
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural supramolecular protein assemblies
Bas J. G. E.
Pieters†
a,
Mark B.
van Eldijk†
b,
Roeland J. M.
Nolte
a and
Jasmin
Mecinović
*a
aInstitute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: j.mecinovic@science.ru.nl; Fax: +31 (0)24 3653391; Tel: +31 (0)24 3652381
bDivision of Chemistry and Chemical Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, USA
First published on 26th October 2015
Abstract
Supramolecular protein assemblies are an emerging area within the chemical sciences, which combine the topological structures of the field of supramolecular chemistry and the state-of-the-art chemical biology approaches to unravel the formation and function of protein assemblies. Recent chemical and biological studies on natural multimeric protein structures, including fibers, rings, tubes, catenanes, knots, and cages, have shown that the quaternary structures of proteins are a prerequisite for their highly specific biological functions. In this review, we illustrate that a striking structural diversity of protein assemblies is present in nature. Furthermore, we describe structure–function relationship studies for selected classes of protein architectures, and we highlight the techniques that enable the characterisation of supramolecular protein structures.
 Bas J. G. E. Pieters | Bas J. G. E. Pieters obtained his BSc in 2009 from Zuyd University, after which he continued his studies at Maastricht University, where he received his MSc in the field of molecular life sciences in 2011. Currently, he is a PhD student at Radboud University in the research group of Dr Jasmin Mecinović. Bas has broad scientific interests and has gravitated towards the multidisciplinary environment, which lies on the border between chemistry and biochemistry. Reflecting these interests, he is presently working on multiple projects, aiming to advance the knowledge in the fields of epigenetics and supramolecular protein assemblies. |
 Mark B. van Eldijk | Mark B. van Eldijk studied molecular life science and received his MSc degree in 2010. Next, he obtained his PhD in 2014 from the Radboud University Nijmegen (The Netherlands) for his work on supramolecular protein chemistry. Under the guidance of Professor Jan C. M. van Hest, he explored the self-assembly of stimulus-responsive protein-based materials. Currently, he is working as a postdoctoral researcher in the group of Professor David A. Tirrell at California Institute of Technology in Pasadena (USA). His current research focuses on bio-orthogonal non-canonical amino acid tagging in complex biological systems. |
 Roeland J. M. Nolte | Roeland J. M. Nolte is Emeritus Professor of Organic Chemistry at Radboud University in Nijmegen, The Netherlands, and former Director of the Institute for Molecules and Materials of this university. He is a member of the Royal Netherlands Academy of Science and holds a special Royal Academy of Science Chair in Chemistry. His research interests span a broad range of topics at the interfaces of Supramolecular Chemistry, Macromolecular Chemistry, and Biomimetic Chemistry, in which he focuses on the design of catalysts and materials. His contributions to science have been recognized with numerous award lectureships, prizes, and a knighthood in 2003. He has served on many editorial boards, including those of the journal Science (Washington) and the RSC journal Chemical Communications (as Chairman). |
 Jasmin Mecinović | Jasmin Mecinović received the BSc degree in chemistry from the University of Ljubljana (2005). He was a Newton-Abraham predoctoral fellow, and received a PhD in organic chemistry and chemical biology from the University of Oxford (2009) under the supervision of Professor Christopher J. Schofield. Subsequently, he was a postdoctoral fellow at Harvard University, where he worked in the group of Professor George M. Whitesides. He is currently an Assistant Professor of Chemical Biology at the Radboud University in Nijmegen. The research in his group is centered around fundamental aspects of molecular recognition in chemistry and biology, with a special focus on the chemical basis of epigenetics and supramolecular protein assemblies. |
Key learning points(1) An astonishing number of structurally diverse supramolecular protein assemblies exists in nature.(2) The topology of multimeric protein assemblies is often highly associated with their biological function. (3) Various bioanalytical, biophysical, and biochemical techniques are employed for the identification and characterisation of supramolecular protein assemblies. (4) Underlying molecular mechanisms for the formation of many well-defined protein assemblies are not fully understood. (5) Natural supramolecular protein assemblies provide a strong base for the design of new biomolecular systems that exhibit novel functions and properties. |
1. Introduction
Proteins – biologically functional molecules – are involved in all fundamental processes in life. From transcription and translation to catalysis, metabolism, transport and structural integrity, proteins have evolved to be part of the sophisticated and highly efficient molecular machinery that controls the function of the cell.1 Nature builds proteins by a bottom-up approach, in which the primary sequence of amino acids largely determines the proteins' tertiary three dimensional structure. Most of the characterised proteins, however, are organised in higher hierarchical quaternary structures, either by forming homo-oligomeric assemblies (i.e. proteins made by identical polypeptide chains) or hetero-oligomeric assemblies (i.e. proteins made by different polypeptide chains).2 Protein homo-oligomerisation exhibits various advantages over the basic monomeric form of proteins, including functional control, allosteric regulation, higher-order complexity, and stability. Hetero-oligomerisation of these biomolecules has the advantage that the distinct functions of individual monomeric forms can be linked and also enables the formation of properly folded well-defined assemblies that would be hardly accessible when all protein subunits are covalently fused. A well-studied example of hetero-oligomeric proteins is hemoglobin, an α2β2 tetrameric assembly that contains four haem prosthetic groups required for binding of molecular oxygen, which efficiently transports oxygen from lungs into tissues via the allosteric regulation mechanism.How can one visualise the architectures of protein assemblies? Over the years, many low- and high-resolution biophysical and bioanalytical techniques for the determination of protein structures have been developed.2,3 The majority of the structures has been solved by X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (EM). A recent examination of three dimensional protein structures showed that out of about twenty thousand protein structures that have been determined by X-ray crystallography, the majority was found to exist in monomeric forms (40%) and as homo-oligomeric assemblies (47%).2 On the other hand, the NMR solution structures that have been determined of over four thousand proteins revealed that monomeric proteins dominate (90%), while only 5% of homo-oligomeric and the same percentage of hetero-oligomeric structures were present. Statistical analyses of the solved EM-based 3D structures of proteins revealed that the majority exist as hetero-oligomers (52%), followed by homo-oligomers (35%), and monomers (13%). As can be concluded from the numbers above, each of the three experimental techniques displays a substantial bias towards the type of the assembly that is found. This is a result of the fact that X-ray crystallography and NMR spectroscopy are more amenable for the determination of 3D structures of small homogeneous proteins, whereas EM is far more suitable for larger protein assemblies, including both homo-oligomeric and hetero-oligomeric complexes. In addition, scattering-based techniques, such as small-angle neutron scattering (SANS), small-angle X-ray scattering (SAXS), and static and dynamic light scattering, also provide precious information about the shape and size of large protein assemblies. Apart from the above mentioned solution and solid state-based techniques, more recently ion mobility spectrometry-mass spectrometry (IMS-MS) has become a valuable method for exploring the protein structures in the gas phase. With the substantial advances of all these techniques, one can expect that several novel structures will be determined in the coming decades, some of them possibly having as-yet unexpected topologies.
Most of the oligomeric proteins characterised to date may be classified as supramolecular assemblies in the broadest sense, because they are typically maintained via several weak non-covalent interactions, sometimes admixed with covalent bonding, as commonly found in small-molecule supramolecular structures. The purpose of this paper is to review supramolecular protein assemblies, and we will focus only on assemblies with topologies that have also been found in the fields of small-molecule supramolecular chemistry and macromolecular chemistry in the past decades, i.e. fibers, helices, tubes, rings, catenanes, knots, and cages.4 In this tutorial review, we aim to illustrate the diversity of naturally-occurring supramolecular protein assemblies and describe recent chemical-biological studies that probe the structure–function relationship in the protein world. In the following sections, we will systematically describe each type of natural supramolecular protein assembly, in the order of increasing complexity. A few exemplary and concise reviews that focus on the design aspects of supramolecular protein assemblies, which are not discussed in this article, are highly recommended for readers with a greater interest in the area of supramolecular protein structures.5,6
2. Linear proteins
Elongated protein structures are prevalent in all forms of life. Several notable examples, including elastin and silk, have not been included in this review due to the restriction of space. Nonetheless, various more prominent structures, such as collagen, actin and amyloids will be briefly addressed in this section.2.1 Collagen
Collagen is the most abundant protein in mammals, and its linear triple helical structure has been well-studied.7 It is the main structural protein in animal connective tissue and a wide variety of collagen types have been characterised. The main identifying feature of collagen is its coiled structure in which three left-handed polyproline II-type (PPII) polypeptide strands form a right-handed triple helix (Fig. 1A). The tight coil structure requires each third amino acid to be a glycine residue, with the first and second residue being any residue, although proline and hydroxyproline are the most common ones. This variability in amino acid sequence results in the possibility for collagen to form both homomeric (i.e. three chemically equal polypeptide chains) and heteromeric triple helices (i.e. individual polypeptide chains in the triple helix are chemically different), with the latter being more prevalent. | ||
| Fig. 1 Linear protein assemblies: (A) collagen (PDB: 3B0S); (B) F-actin, Reproduced with permission of Annual Review of Biophysics of Dominguez et al.8 © 2011 by Annual Reviews, http://www.annualreviews.org; (C) amyloids (PDB: 2MXU). | ||
The collagen structure is stabilised via one single hydrogen bond per amino acid triplet, namely between glycine's amide NH and a neighbouring strands' backbone carbonyl group. Proline and hydroxyproline residues, located on the first and second position of the triplet, enable preorganisation of each individual strand in its PPII conformation, thus resulting in a thermodynamically more favourable collagen triple helix folding. The hydroxyproline residues on the second position are of major influence in collagen stability as the 4R-hydroxyl group imposes a C4-exo conformation on the pyrrolidine ring via the gauche effect, while prolines on the first position of the triplet usually display a C4-endo conformation, which is also believed to help stabilising the triple helical structure.7
The collagen triple helix that is initially formed in vivo is referred to as procollagen. N- and C-proteinases cleave the procollagen at both termini in order to form tropocollagen (TC), which is flanked by short non-helical telopeptides. These TCs can self-assemble into higher-order structure, which has an even further stabilising effect on the triple helices. The telopeptides undergo lysyl oxidase-catalysed cross-linking and become covalently connected during fibril assembly, both within and between the microfibrils. This structure endows collagen with the unusual stability that is required for its function in tissues, such as cartilage or tendon.
2.2 Actin
Actin is one of the most abundant proteins in eukaryotic cells, comprising an estimated 15% of the total protein content in muscle cells. It is involved in a wide variety of cellular functions, such as maintaining the cytoskeleton or muscle contraction, where actin is part of the myofibril structure. Actin can be found as two distinct forms: monomeric globular (G-actin) and as a homopolymeric linear filament (F-actin).G- and F-actin structures are, although related, not identical. The crystal structure of G-actin has been well-defined; it is a 55 Å × 55 Å × 35 Å structure, which is folded into two α/β-domains.8 Crystallisation of the filamentous structure has not been possible, therefore, the F-actin structure has been elucidated using X-ray diffraction data from concentrated F-actin solutions, and electron microscopy (EM), although it should be noted that these structures are still slightly speculative. These studies have shown F-actin to be a single left handed helical structure consisting of approximately 13 monomeric units per 6 turns (36 nm, Fig. 1B), with an extensive amount of longitudinal interactions mainly electrostatic and hydrophobic in nature, where several loop structures form intermolecular interactions. Lateral interactions are not as pronounced, however, and are governed by a structure dubbed a “hydrophobic” plug and a variety of hydrogen bonds.
2.3 Amyloids
Another interesting and biologically relevant structure is the amyloid fibril.9 Although opposed to the other structures, which have been discussed, these fibrils are detrimental, rather than useful. Amyloid fibrils are rigid, highly structured protein polymers, which have been associated with a variety of diseases, including Alzheimer's and Huntington's disease. No apparent similarities in primary or higher order structures have yet been identified for amyloid associated proteins and it is postulated that any protein may be susceptible to amyloid formation. Even though a variety of proteins can form amyloid fibrils, the fibril structures are remarkably similar, generally being unbranched filaments several nanometers in diameter and up to micrometers in length. The fibrils are highly ordered and tightly packed structures, which are mainly composed of cross-β-sheets (Fig. 1C). It is the peptide backbone, which enables the formation of the rigid β-sheet structures that encompass the majority of the fibrils via inter-backbone hydrogen bonds. The space between the opposing β-sheet is mainly determined by the side chains of the, now non-native, proteins that make up the amyloid fibril.An interesting feature regarding amyloids is that increasing evidence points towards the notion that the amyloid state of a protein might actually be thermodynamically more stable than its natively folded counterpart.9 This stability increases with protein concentration and it may be that the reason why proteins tend not to form amyloid structures under physiological conditions is due to kinetic barriers, which prevent amyloid formation. When this kinetic barrier is breached, however, toxic, self-propagating oligomeric assemblies can form, thus leading to protein metastasis and disease progression.
3. Ring proteins
The function of numerous ring-shaped proteins (or toroidal proteins) is highly connected with manipulations on the double-stranded helical structure of DNA. Most notably, ring proteins are involved in fundamental biochemical processes in living organisms, including the enhancement of the processivity of DNA polymerases, unwinding of DNA to generate single stranded DNA, homologous DNA recombination, unwinding of supercoiled DNA and DNA transport (Fig. 2).10 Although many ring proteins have an unrelated primary sequence of amino acids and have highly distinguished biological functions, the widespread appearance of this well-defined quaternary structure highlights its intrinsic ability to efficiently encode and repair the genetic material of prokaryotes, archaea and eukaryotes required for the organisms' viability. In this section, we will describe the role of several ring-shaped DNA-binding proteins, with a focus on protein clamps, which are involved in bioprocessive catalysis, and thus resemble the very popular and extensively studied rotaxane structures in the field of supramolecular chemistry.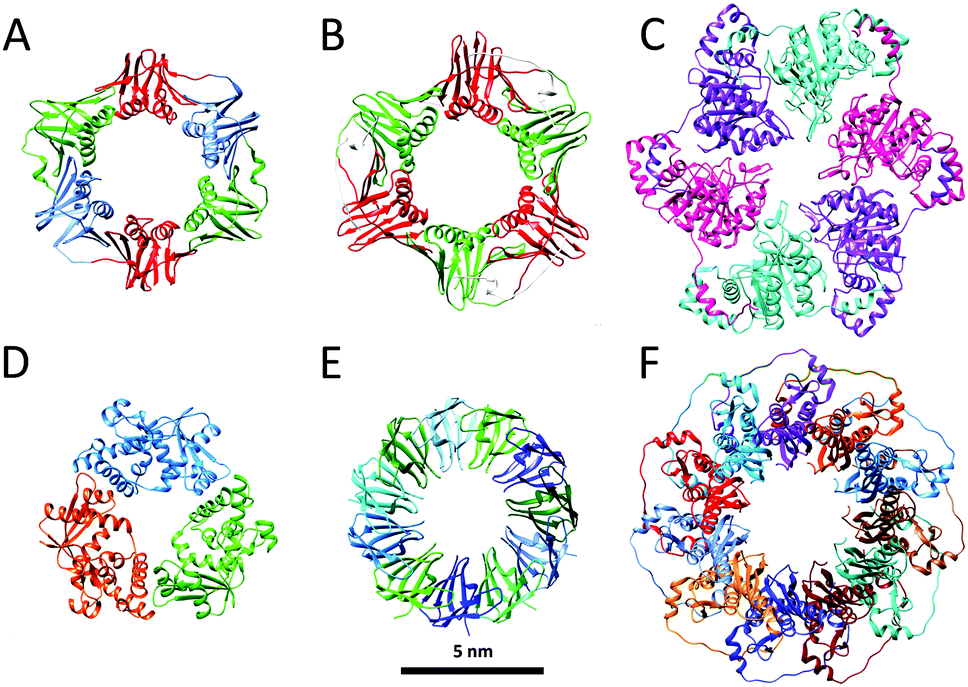 | ||
| Fig. 2 Ring proteins: crystal structures of (A) the β-clamp of E. coli (PDB: 2POL); (B) the proliferating cell nuclear antigen PCNA of H. sapiens (PDB: 1AXC); (C) bacteriophage T7 gp4 helicase (PDB: 1E0K); (D) bacteriophage λ exonuclease (PDB: 1AVQ); (E) TRAP (PDB: 1QAW); (F) RAD52 (PDB: 1KN0). Structures are at the same scale (scale bar 5 nm). | ||
3.1 DNA clamps
Reproduction is essential to the survival of an organism and in order to effectuate this a large series of complex chemical events are needed of which the faithful copying of its DNA is one of the most important steps. The enzymes responsible for this copying process are the DNA polymerases, which by themselves are distributive enzymes,11 meaning that they synthesise only a limited number of nucleotides of complementary DNA before they dissociate from the DNA template. In order to allow to this process to proceed for longer periods of time, i.e. to achieve multiple rounds of catalysis, the polymerase enzyme has to remain in contact with the DNA strand. During evolution nature has solved this problem by linking the polymerase to a ring-shaped protein, called clamp, which encircles the DNA template and freely slides along it. In this way the enzyme remains attached and the replication process continues to go on making it processive. The first crystal structure of such a ring-shaped protein, the sliding β clamp of Escherichia coli (E. coli) was reported in 1992.12 It consists of two semi-circular protein subunits, which are aligned head-to-tail forming a ring with an inner diameter large enough to hold a duplex DNA chain (Fig. 2A). Each subunit consists of three independent domains connected by long loops, providing the clamp with a pseudo-sixfold symmetry. The overall charge of the clamp is negative, but its inner surface, which is lined by α-helices, has a positive charge and interacts with the DNA chain via water molecules. The outer surface is covered by β-sheets. Association of the polymerase with the clamp considerably enhances the replication speed from approximately 20 nucleotides per second to 750 nucleotides per second, while the processivity increases from some 10 base pairs to more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 base pairs.
000 base pairs.
The fact that the ring-shaped clamp protein is essential for DNA replication and hence for the survival of the organism follows from the fact that its circular architecture and function have been retained during evolution: it is found in various species ranging from bacteria and yeasts to Homo sapiens (H. sapiens). In eukaryotes the functional equivalent of the β-clamp is the proliferating cell nuclear antigen PCNA (Fig. 2B). Unlike the β-clamp, this circular protein is a homotrimer of head to tail aligned monomers each containing two domains, overall forming a six-domain ring.13 The same circular trimeric structure is found in other species, such as the archaeal Pyrococcus furiosus and Sulfolobus solfataricus. Despite this similarity in circular shape there is little similarity (<10%) in the sequence of the amino acids of the constituting proteins. Apparently, these different sequences can lead to folding patterns that result in the same overall architecture.
The stabilities of the clamps are regulated by intermolecular interactions between the subunit interfaces. Despite the fact that protein rings are under spring tension due to the bending of the subunits, they remain in most cases stable in solution and only open in the presence of a clamp loader.13 Detailed mechanistic studies have shown that this opening and the subsequent loading of the clamp onto the DNA chain is an ATP-driven process, which is highly conserved during evolution. In the absence of ATP clamp loaders have only a weak interaction with the clamp. On binding ATP, the clamp loader undergoes a conformational change allowing a kind of “hydrophobic plug” to wedge into a hydrophobic pocket of the clamp forcing it to open (Fig. 3). This process is facilitated by the release of spring tension in the clamp. Once formed the clamp loader-open clamp complex recognises a special site (PT junction) on DNA, while adopting a conformation (notched screw cap arrangement) that matches the helical geometry of the DNA duplex. On hydrolysis of the bound ATP the clamp loader loses its affinity for the clamp-DNA complex and is ejected, after which the electrostatic interactions between the positively-charged inner surface of the clamp and the negatively-charged DNA lead to closure of the open sliding clamp around DNA. The replicative polymerase enzyme subsequently associates with this complex and the process of DNA replication starts.
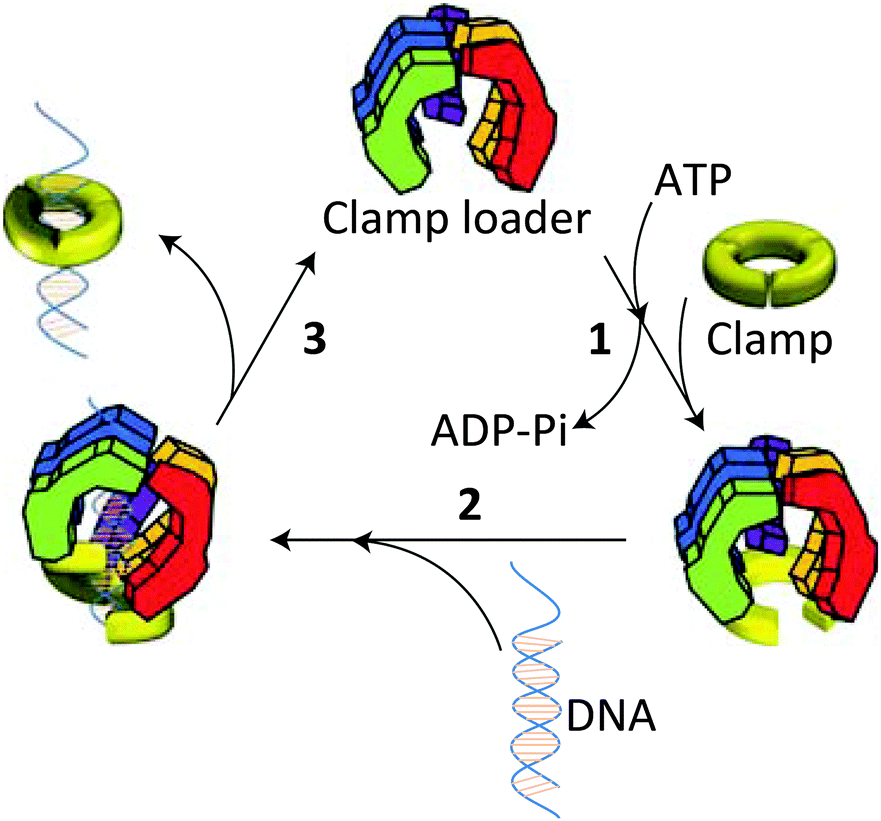 | ||
| Fig. 3 Opening and loading of the clamp with the help of a clamp loader: (1) binding of ATP induces a conformational change in the clamp loader allowing it to associate with the clamp and forcing the latter to open. (2) The clamp loader-clamp complex recognises a PT site on DNA and loads onto it. (3) Hydrolysis of ATP weakens the interaction of the clamp loader with the DNA-clamp complex leading to its ejection and closure of the clamp around DNA. Hereafter, the replicative polymerase enzyme adheres to the clamp, after which the replication process starts. Reproduced from ref. 13 with permission from Cold Spring Harbor Laboratory Press. | ||
3.2 Helicases
Interestingly, nature also makes use of ring-shaped protein architectures for separating the double helix of DNA. Hexameric helicases are highly conserved proteins that have been found in bacteriophages, viruses, bacteria and eukaryotes, and are involved in DNA replication, repair and recombination.10 Bacteriophage T7 helicase, for example, has an inner diameter of ∼20 Å and encircles the single stranded DNA on the 5′ strand during unwinding the DNA in the 5′–3′ direction (Fig. 2C).Helicases also take part in replication of prokaryotes. Replication is initiated at a site on DNA called ori (origin of replication) and involves a series of proteins, i.e. an initiator protein, a helicase, a helicase loader, and a primase. The latter protein synthesises the short RNA fragment (primer) that serves as the starting point for DNA synthesis. The crystal structure of the helicase of Bacillus stearothermophilus in complex with the loader protein of Bacillus subtilis, and the helicase-binding domain of Bacillus stearothermophilus has been solved recently (Fig. 4).14 The helicase has a ring-shaped hexameric structure and interacts with the other proteins with a stoichiometry of one helicase ring binding to three loader protein dimers and to three helicase binding domains of the primase. The overall architecture is that of a three-layered ring system, displaying a height of 130 Å, an outer diameter of 126 Å, and an inner diameter of 50–55 Å, which is large enough to allow a double-stranded or single-stranded DNA chain to pass. The study reveals that the helicase ring is not completely planar, but has a helical spring lockwasher structure, which is important for the translocation process. It, furthermore, suggests that the helicase undergoes a transformation from an open ring state to an open-spiral state and finally to a closed spiral state during the process of loading to the ori-site on DNA. This eventually results in the dissociation of the loader protein ring from the complex and the start of the replication cycle.
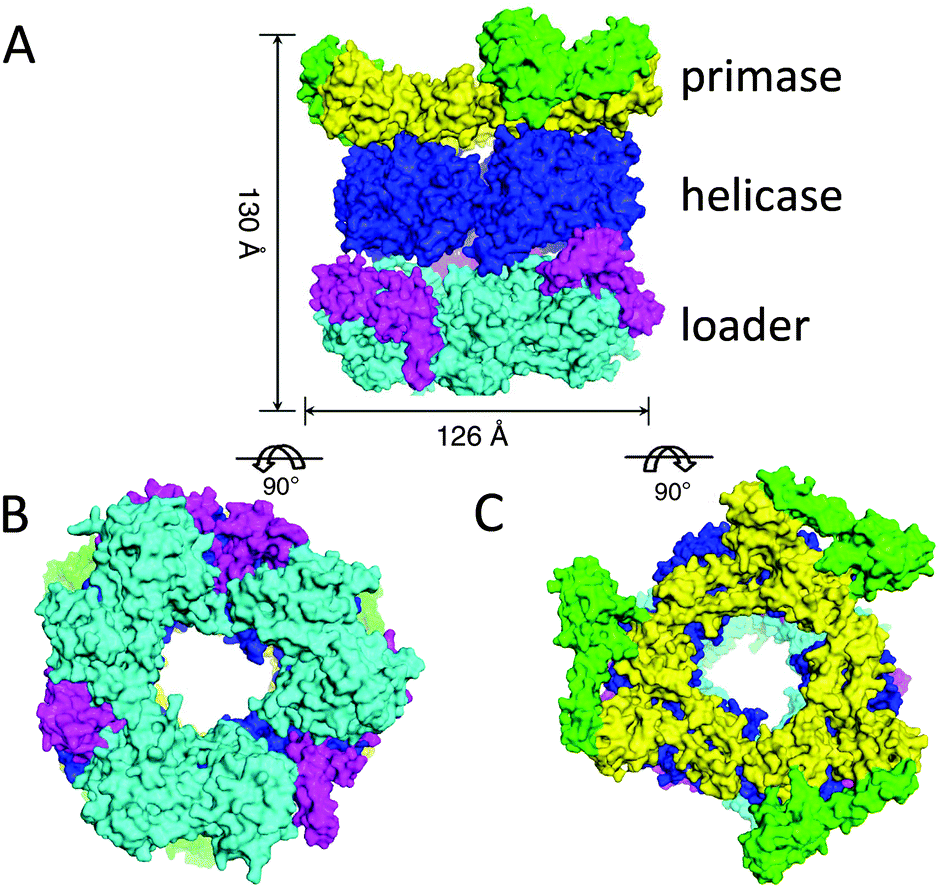 | ||
| Fig. 4 Structure of the complex between the helicase, the helicase loader, and the primase proteins: (A) side view, (B) bottom view, (C) top view. Adjusted from Liu et al.14 licensed by http://creativecommons.org/licenses/by/3.0/. | ||
3.3 Nucleases
DNA damage occurs frequently in cells and is a phenomenon that must be carefully managed in order to maintain the integrity of the genome. Nucleases are enzymes that are involved in DNA cleavage and as such participate in the processes that are used by the cell to repair its nucleic acid damages. Many of the nucleases have a ring-shaped structure. An example is the bacteriophage λ exonuclease, which binds to double-stranded DNA and processively digests the 5′-strand into mononucleotides.15 This nuclease activity is a metal-dependent process with a strong preference for Mg2+ as the metal ion. The X-ray structure of the λ-exonuclease revealed that this enzyme is a toroidal-shaped homotrimer possessing a central funnel-shaped channel for tracking along the DNA chain (Fig. 2D). This channel is at one side wide enough to allow double-stranded DNA to enter, but narrows at the other end, such that only single-stranded DNA can pass. Mechanistic studies have revealed that the cleavage of DNA proceeds via an SN2 mechanism involving one or more metal-bound water molecules that act as nucleophiles for the splitting of the phosphate backbone and as proton donors for the leaving groups.To date, several other ring proteins, which are associated with DNA and RNA, have been characterised.10 For example, trp RNA-binding attenuation protein (TRAP) from Bacillus subtilis binds single stranded RNA and is involved in termination of transcription of the trp operon. Structural analyses revealed that TRAP forms an undecameric ring structure with a 25 Å diameter of the central hole (Fig. 2E). Non-denaturing mass spectrometric studies on TRAP showed that the 11-mer ring assembly also persists in the gas phase, in the absence of bulk water.16 These studies demonstrated that the ring structure becomes more stable in the presence of tryptophan, and that binding of RNA substrate additionally stabilises TRAP. Another example of ring proteins is human RAD52, which is involved in homologous recombination of DNA; the full length protein forms a heptameric assembly, whereas the catalytically active N-terminal half exists in the undecameric form (Fig. 2F).
4. Tubular proteins
The simplest organisms such as viruses make use of tubular structures for a variety of purposes, including host infection and storage of their genetic material. The more complex eukaryotes use these structures to create cellular pores through which molecules can be actively shuttled in and out of a cell. In order to facilitate such a variety of functions, a strikingly large number of tubular structures has evolved. Here we take a closer look at a selection of such structures found in various organisms. For the purpose of this review, we define tubes as naturally-occurring elongated structures with a “hollow” core.4.1 Tobacco mosaic virus
One of the best studied tubular protein structures is the tobacco mosaic virus (TMV) capsid.17 In the late 19th century, the TMV virus was the first virus to be discovered by Beijerinck, when an infectious agent was identified by him as the cause for disease in tobacco plants. Its general structure had already been postulated as early as the 1950's by Rosalind Franklin, and consists of a helical coat structure encapsulating a single stranded RNA molecule. The TMV helical coat structure is composed of 2130 identical coat particles (CP), which self-assemble into a right-handed helix; 300 nm in length, 18 nm in diameter with a 4 nm inner channel (Fig. 5A). | ||
| Fig. 5 Tubular protein assemblies: (A) TMV (PDB: 4UDV). The side view only displays part of the length of the helical TMV assembly; (B) α-hemolysin pore complex (PDB: 7AHL); (C) antrax protective antigen pore (PDB: 3J9C); (D) PhiX174 bacteriophage tail (PDB: 4JPP); (E) Hcp1 from P. aeruginosa (PDB: 1Y12). | ||
It is assumed that assembly starts when a two-layered disk (named 20S disk after its sedimentation coefficient) comprised of two rings of 17 CPs each recognises an RNA sequence. This recognition sequence facilitates a hydrogen-bonding pattern between the RNA bases and the RNA base binding sites of the 20S aggregate causing the RNA loop to fold. The RNA loop is then pulled through the TMV coat particle as additional short helix aggregates stack on top of the nucleation disc. TMV has a remarkably robust structure, displaying stability at temperatures as high as 90 °C, remaining stable in a pH range of 3.0–9.0 and it shows resilience to organic solvents such as acetone, ethanol, and water rich THF and DMSO. Interestingly, the TMV particles are also able to aggregate into larger units, such as fibers, which can be induced in vitro by increasing the particle and/or ion concentration.
4.2 α-Hemolysin
Another well-known tube-like protein is α-hemolysin, a homo-oligomeric heptameric protein complex secreted by Staphylococcus aureus. It forms a mushroom-shaped structure composed of a cap, rim and stem-like architecture (Fig. 5B).18 The structure in total is 100 Å in length, with the stem being 52 Å and the cap 70 Å in height. Its width spans 100 Å at its maximum, whereas the solvent filled channel, which runs in the longitudinal direction of the pore has a diameter of 14–46 Å and the stem consisting of 14 β-strands, which possess a β-barrel structure, is 26 Å in diameter. The stem domain of α-hemolysin forms the transmembrane channel, whereas the cap and portions of the rim domain protrude from the phospholipid bilayer. The inside of the stem is composed of charged residues, alternated with several rings of hydrophobic residues. Its outside, on the other hand, is lined with hydrophobic amino acid residues in order to facilitate interactions with the phospholipid bilayer. Additionally, regions of the stem and rim domains are believed to interact with the head groups of the phospholipids, allowing for the binding of α-hemolysin to cell membranes. Recently, this protein complex has received a particular interest because of application as nanopore for DNA sequencing.4.3 Anthrax protective antigen pore
One of the more aesthetic structures presented in this review is the anthrax protective antigen pore (PA pore); indeed, its structure has been compared to a flower (Fig. 5C).19 Within this analogy its individual monomers consist of 4 domains representing the flower's corolla (domain 1, 3 and 4), calyx and stem (domain 2). The PA pore is a homo-heptamer consisting of 63 kDa subunits, which assemble into a tubular structure, 180 Å in height, 160 Å in width at its widest section and 27 Å diameter wide β-barrel with a predominantly hydrophobic outer surface. Upon endocytosis, the pore functions as a translocation channel through which the toxic lethal factor and edema factor enzymes can be unfolded, translocated and refolded, enabling the efficient delivery of the toxins into the cytosol. This translocation is mediated via the predominantly hydrophilic and negatively-charged inner surface. Although the opening of the pore is 30 Å in width, the ϕ-clamp located below this opening is only 6 Å in diameter, and may only be able to accommodate the translocation of unfolded primary protein structures. Below the ϕ-clamp lie an enlarged chamber and the β-barrel tube, which are large enough to hold secondary protein structures and may facilitate refolding of the translocated toxins.4.4 PhiX174 bacteriophage tail
A more recently discovered protein tube is the PhiX174 bacteriophage tail structure. Viruses have developed a variety of mechanisms through which they can infect a host. One of these mechanisms is the formation of tube-like structures capable of penetrating the hosts' cell membrane allowing the genetic material of the virus to be injected into the host.The α-helical barrel structure allows the bacteriophage to infect cells using the PhiX174 DNA pilot protein H in order to form a DNA translocating channel.20 The protein H has been shown to exist of 10 α-helical structures, which form a 170 Å long and 48 Å wide decameric coiled-coil structure (Fig. 5D). Because each monomer is kinked at residues Tyr193–Ala194–Gln195, the assembly can be divided into domains A and B, each with a specific twist and diameter. Domain A has a 22 Å diameter and a slight right-handed twist, whereas domain B has an inner diameter of 24 Å and a less steep left-handed twist. Due to the spatial organisation of the protein H coiled-coil tube, each monomer interacts with its two neighbouring monomers mainly via hydrogen bonds. The inside of the tube is predominantly negatively-charged, but also contains several glutamine, asparagine and arginine residues, thus presumably facilitating the transport of DNA across the bacterial membrane during infection.
4.5 Hcp1
The Hcp1 protein that has been found in Pseudomonas aeruginosa is a circular homo-hexamer postulated to be involved in the bacterial type VI secretion system.21 Its individual subunits are only 17.4 kDa in mass and assemble into a ring structure of 90 Å × 44 Å in dimension with a central hole of 40 Å in size. It was observed that these structures were able to further self-assemble into elongated tubular structures. This structure was artificially stabilised by introducing cysteine residues at residues Gly90 and Arg157, allowing for covalent linking of the individual ring structures (Fig. 5E). Optimisation of assembly conditions allowed for the generation of reasonably long (up to 0.1 μm) nanotubes which can be capped on both ends. The Hcp1 protein tube does illustrate that subtle mutations may lead to new protein topologies and suggest that these structures have interesting potential in the field of protein engineering. Such structures could serve as carriers for small molecules and may have an ability to be used as biologically inspired drug delivery systems.5. Catenane proteins
Catenanes, one of the most widely studied mechanically-interlocked molecular topologies in chemistry, have been established as intriguing biomolecular architectures in nature since their appearance in the literature in 1960s. Pioneering work by Vinograd and coworkers demonstrated that catenated DNA structures exist inside living cells. On the other hand, only few examples of proteins that occur in interlocked catenane form in prokaryotes, archaea and eukaryotes have been reported in the past twenty years, most of them being discovered unexpectedly. The majority of characterised catenane proteins can be classified as [2]catenanes. Two catenane protein subfamilies include the covalently maintained catenanes (i.e. there is a covalent linkage between monomers within each of the individual rings) and the non-covalently maintained catenanes (i.e. individual rings are assembled solely via non-covalent bonds). Herein, we describe examples of both types.5.1 Covalent catenanes
Determination of the crystal structure of lysyl oxidase derived from Pichia pastoris yeast also revealed the presence of the catenane topology. Similarly to PaCS, the dimeric protein structure is maintained via two disulfide bridges between Cys45 and Cys756 within each of the two polypeptide chains (Fig. 6C). The functional advantage of the catenated lysyl oxidase remains to be established, but it is likely that it exhibits an increased thermal stability when compared to a putative noncross-linked protein.
5.2 Non-covalent catenanes
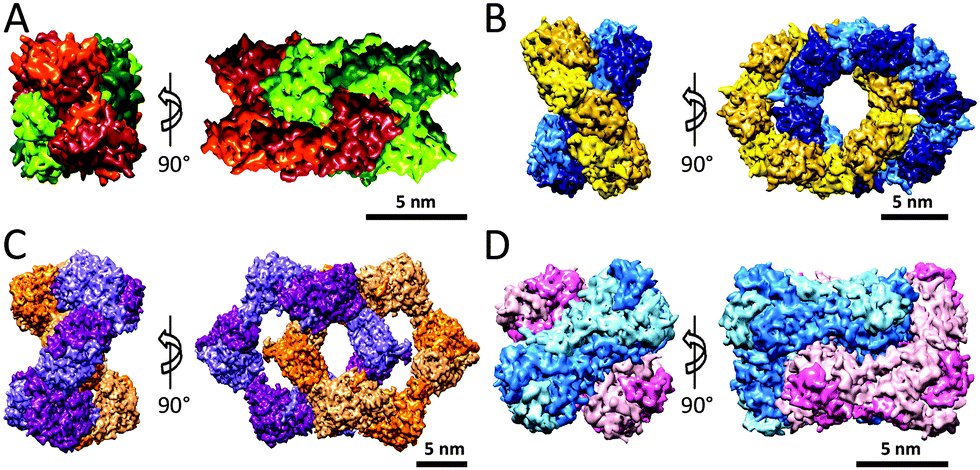
Unambiguous experimental evidence for the existence of the catenane form of CS2 hydrolase in solution has been provided recently.28 A combination of size exclusion chromatography and non-denaturing mass spectrometry revealed that the ratio between catenane and ring forms remained unaltered in the concentration range of 0.1–10 mg mL−1, highlighting that the unique interlocked catenane structure persists in very dilute solutions in the absence of precipitating agents. Subsequently, transmission electron microscopic (TEM) studies on individual assemblies of CS2 hydrolase provided visual evidence for the protein topologies in high resolution. TEM-based reconstructions of the catenane and ring assemblies of CS2 hydrolase are in good agreement with their structures, as obtained by X-ray crystallography.29 It should be noted that, in contrast to the catenane assembly observed in the highly crowded crystalline state, the TEM structures were solved at dilute protein concentrations and without potential crystal contacts between monomers/oligomers that may influence the spatial organisation of the protein assembly, thus additionally confirming the appearance of the catenane form of CS2 hydrolase under a wide range of experimental conditions. The ability to separate and purify individually the catenane and ring forms of CS2 hydrolase also allowed to comprehensively examine this special case of the topology-function relationship in the protein world.30 Asymmetric flow field-flow fractionation coupled to multi-angle laser light scattering (AF4-MALLS) and native mass spectrometric studies on individual assemblies demonstrated that the catenane form is converted into the ring form at elevated temperatures and that this equilibrium is completely on the side of the ring. Compelling experimental data suggest that the ring assembly is the thermodynamically more stable form of the two assemblies, whereas the catenane is a kinetically trapped form. The catenane and ring forms of CS2 hydrolase not only differ in their thermal stability, but also in the catalytic efficiency for the conversion of carbon disulfide into carbonyl sulfide and hydrogen sulfide. Enzyme kinetics studies showed that the hexadecameric catenane assembly is enzymatically more active than the octameric ring assembly, but that the ring form is more active than the catenane form when calculated as the enzyme efficiency per monomer.30 It is possible that, in contrast to the fully accessible eight active sites of the ring assembly, the topologically more complex catenane assembly in which both interlocked rings are tightly packed, has partially hindered access for CS2 substrate to some of the sixteen active sites, which in turn results in lowered kcat/Km values for the catenane relative to the ring.
6. Knot proteins
Another interesting and quite newly discovered type of protein structure is found in the class of knotted proteins. It has only recently come to light that proteins are able to display an intriguing knotted arrangement; that is, a tertiary structure which does not disentangle when the protein is pulled at both N- and C-termini at the same time.31,32 The functional implications of knotted structures are not fully understood from both chemical and biological perspectives, but such structures do provide us with interesting models, which can be used to study the process of protein folding. Due to the complexity of identifying knots in proteins, however, traditional approaches such as inspection of structures obtained by X-ray crystallography are impractical, since all protein structures would need to be analysed manually. Especially for larger protein structures, the visual identification of knots becomes increasingly difficult; therefore, more computational approaches need to be employed in the discovery of such biomolecular structures.33 The computational methods that have been developed can be divided into two main categories, namely mechanically and knot theory-based approaches, although neither of them is perfect.32In the past decade, various bioanalytical, biophysical, and bioinformatics tools have been developed in order to study knotted proteins in vitro and in silico.31 For example, atomic force microscopy (AFM) can be employed to physically stretch and untangle isolated proteins. By employing in vitro transcription–translation (IVTT) in cell-free expression systems, knotted proteins can be expressed in the absence of chaperonins, which gives the opportunity to examine the protein's intrinsic folding properties. In addition, recombinantly produced proteins can be chemically unfolded and refolded in order to investigate their folding pathways using urea- and/or pH-jump experiments. An approach has been devised in which denatured proteins are cyclised using disulfide bonds, thus allowing to determine whether proteins can exist in knotted conformations under denatured conditions. Supporting techniques, such as fluorescence spectroscopy, circular dichroism, and cofactor binding assays can be employed to determine correct (re-)folding of the knotted protein structures.34
To date, four types of knots have been identified in various protein structures: namely trefoil knots (also referred to as 31 knot), figure-of-eight knots (41), a five-crossings knot (52) and a six-crossings knot (61).31 Additionally, such knotted structures can be further subdivided into shallow and deep knot structures. Here the slightly arbitrary denotations shallow and deep refer to the number of amino acid residues that have to be cleaved from the proteins termini before the knotted structure untangles. Finally, knots display chirality, i.e. a certain type of handedness determined by the direction of each crossing of the protein backbone, although no apparent preference for knots to fold into a specific type of handedness has been identified.
6.1 31 knots
The first protein that was identified as a knotted protein is the well-known carbonic anhydrase.31 Carbonic anhydrase is an enzyme capable of converting carbon dioxide and water into bicarbonate and protons; this function is critical in maintaining the physiological bicarbonate buffer system in biological media. The protein is a trefoil knot, shallowly knotted at its C-terminus, where the thread is part of a β-sheet structure (Fig. 8A). Work in which atomic force microscopy (AFM) was employed in order to unfold single molecules of carbonic anhydrase has shown that when the protein is unfolded, by pulling at both termini, the knot tightens causing the protein to stretch over a shorter distance than would be expected for the unknotted linear structure. This implies that the knotted structure persists during this type of mechanical unfolding and would be supportive of the suggestion that knot structures may provide some kind of protection, increasing the protein's stability.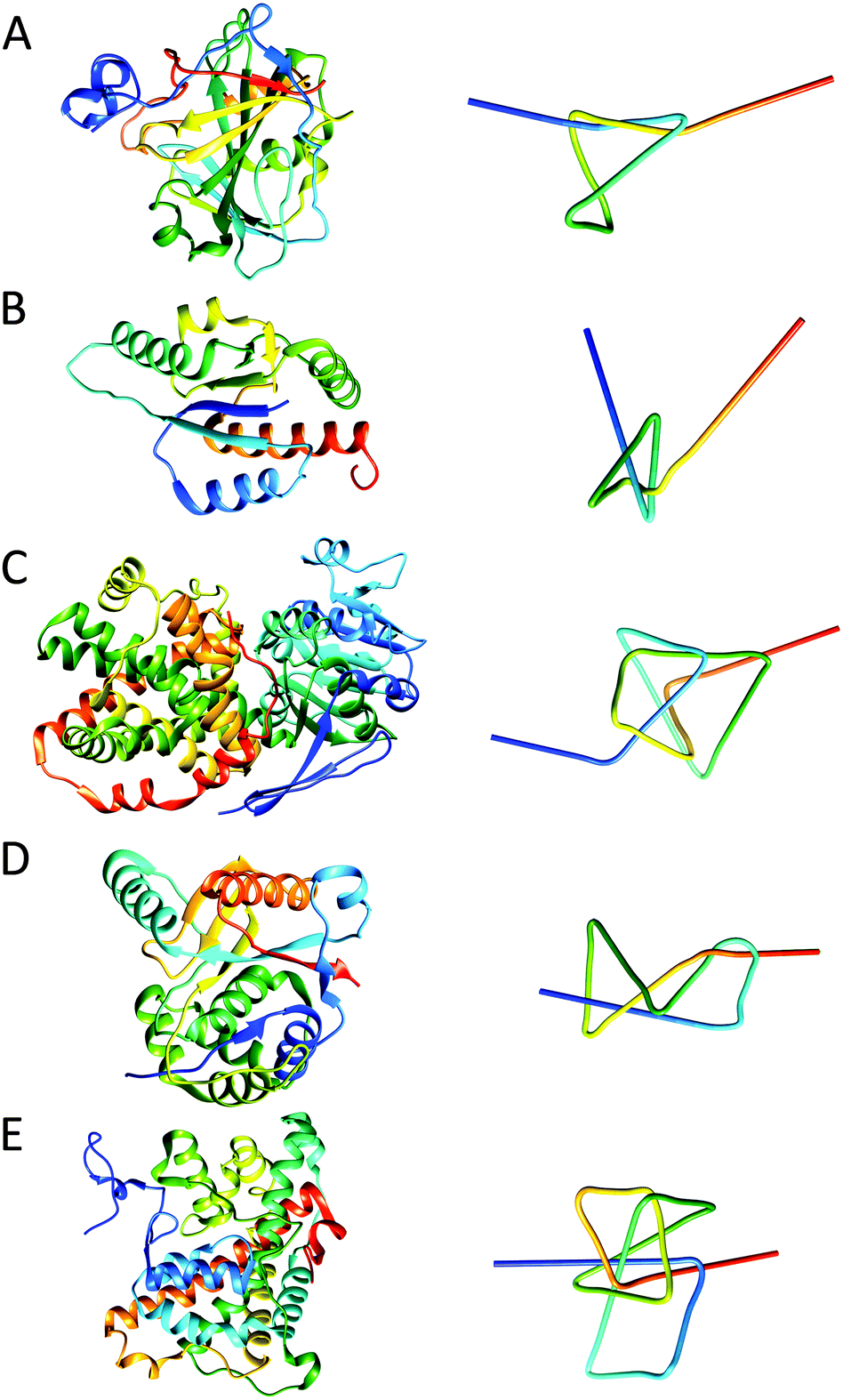 | ||
| Fig. 8 Structures of various protein knots and their simplified visualisations generated by protein knot server:35 (A) trefoil knot in carbonic anhydrase (PDB: 1CA2); (B) 31 α/β-knot structure in RNA methyltransferase YbeA from E. coli (PDB: 1NS5); (C) ketol-acid reductoisomerase with 41 knot (PDB: 3FR8); (D) 52 knot in ubiquitin hydrolase UCH-L3 (PDB: 1XD3); (E) Stevedore's protein knot (61) in α-haloacid dehalogenase (PDB: 3BJX). Note the corresponding colour gradient from blue (N-terminus) to, cyan, green, yellow and red (C-terminus) between the protein structure and simplified visualisation. | ||
The most studied knotted proteins, however, do not belong to the carbonic anhydrase fold family. Instead the Haemophilus influenzae YibK and E. coli YbeA proteins, which belong to the RNA methyltransferase family, have been most thoroughly investigated.31 They were shown to possess a deep 31 α/β-knot structure. Despite having only 19% sequence similarity, both proteins do have common characteristics; YibK and YbeA are both homodimeric proteins of 160 and 155 residues, respectively, and both form the trefoil knot by threading their C-terminal residues. For YibK the final 40 residues are threaded through a knotting loop, whereas YbeA threads its final 35 residues (Fig. 8B). Both proteins fold via folding intermediates before finally dimerising, with YibK appearing to have a more complex folding pathway with three folding intermediates, whereas YbeA has only one intermediate. Interestingly, both proteins can be fully unfolded (although they retain a knotted “primary” structure) with urea after which they can readily be refolded into their native states without the assistance of chaperone proteins (Fig. 9). This observation indicates that the knotted structure does not impair proper protein folding, though it has been shown that the presence of bacterial GroEL–GroES chaperonin does accelerate the folding process significantly, at least 20-fold at the experimental conditions used for the examination of the refolding process.36
 | ||
| Fig. 9 The mechanism for the formation of knotted proteins. Reprinted by permission from Macmillan Publishers Ltd: Nature Chemical Biology, Mallam et al.36 © 2012. | ||
6.2 41, 52 and 61 knots
Ketol-acid reductoisomerase (KARI) is the most deeply knotted protein identified to date, having a 41 knot, which can be retained until about 240 N-terminal and 60 C-terminal amino acids have been cleaved off (Fig. 8C).31,37 It is a protein found in plants, such as spinach and rice, where it is a part of the branched chain amino acid synthesis pathway, as it is capable of catalysing the conversion of 2-aceto-2-hydroxybutyrate into (2R,3R)-2,3-dihydroxy-3-methylvalerate or 2-acetolactate into (2R)-2,3-dihydroxy-3-isovalerate. Via this pathway, the amino acids valine, leucine and isoleucine are synthesised, and all are classified as essential amino acids for humans.Another protein, ubiquitin hydrolase UCH-L3 contains an example of a 52 knotted structure (Fig. 8D).38 It is a 26 kDa hydrolase believed to be involved in the maintenance of cellular ubiquitin concentrations and has been linked to diseases ranging from breast cancer to Parkinson's disease. Despite its complexity, this protein like YibK and YbeA, is able to fold properly into its native state without the aid of chaperones. It is proposed that the knotted topology may provide the ubiquitin hydrolase with a degree of resistance against degradation by the 26S proteasome.
The most complex knot identified to date is the Stevedore's protein knot (61) in α-haloacid dehalogenase (DehI).39 DehI is an enzyme, found in a Pseudomonas putida strain, capable of catalysing the cleavage of carbon–halogen bonds of halogenated organic compounds. The knot present in this protein is relatively deep, and over 65 residues on the N-terminus and 20 residues on the C-terminus can be deleted before the knot structure is abrogated (Fig. 8E). When the knotted structure is examined more closely, it is evident that it consists of two regions with similar structures and approximately 20% sequence identity, each ∼130 amino acid residues in length. Individually, these two regions are unknotted, it is only when taken together that a knotted structure is formed.
Given the tutorial nature of this review, we are not able to discuss all aspects of knotted proteins here. Suffice to say that the scientific literature provides valuable information on other knotted, knot-like, and knot-related structures. For instance, the cysteine knot superfamily encompasses various polypeptide toxins, such as Kalata B1, which has anti-microbial activity. In addition, knot-related slipknots, including alkaline phosphatase and thymidine kinase, are strictly speaking not knotted structures, but can become knotted when one of the termini is deleted.32 It is envisioned that future studies of knotted protein structures undoubtedly will greatly contribute to the scientific understanding of the fundamental biological principles behind protein folding.
7. Protein cages
Compartmentalisation is crucial to life by providing spatial control to biological processes and shielding compartmentalised compounds from the environment and, in turn protecting the environment from unstable and toxic intermediates. The importance of compartmentalisation is observed on different levels of complexity, ranging from the cellular level, as well as the level of organelles, down to the molecular level of protein compartments. These protein compartments have been found both in bacteria and in eukaryotic cells, but also viruses make use of a protein capsule to encapsulate their genetic material.These various types of protein cages form a very interesting class of supramolecular protein complexes with inspiring properties. Many of the known naturally-occurring protein cages have a spherical shape, although some more irregular shapes have also been observed. Most of these sphere-like protein capsules have the symmetry of icosahedrons. The materials science community has demonstrated particular interest in protein cages because of their potential applicability as drug delivery vehicles. This has also resulted in engineering of naturally-occurring capsules to optimise their properties for specific applications. In this section we will discuss a number of well-studied naturally-occurring protein cages, with a focus on homomultimeric protein cages.
7.1 Ferritins
Ferritin protein cages are ubiquitous in almost all forms of life, and can be divided into three subcategories: classical ferritins, bacterioferritins and DNA-binding proteins from starved cells (named Dps). Ferritins are members of ferroxidase family of enzymes and can sequester iron by concentrating it in their internal cavities for storage and detoxification. These protein cages assemble from four-helix bundle monomers (four α-helices, labeled A, B, C and D). Interestingly, two distinctly different ferritin architectures with different symmetries and oligomerisation states have been characterised in different organisms, maxi-ferritins and mini-ferritins (Fig. 10A and B, respectively). Classical ferritins and bacterioferritins are maxi-ferritins and are found in animals, plants and bacteria and form capsules of 24 subunits (480 kDa) and can accommodate about 4500 iron ions, whereas the Dps proteins, that are present in bacteria, are mini-ferritins, consisting of 12 subunits (240 kDa).40 Although the DNA and protein sequences of the different ferritins vary considerably, the secondary and tertiary structures have been found to be highly conserved. Superimposing the tertiary structures of the ferritin subunits from mini- and maxi-ferritin architectures also shows high degree of similarities (Fig. 10C). However, there also are several key differences; for example, in maxi-ferritin the ferroxidase active site is in the centre of the helix-bundle of each subunit, while the catalytic sites of mini-ferritins are between two subunits. In addition, the small helix in the loop between the B and C helix is only found in mini-ferritin. Furthermore, the fifth helix (E-helix) in the maxi-ferritin is not present in mini-ferritin and is responsible for the four-fold symmetry axis which is important for the formation of the 24-mer capsule. The strong interactions between the helices in the helix-bundle motif and between subunits in the assembled ferritins result in a highly stable protein structure that can be heated up to 80 °C or exposed to denaturing conditions such as 6.0 M guanidine at neutral pH without disassembly.41 | ||
| Fig. 10 Examples of protein cages: (A) maxi-ferritin (PDB: 1BFR); (B) mini-ferritin (PDB: 1DPS); (C) superimposition of ribbon structures of mini-ferritin (light) and maxi-ferritin (dark); (D) a surface view of the rat vault shell (PDB: 4HL8); (E) ribbon structure of the major vault protein monomer, showing the structural repeat domains (green), the shoulder domain (blue), the cap-helix domain (red), and the cap-ring domain (magenta); (F) surface-structure of triskelion and zoom showing the α-helical zigzags (PDB: 1XI4); (G) structure of hexagonal barrel. Partially reprinted by permission from Macmillan Publishers Ltd: Nature, Fotin et al.42 © 2004. | ||
The assembly mechanism of maxi-ferritin has been studied most thoroughly and is quite well understood, whereas the assembly of mini-ferritins is relatively poorly understood. The assembly of the apo form of maxi-ferritin from horse spleen has been shown to go through a number of concentration-dependent association steps, which occur after folding of the unstructured monomer (M1*). Through chemical crosslinking, intrinsic fluorescence emission spectroscopy experiments, gel permeation chromatography and ultracentrifugation it was shown that assembly of the 24-mer maxi-ferritin (M24) proceeds via several intermediates involving structured monomers (M1), dimers (M2), trimers (M3), hexamers (M6) and dodecamers (M12). The hexamer (M6) is a transient intermediate as it could only detected in small quantities. This assembly scheme was further confirmed via reassociation studies of isolated intermediates that could be obtained upon reversible chemical dissociation with 2,3-dimethylmaleic anhydride. Altogether, these experiments resulted in the following self-assembly scheme:41
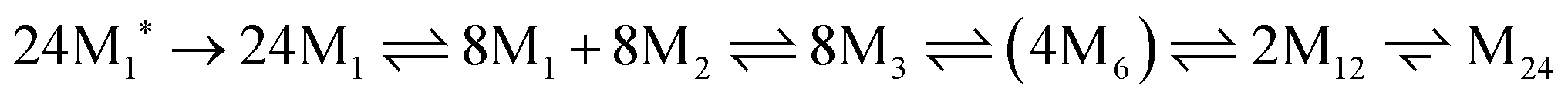
7.2 Vaults
Vaults are ribonucleoprotein cages of 13 MDa which have been found in various eukaryotic species. Depending on the type of tissue, up to several thousands of copies can be found per cell. Even though vault proteins have been a topic of investigation since their discovery in 1986, their exact function still remains unclear. It has been suggested that vaults are involved in basic cellular activities, such as transport, signal transmission and immune response, because they are highly evolutionary conserved and because they are present in many different tissue types.The vault nanocapsule consists of untranslated RNA and multiple copies of three different proteins; the major vault protein (MVP) and two minor vault proteins (VPARP and TEP1).43 The assembly of the MVP alone is already sufficient for the formation vault-like particles. Multiple copies of the two other vault proteins and the small vault RNAs are packaged inside the MVP shell. X-ray crystallography of rat liver vault showed that the vault particles exhibit 39-fold dihedral symmetry, which means that each half-vault consists of 39 MVP monomers (Fig. 10D). Each monomer folds into 12 domains: 9 structural repeat domains, a shoulder domain, a cap-helix domain, and a cap-ring domain (Fig. 10E). Most of the interactions that are suggested to drive the assembly of subunits of one half-vault are located in the cap-helix domain. The basis of the interaction between the two half-vaults is a relatively weak anti-parallel β-sheet. Interestingly, the disruption of the assembled vault therefore results in the formation of half-vaults.43 Recently, a unique assembly mechanism was proposed, in which clusters of cytoplasmic ribosomes (called polyribosomes) template the vault assembly by directing nascent MVP polypeptides to participate in assembly via spatially coordinated synthesis.44 It was suggested that this assembly mechanism in which the polyribosomes act as a 3D nanoprinter might also be involved in the formation of other homomultimeric protein complexes.
7.3 Clathrin cages
Clathrin-coated vesicles are important vehicles for intracellular trafficking and are found in all eukaryotic cells. Clathrin cages differ from the other protein capsules discussed in this review because in this case the proteins form a lattice coat surrounding a membrane vesicle. The coat is constructed from three-armed assembly units that radiate from a central hub. Such a triskelion consists of three heavy chains (180 kDa) and three light chains (25 kDa), forming legs from α-helical zigzags (Fig. 10F). Triskelions assemble to form a lattice structure with open hexagonal and pentagonal faces. Clathrin can form structures ranging from small coats of 28 and 36 assembly units, named mini-coats and hexagonal barrels (Fig. 10G), up to extended hexagonal arrays. The size of the clathrin-coated vesicles is dictated by the cargo's diameter, and the hexagonal barrel (700 Å × 800 Å) is probably the smallest polyhedron that can enclose a transport vesicle.427.4 Chaperonins
Chaperonins are cylindrically-shaped protein assemblies which are members of a set of protein families named molecular chaperones. Like other members of this family, they assist in correct protein folding and proteome maintenance. Chaperonins can be divided into two subtypes; group I is composed of the cylinder and a detachable cap and is found in bacteria (GroEL–GroES) as well as in endosymbiotic organelles such as mitochondria and chloroplasts (HSP60–HSP10).45 Group II chaperonins have a built-in lid and are found in archaea and in the cytosol of eukaryotic cells.45Both groups of chaperonins consist of two back-to-back stacked rings. However, in group I the rings are heptameric, comprised of identical subunits of 60 kDa, whereas the two rings in group II are more complex eight or nine membered heteromultimeric assemblies. The bacterial GroEL–GroES complex (Mw ≈ 1 MDa) is the most studied and its very interesting mechanism of action has been unraveled (Fig. 11A and B). The open internal cavities of the two rings are 5 nm in diameter and are surrounded by a hydrophobic surface. In this state the rings are ready to capture polypeptides with exposed hydrophobic surface. Then upon ATP binding, the GroEL ring recruits a GroES cap, a protein ring comprised of seven identical 10 kDa subunits. This initiates a dramatic structural transformation, in which the open cavity changes into an enclosed chamber with hydrophilic lining. Folding of the protein can occur in this protected environment, after which the chamber is re-opened to release the protein (Fig. 11C).
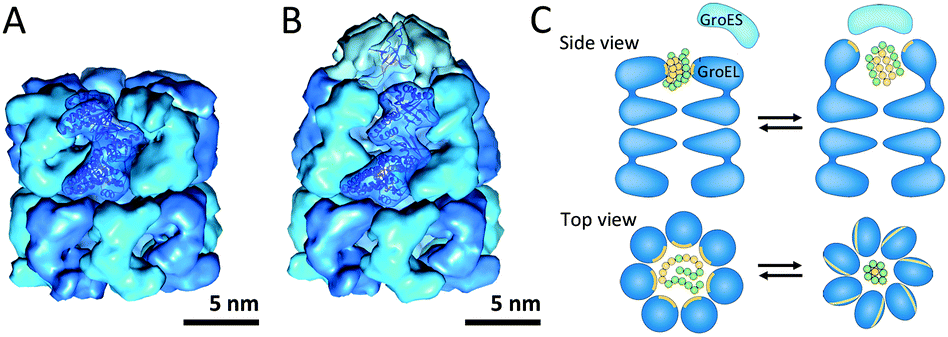 | ||
| Fig. 11 Chaperonin group I: (A) uncapped GroEL (PDB: 1OEL); (B) GroEL–GroES complex (PDB: 1SVT); (C) schematic overview of structural changes during GroEL-assisted protein folding in which the hydrophobic surfaces and residues are shown in yellow and the polar residues are displayed in green. Reprinted by permission from Macmillan Publishers Ltd: Nature Review Molecular Cell Biology, Saibil45 © 2013. | ||
7.5 Viruses
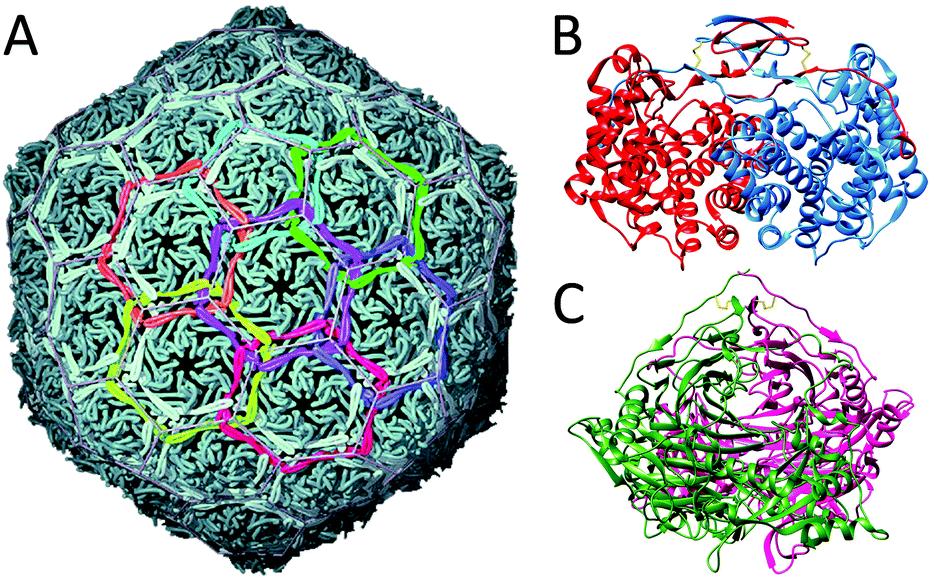
Viruses need to be robust in order to protect their genomic cargo from changes in the environment (temperature, pH and ionic strength), but yet they need to be fragile enough to dissociate and release the cargo. Virus particles are perhaps the largest class of protein-based nanocages. In approximately half of them, the capsid is spherical, and most commonly their coat proteins are arranged with icosahedral geometry. Even though rod-like viruses, such as TMV, are also examples of protein cages, we have discussed these in the section about tubular protein assemblies. Many different families of spherical virus capsules are known, but structure-wise viruses can be organised in two different types: nucleocapsids with and without a proteoglycan layer.Virus assembly has been studied thoroughly, as understanding of the assembly mechanism could facilitate the design of antiviral agents. The cowpea chlorotic mottle virus (CCMV) was the first icosahedral virus that was assembled in vitro and has served as a model system for investigation of capsid assembly, disassembly and stability.46 Some viruses require a nucleic acid scaffold or a scaffolding protein for correct assembling process. Interestingly, the 180-mer CCMV capsid can also be formed in the absence of its nucleic acid cargo. Assembly of this virus has been shown to start with the formation of a pentamer of dimers which subsequently associates with free dimers to form the completed capsid (Fig. 12A). In general, assembly of a population of virus capsids is characterised by concurrent reactions for nucleation, elongation and capsid completion. Furthermore, the subunit–subunit association is weak and assembly goes through numerous reversible reactions, which allow for correction of assembly mistakes by dissociation (Fig. 12B). For several viruses it has been shown that increasing of the association energy results in kinetic traps and defects in the assembly.46
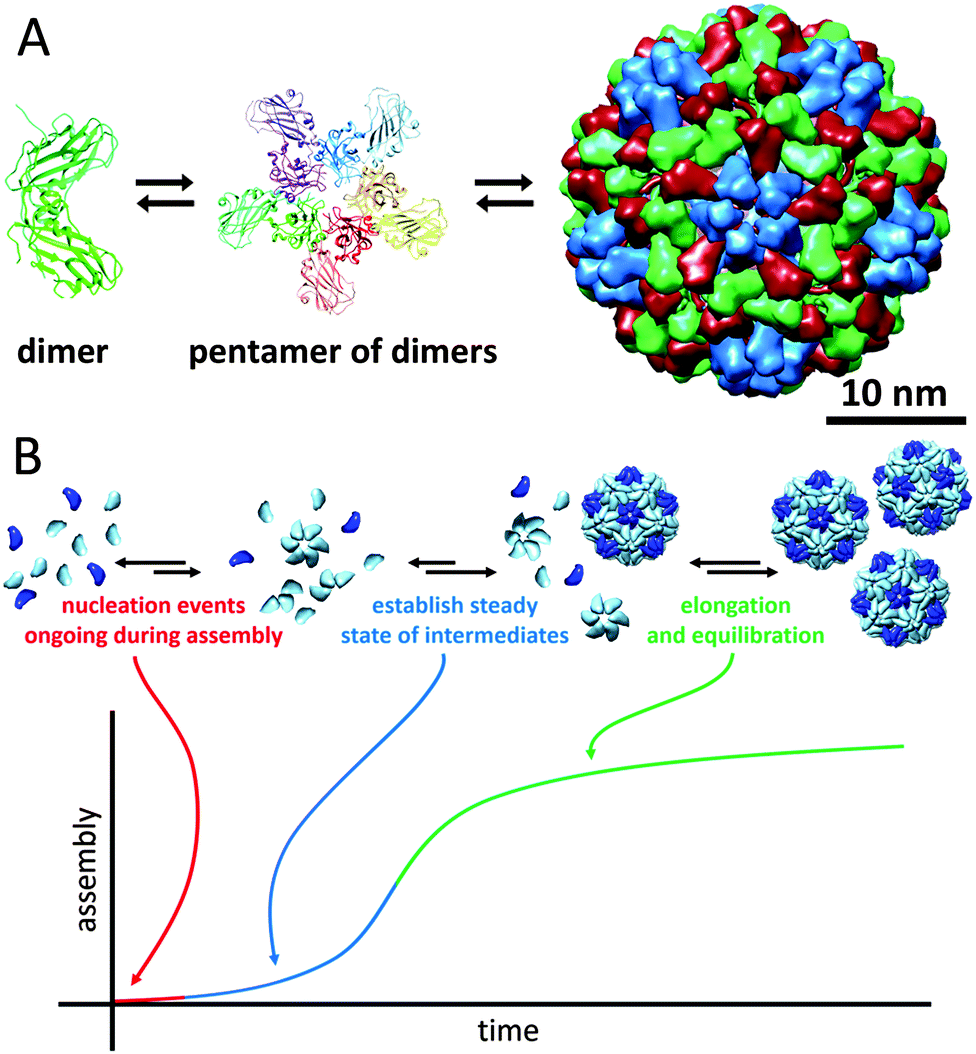 | ||
| Fig. 12 The cowpea chlorotic mottle virus: (A) assembly pathway of CCMV. Assembly is initiated by formation of a pentamer of dimers, which then associates with other dimers to form the complete 180-mer capsid (PDB: 1CWP); (B) assembly kinetics of capsids, in which each individual capsid must arise from an individual nucleation event (figure based on ref. 46, Viral Nanotechnology). | ||
7.6 Bacterial compartments
Bacterial nano- and microcompartments are reaction chambers that enclose enzymes and other proteins. These supramolecular architectures protect the bacterial cell by sequestering toxic intermediates and by shielding delicate processes from the environment. Encapsulation of enzymatic cascades can also be advantageous because it can result in increased reaction efficiencies. In this section we discuss several examples of proteinaceous bacterial compartments that do not have eukaryotic counterparts.Lumazine synthase is an enzyme that occurs in different topologies. In Bacillaceae, it forms a unique core–shell complex, consisting of an icosahedral shell of 60 lumazine synthase subunits and a core of three riboflavin synthase subunits, with a diameter of 16 nm (Fig. 13A). This supramolecular enzyme complex is involved in the catalysis of the final steps in the synthesis of riboflavin (vitamin B2). This compartmentalisation is suggested to offer protection to intermediates and provide a reaction rate enhancement as a result of substrate-channelling between the active-sites, which are in optimal proximity on the internal surface of the capsid. However, this does not fully explain the structural complexity, because in many other organisms lumazine synthase exists as an empty capsule or as a pentameric or decameric complex, indicating that this remarkable organisation is not required for the function of these enzymes. Furthermore, the recombinantly expressed lumazine synthase from hyperthermophilic bacterium Aquifex aeolicus has been studied for its thermostability, and with an apparent melting temperature of 120 °C it is one of the most thermostable proteins known.47
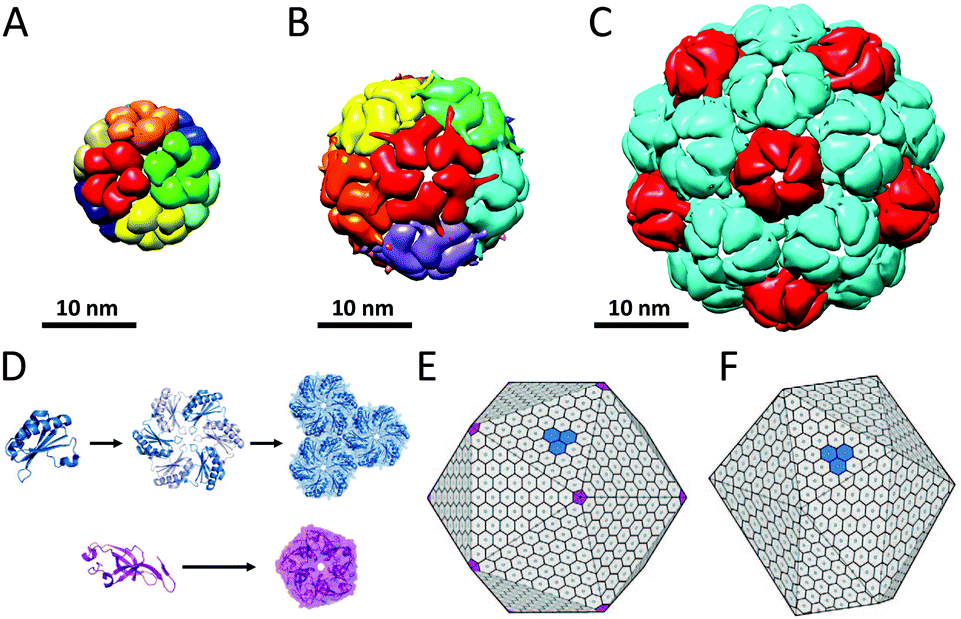 | ||
| Fig. 13 Bacterial nano- and microcompartments: (A) lumazine synthase (PDB: 1RVV); (B) encapsulin 60-mer (PDB: 3DKT); (C) encapsulin 180-mer (PDB: 4PT2); (D) assembly of monomers of the carboxysome shell bacterial microcompartment (BMC). In carboxysomes, the BMC shell proteins assemble into hexamers (blue), which are the main building blocks of the shell, whereas pentameric protein subunits (purple) form the vertices of the shell assemble. Carboxysomes closely resemble a regular icosahedron (E), while pdu microcompartments are less geometrically regular (F). Reprinted from Current Opinion in Structural Biology, Yeates et al.50 © 2011, with permission from Elsevier. | ||
Encapsulins form another example of a widespread class of conserved archaeal and bacterial proteins that assembly into a nanocompartment for the encapsulation of enzymes. These homomultimeric reaction chambers arise from encapsulin monomers that were shown to assemble around proteins involved in oxidative stress. Encapsulin nanocompartments consisting of 60 and 180 monomers forming thin icosahedral shells with a diameter of 24 and 32 nm, respectively, have been reported (Fig. 13B and C).48 Packaging of enzymes is directed by their C-terminal peptide sequence that anchors the enzymes to the interior of the capsules.49 Despite the very different functions of encapsulins and viral capsid proteins, there is a striking similarity in the tertiary structure of the HK97 family capsid proteins and the encapsulin monomers, which suggest a common ancestor, although the unique catenation of HK97 bacteriophage has not been observed.
In addition to these well-defined bacterial nanocompartments, nature also produces larger very sophisticated proteinaceous microcompartments, which accommodate two or more sequentially acting enzymes. Carboxysomes (Fig. 13E), propanediol utilisation operon (pdu, Fig. 13F) and ethanolamine utilisation (Eut) microcompartments are well-studied examples of these protein-based organelles that have been reviewed.50 The Eut microcompartment, for example, sequesters the metabolic pathway for ethanolamine to prevent exposure of the bacterial cytosol to the reactive acetaldehyde intermediate.51 Bacterial microcompartment (BMC) shells range from 100 to 150 nm and are composed of a few thousand BMC shell proteins. BMC monomers assemble into hexameric discs that form the basic building block of the shell. In carboxysomes, BMC shell proteins forming pentamers have been found that occupy the vertices of an icosahedral shell (Fig. 13D and E).50 In contrast to the encapsulin family, no viral counterparts are known for this class of bacterial protein cages.
8. Conclusion and outlook
Nature has built with high precision an astonishing number of versatile and functionally well-defined protein architectures. The knowledge on how multimeric protein assemblies spatially arrange and how the three dimensional structure of a protein affects its function may stimulate new research endeavours of the next generation of supramolecular chemists and chemical biologists. Reflecting on the discoveries in the past few decades, it is striking to see how many wonderful architectures nature has produced and one can envision, that with the expected significant advances in chemical and biological research in the coming decade we will discover more intrinsically novel and perhaps unimaginable architectures than currently known in the protein world. The supramolecular protein assemblies presented in this review already exhibit a high level of complexity, but there are even more complex assemblies that are built on proteins only (such as rotary motors, e.g. ATP synthase) or on protein–DNA and protein–RNA complexes (such as the nucleosome and the ribosome).In science the field of supramolecular protein assemblies has successfully passed its first vital phase, in which fundamental biochemical discoveries highlight the great natural diversity of protein structures and a phase in which the examination of the structure–function relationship has led to a deeper insight in the mechanism of enzyme action and the properties of protein materials. It has now entered the era in which our fundamental understanding of protein assemblies will deepen, such that it can be used for the rational design of new biomolecular systems that exhibit completely novel functions and properties. The research on supramolecular protein assemblies has typically been carried out in simple buffer solutions at low protein concentrations, whereas the ‘living environment’ of proteins is the crowded habitat of the cell. It remains to be established whether the results and conclusions from in vitro experiments on protein assemblies reflect the formation of these assemblies in the natural cellular environment, in which concentrations are high and the medium is much more complex containing many different species that all interact with each other.3 Moreover, it is currently not clear whether the formation of multisubunit protein assemblies typically occurs cotranslationally, although there is some evidence that this might be the case for some of them. Nