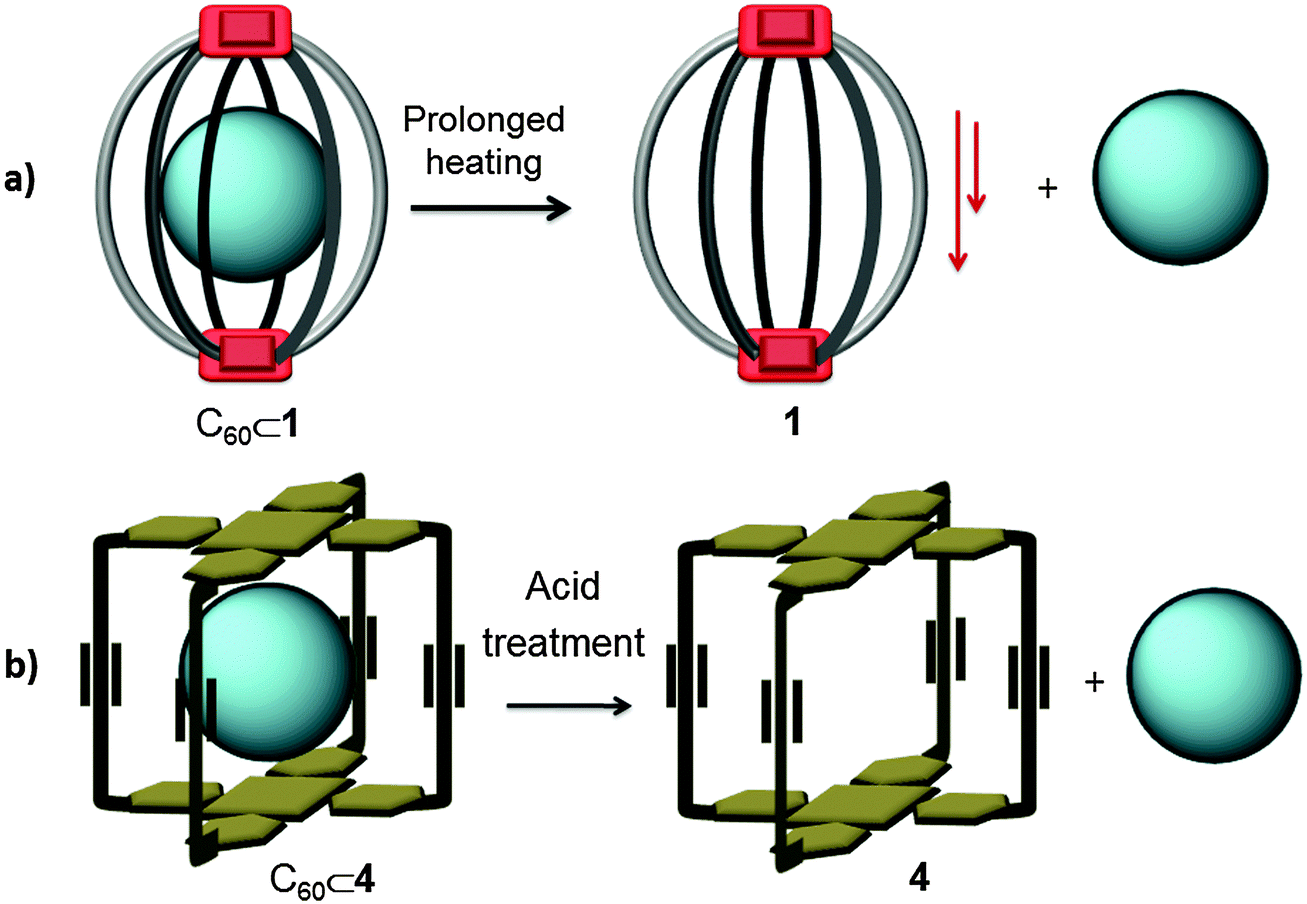 | ||
| Fig. 23 Reported strategies for fullerene release from covalent molecular receptors. (a) Prolonged heating causing the precipitation of the host.16 (b) Protonation and deprotonation of porphyrin.18 | ||
A different strategy was followed by Zhang, who demonstrated that receptor 4 (Fig. 2) presents a much higher association constant for C70 compared to C60 (Ka(C70)/Ka(C60) > 1000).18 Considering the difference in binding affinity the authors worked on the controlled selective separation of C70 from fullerene mixtures. The porphyrin–fullerene interaction could be easily tuned just by protonation of the non-metallated porphyrin. The protonation of the porphyrin reduces its electron-donating ability, and thus the porphyrin–fullerene interaction is weakened. Therefore, the fullerene association–dissociation was reversible and controlled by changing the pH of the media (Fig. 23b). The authors optimized the isolation process in three steps: (1) the cage is added to a C60/C70 (91% C60 and 9% C70) solution in CS2 and sonicated for 30 seconds; (2) then the host–guest adducts are collected in a chloroform solution, whereby upon addition of 100 equivalents of trifluoroacetic acid (TFA) to the chloroform solution, the fullerene molecules (mainly C70) are released as a black precipitate and removed. Once a cycle of the isolation process was completed, the abundance of C70 in the sample increased from 9 mol% to 79 mol%. (3) In the final step the nanocapsule was recovered by the addition of 100 equivalents of triethylamine (TEA) to the remaining chloroform solution. Remarkably, the efficiency of the protocol was demonstrated by UV-Vis experiments. The regeneration of the C70⊂4 adduct by the addition of 100 eq. of TEA to the mixture of protonated recycled 4 and free C70 was evidenced by a UV-Vis band that was almost identical than the one belonging to the initial pure C70⊂4 complex. The observation of an almost identical UV-vis spectra indicated that nearly all the cage can be recovered after fullerene release; the cycle was repeated up to 5 times.
4.2 Supramolecular receptors
Despite the fact that covalent receptors for fullerenes allow for their reversible encapsulation, generally the experimental protocols required to recover the fullerenes and the receptors are quite tedious (Section 4.1). In the case of supramolecular receptors, these can easily change their conformation, geometry (especially in the case of coordination capsules), and even molecular structure, commonly in a reversible fashion. This greater versatility (tunable capsule geometry, electronic complementarity and symmetry) facilitates the release of the entrapped fullerenes.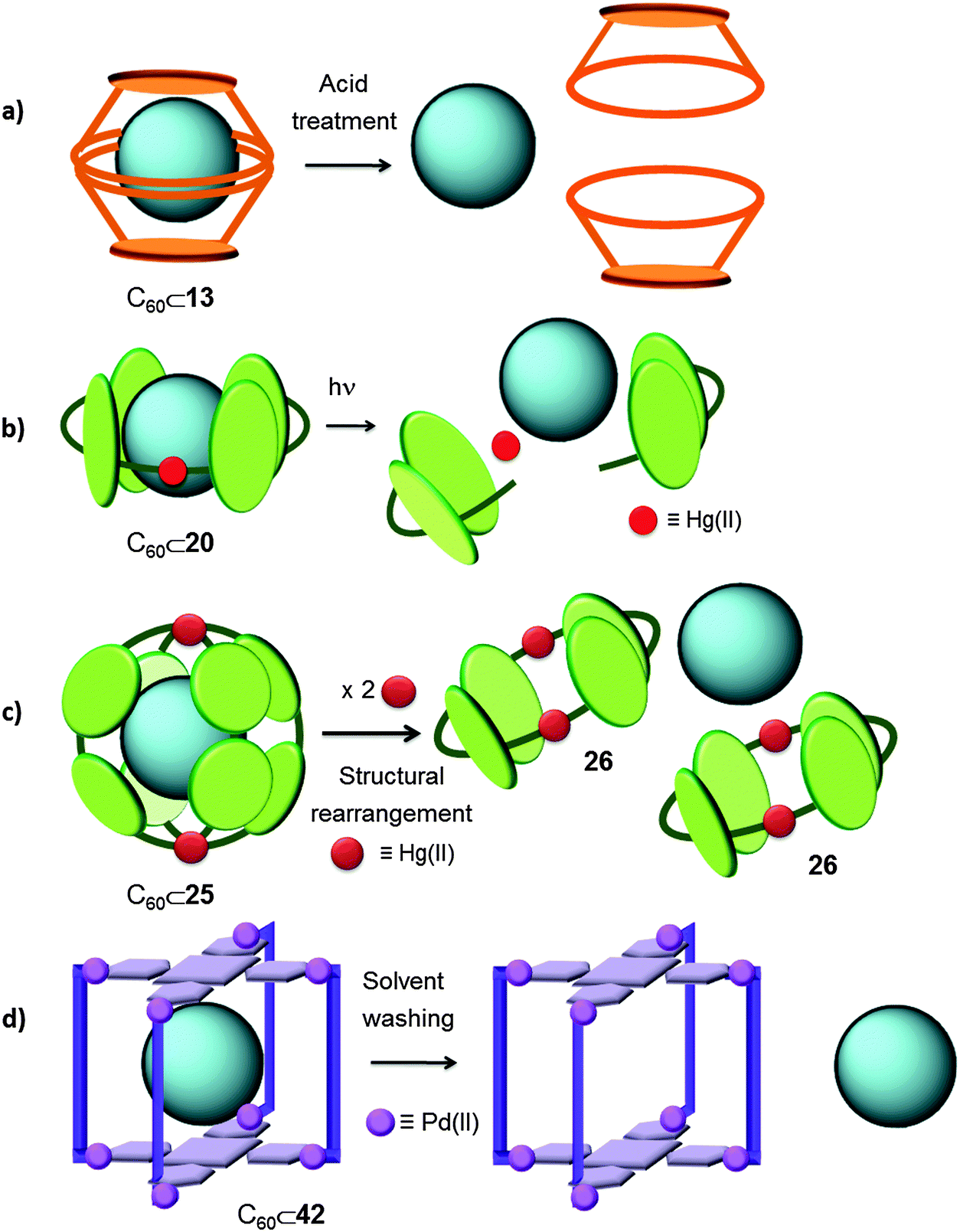 | ||
| Fig. 24 Reported strategies for fullerene release from supramolecular capsules. (a) Acid or base treatment to provoke a structural rearrangement of the host.30 (b) Photoreduction of metal ions to disassemble the metal-coordination-based supramolecular host.36 (c) Transformation of the shape of the cage in response to an external stimulus.40 (d) Solid–liquid solvent washing strategy.48 | ||
4.2.2.1 Transformable cages. Strikingly, Yoshizawa and co-workers were able to release C60 from their tubular receptor 20 by using light as a non-invasive stimulus (Fig. 10 and 24b).36 Interestingly, when a red-purple solution containing C60⊂20 was irradiated with a 36 W incandescent light (UV-vis irradiation, acetonitrile, room temperature, 1.5 h) the solution turned yellow and a precipitate corresponding to C60 and metallic silver appeared. The released C60 was extracted with toluene and recovered as a brown solid (68%). Finally, UV-vis irradiation of the remaining yellow solution containing ligand 19 in the presence of C60 and AgNO3 allowed recovery of the host–guest adduct C60⊂20 in ∼60% yield. As it has been mentioned, tube 20 displayed better affinity for C60 than for C70. Hence, when it was used to encapsulate fullerenes from commercially available fullerene soot containing only 5% C60, the designed catch-and-release procedure provided pure C60 in 63% yield based on 20.
The same authors also took advantage of the structural rearrangement strategy to liberate fullerenes from their Hg(II)-based 3D cage (25) (Fig. 13 and 24c),40 since the tube-like receptor 26 displayed no interaction with C60 or C70 while cage 25 displays affinity for both species. The stronger binding affinity of capsule 25 was ascribed to its greater number of anthracene panels interacting with the fullerene molecule and full enclathration in all the directions. Interestingly, all the fullerenes embedded in cage 25 were quantitatively liberated upon capsule-to-tube conformational transformation induced by the simple addition of metal ions (from 25 to 26). When 2 equivalents of Hg(CF3SO3)2 were added to a blue-violet solution of C60⊂25 in acetonitrile, the solution turned pale yellow, and a brown suspension of C60 was formed upon guest expulsion, which was recovered by centrifugation. Hence the conformational change and the fullerene release phenomenon could be detected by the naked eye on the basis of the change in colour. The encapsulation and release of C70 was also successfully performed using the same reaction conditions.
4.2.2.2 Solid washing strategy. Capsule 42 can encapsulate fullerenes when it is soaked as a solid in fullerene containing toluene, enabling the authors to envision that a solid washing strategy might be appropriate in order to design a straightforward experimental protocol to release the sequestered fullerenes in a selective fashion (Fig. 21 and 24d).48
The encapsulation–release processes were first explored for C60 and monitored by HRMS (Fig. 25). The designed experimental protocol consists of two main stages: (1) a solid sample of 42 was charged in a small column of Celite®. Then a solution of C60 in toluene was passed through the column until full encapsulation which was confirmed by HRMS (Fig. 25). (2) Afterwards, release of C60 was achieved by consecutive washings of the solid sample of C60⊂42 retained on the column, with a mixture of 1,2-dichlorobenzene/CS2 (1/1 v/v mixture). This solvent mixture fulfilled the requirements of both the high solubility for the fullerene and the low solubility for the 42 receptor and the C60⊂42 adduct. Washing the solid containing the fullerene five times with the chosen solvent mixture (∼1.3 mL mg−1) was sufficient to liberate all of the encapsulated C60 as confirmed by HRMS. Delightfully, the solid remaining on the column was the pure empty capsule 42, which was ready to be filled again with C60 and to repeat the cycle. The solid–liquid encapsulation–release protocol was repeated up to 5 times and finally the capsule which was retained on the column was recovered by dissolving it in acetonitrile. After 5 encapsulation–release cycles, 91% of the initial capsule was recovered which makes this strategy very efficient. Note that Fujita's MOF sponge could only liberate fullerenes by deconstruction of the crystals by acid treatment.44
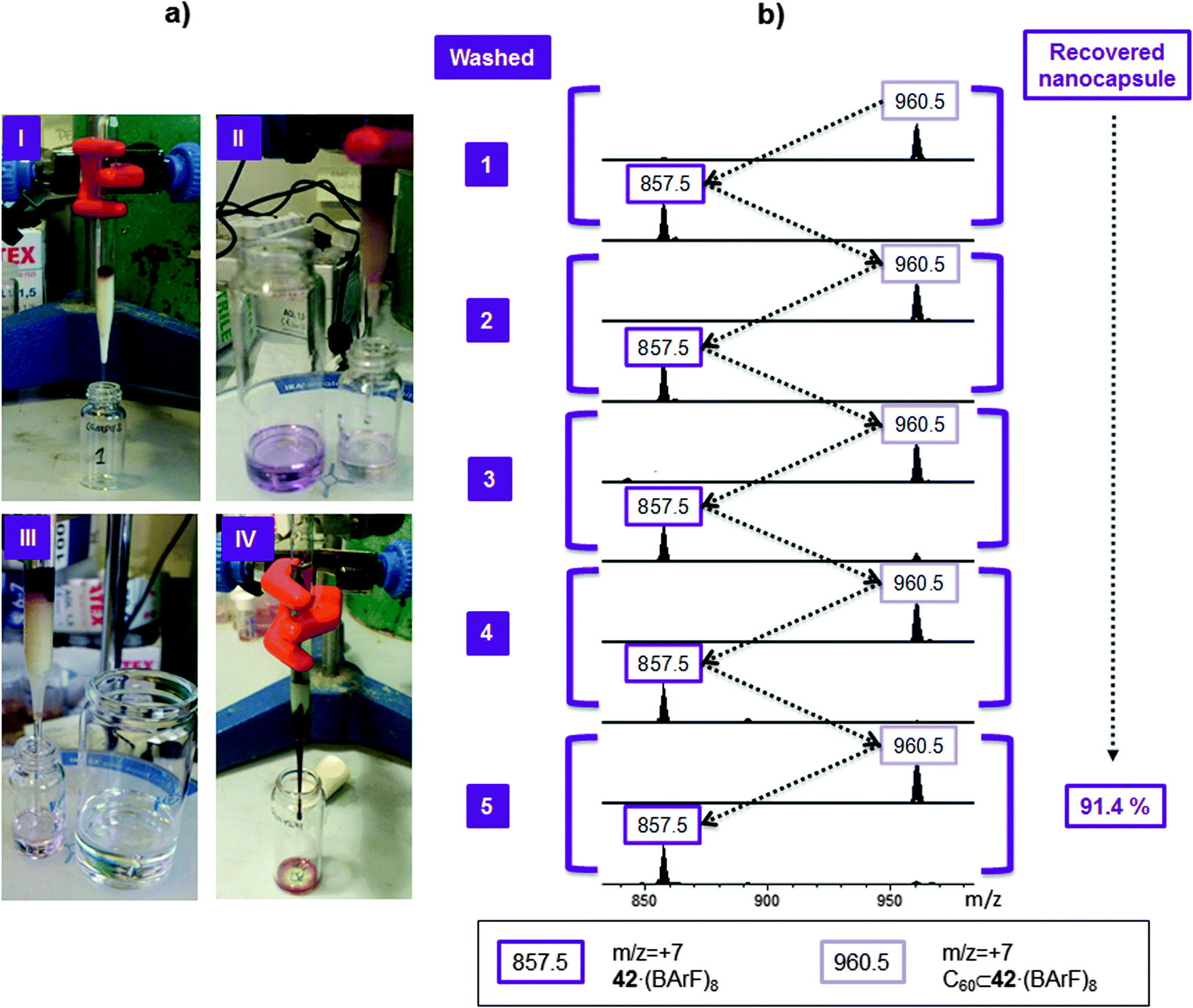 | ||
| Fig. 25 (a) Images of the experimental protocol used for the solid–liquid encapsulation and release of C60 fullerene from the solid nanocapsule 42 (I. Solid capsule retained in the Celite® column, II. A solution of C60 in toluene is passed through the column, III. C60 is released by consecutive washing with a 1/1 mixture of 1,2-dichlorobenzene/carbon disulphide and IV. Cage recovery by solving it in CH3CN). (b) HRMS monitoring of the C60 extraction washing-protocol using pure C60 encapsulated within capsule 42 in the solid state.48 | ||
The same solid washing strategy allowed selective separation of C60 from the fullerene extract (C60 70%, C70 28%, and higher fullerenes 2%). However, an ineffective mixing of the solid cage sample and the solution containing the mixture of fullerenes in the column resulted in an inhomogeneous distribution of the latter in the solid. The authors therefore decided to perform the encapsulation in solution (the capsule in CH3CN and the fullerene extract in toluene were mixed), and the fullerene containing cage was precipitated by addition of diethyl ether and re-introduction in the column. In the first encapsulation–release cycle the ratio of C60⊂42/C70⊂42 was 4/1, and exclusively C60 was released from the capsule after washing the inclusion compound with a 1:1 mixture of 1,2-dichlorobenzene/CS2. When the cycle was repeated up to 7 times (∼10% mass loss was observed after each cycle), the capsule was enriched with mainly C70 and other higher fullerenes. Concerning the recovery of the retained higher fullerenes (mainly C70 and C84), different solvent mixtures were tested but no significant release was achieved. Deconstruction of the nanocapsule could be then achieved by addition of TFA, releasing C70 and other higher fullerenes sequestered within the saturated cage retained in the column. Finally, the capsule could be partially re-constructed by treatment with TEA in DMF under reflux.
5. Summary and future prospects
Fullerenes have potential applications in multiple research fields such as materials science and medicine. However, these applications are limited in origin by the cost of purification of fullerenes. Despite the fact that fullerene extracts are easily available in macroscopic quantities from fullerene soot, finding an efficient strategy to obtain these molecules in a pure form remains elusive, especially for higher fullerenes (Cn, n > 70). Nowadays, efficient chromatographic techniques are available for the purification of fullerenes, but still more sustainable, selective and versatile methodologies are required.In this Tutorial Review the use of molecular receptors for the separation of fullerenes by means of host–guest interactions has been covered. Some insight has been given about the key factors that need to be considered when designing a fullerene molecular receptor: structural complementarity, electronic complementarity and receptor tunability.
Although very elegant examples of covalent and hydrogen-bonded fullerene hosts have been described in the literature, in this review attention has been focused on coordination supramolecular receptors. The synthesis of covalent receptors commonly follows tedious and low yielding experimental protocols, whilst in the case of hydrogen-bonded structures the calculated values of the binding constants are low in contrast to the ones achieved with covalent hosts (KH-bonded (105 M−1) ⋘ KCovalent (109 M−1)). The metal–ligand coordination approach emerged as a promising alternative to solve the synthetic cost of covalent strategies while increasing the association constants of hydrogen-bonded structures. Due to the diversity of organic ligands (linkers) and transition metals available, the metal–ligand coordination approach allows for the generation of a manifold number of supramolecular structures. This approach offers a versatile and controlled strategy to design fullerene receptors due to the predictable metal ion coordination environment, and has tunability advantages with respect to MOFs. Moreover, the latter are often limited to slow solid–liquid host–guest interactions. Besides the greater robustness of coordination structures in comparison with other supramolecular structures (i.e. hydrogen bonded receptors), these also present a flexible and dynamic structure which facilitates and improves the interaction with the fullerene molecule. Binding constants of up to 108 M−1 have been achieved by using this approach (KH-bonded ∼ 105 ≪ Kcoordination ∼ 108 < Kcovalent ∼ 109). Throughout this review the advantages of this approach have been discussed and relevant examples of coordination polygons and cage-like structures with the ability to accommodate fullerenes have been detailed.
In the last part of the review, it has been shown that efficient recovery of the encapsulated fullerenes is possible, with an impact on the practicality of fullerene purification methodologies. Strategies ranging from thermal or acid treatments to conformational and structural rearrangements are the most promising ones since the host can be partially recovered. Strikingly, very recently a solid-washing strategy has been described in which fullerene C60 can be reversibly encapsulated/released from a coordination capsule (42), without the need to disassemble the cage or do any further treatment to recover the recycled host. Moreover the receptor was re-used up to 5 times, and finally >91% of the capsule was recovered.
The application of molecular receptors for the purification of fullerenes is in its infancy, and still a lot of effort needs to be devoted to the development of new methodologies. Nevertheless, this field is rapidly evolving and more convenient solutions to the current tedious, time- and energy-consuming chromatographic methods might appear in the near future.
Acknowledgements
We thank the European Research Council (ERC-2011-StG-277801 to X.R., ERC-2009-StG-239910 to M.C.), the Spanish MINECO (Consolider-Ingenio CSD2010-00065, INNPLANTA project INP-2011-0059-PCT-420000-ACT1, CTQ2012-32436, CTQ2013-43012-P and CTQ2013-50306-EXP), and Generalitat de Catalunya (2014 SGR 862 and PhD grant to C.G.S.). X.R. and M.C. are also grateful for ICREA-Acadèmia awards.References
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- Y. Iwasa, Nature, 2010, 466, 191–192 CrossRef CAS PubMed.
- G.-F. Liu, M. Filipović, I. Ivanović-Burmazović, F. Beuerle, P. Witte and A. Hirsch, Angew. Chem., Int. Ed., 2008, 47, 3991–3994 CrossRef CAS PubMed.
- C. Yeretzian, J. B. Wiley, K. Holczer, T. Su, S. Nguyen, R. B. Kaner and R. L. Whetten, J. Phys. Chem., 1993, 97, 10097–10101 CrossRef CAS.
- A. Hirch and M. Brettreich, Fullerenes: chemistry and reactions, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- D. Canevet, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248–9259 CrossRef CAS PubMed.
- P. D. W. Boyd and C. a. Reed, Acc. Chem. Res., 2005, 38, 235–242 CrossRef CAS PubMed.
- J. Effing, U. Jonas, L. Jullien, T. Plesnivy, H. Ringsdorf, F. Diederich, C. Thilgen and D. Weinstein, Angew. Chem., Int. Ed., 1992, 31, 1599–1602 CrossRef.
- T. Andersson, K. Nilsson, M. Sundahl, G. Westman and O. Wennerström, J. Chem. Soc., Chem. Commun., 1992, 604 RSC.
- Y. Liu, H. Wang, P. Liang and H.-Y. Zhang, Angew. Chem., Int. Ed., 2004, 43, 2690–2694 CrossRef CAS.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229–231 CrossRef CAS.
- Z. Yoshida, H. Takekuma, S. Takekuma and Y. Matsubara, Angew. Chem., Int. Ed., 1994, 33, 1597–1599 CrossRef.
- Y. Nakanishi, H. Omachi, S. Matsuura, Y. Miyata, R. Kitaura, Y. Segawa, K. Itami and H. Shinohara, Angew. Chem., Int. Ed., 2014, 53, 3102–3106 CrossRef CAS PubMed.
- M.-J. Li, C.-H. Huang, C.-C. Lai and S.-H. Chiu, Org. Lett., 2012, 14, 6146–6149 CrossRef CAS PubMed.
- M. Ku, S.-J. Huang, S. Huang, Y. Liu, C.-C. Lai, S.-M. Peng and S.-H. Chiu, Chem. Commun., 2014, 50, 11709–11712 RSC.
- C. Zhang, Q. Wang, H. Long and W. Zhang, J. Am. Chem. Soc., 2011, 133, 20995–21001 CrossRef CAS PubMed.
- Y.-B. Wang and Z. Lin, J. Am. Chem. Soc., 2003, 125, 6072–6073 CrossRef CAS PubMed.
- K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC.
- L. P. HernándezEguía, E. C. Escudero-Adán, I. C. Pintre, B. Ventura, L. Flamigni and P. Ballester, Chem. – Eur. J., 2011, 17, 14564–14577 CrossRef PubMed.
- G. Gil-Ramírez, S. D. Karlen, A. Shundo, K. Porfyrakis, Y. Ito, G. A. D. Briggs, J. J. L. Morton and H. L. Anderson, Org. Lett., 2010, 12, 3544–3547 CrossRef PubMed.
- J. Song, N. Aratani, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2010, 132, 16356–16357 CrossRef CAS PubMed.
- K. Tashiro, T. Aida, J.-Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 1999, 121, 9477–9478 CrossRef CAS.
- J.-Y. Zheng, K. Tashiro, Y. Hirabayashi, K. Kinbara, K. Saigo, T. Aida, S. Sakamoto and K. Yamaguchi, Angew. Chem., Int. Ed., 2001, 40, 1857–1861 CrossRef CAS.
- M. Yanagisawa, K. Tashiro, M. Yamasaki and T. Aida, J. Am. Chem. Soc., 2007, 129, 11912–11913 CrossRef CAS PubMed.
- Y. Shoji, K. Tashiro and T. Aida, J. Am. Chem. Soc., 2004, 126, 6570–6571 CrossRef CAS PubMed.
- F. Hajjaj, K. Tashiro, H. Nikawa, N. Mizorogi, T. Akasaka, S. Nagase, K. Furukawa, T. Kato and T. Aida, J. Am. Chem. Soc., 2011, 133, 9290–9292 CrossRef CAS PubMed.
- L. P. Hernández-Eguía, E. C. Escudero-Adán, J. R. Pinzón, L. Echegoyen and P. Ballester, J. Org. Chem., 2011, 76, 3258–3265 CrossRef PubMed.
- E. Huerta, G. a. Metselaar, A. Fragoso, E. Santos, C. Bo and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 202–205 CrossRef CAS PubMed.
- E. Huerta, E. Cequier and J. de Mendoza, Chem. Commun., 2007, 5016–5018 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- M. Schmittel, B. He and P. Mal, Org. Lett., 2008, 10, 2513–2516 CrossRef CAS PubMed.
- S. Goeb, S. Bivaud, P. I. Dron, J.-Y. Balandier, M. Chas and M. Sallé, Chem. Commun., 2012, 48, 3106–3108 RSC.
- S. Shanmugaraju, V. Vajpayee, S. Lee, K. W. Chi, P. J. Stang and P. S. Mukherjee, Inorg. Chem., 2012, 51, 4817–4823 CrossRef CAS PubMed.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976–12979 CrossRef CAS PubMed.
- A. Ikeda, M. Yoshimura, H. Udzu, C. Fukuhara and S. Shinkai, J. Am. Chem. Soc., 1999, 121, 4296–4297 CrossRef CAS.
- I. Sánchez-Molina, B. Grimm, R. M. Krick Calderon, C. G. Claessens, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 10503–10511 CrossRef PubMed.
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem. – Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CrossRef CAS PubMed.
- T. K. Ronson, A. B. League, L. Gagliardi, C. J. Cramer and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 15615–15624 CrossRef CAS PubMed.
- Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed.
- K. Suzuki, K. Takao, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2544–2545 CrossRef CAS PubMed.
- T. Nakamura, H. Ube, R. Miyake and M. Shionoya, J. Am. Chem. Soc., 2013, 135, 18790–18793 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef PubMed.
- A. Ikeda, H. Udzu, M. Yoshimura and S. Shinkai, Tetrahedron, 2000, 56, 1825–1832 CrossRef CAS.
- C. G. Claessens and T. Torres, Chem. Commun., 2004, 1298–1299 RSC.
- Z. Wu, X. Shao, C. Li, J. Hou, K. Wang, X. Jiang and Z. Li, J. Am. Chem. Soc., 2005, 127, 17460–17468 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |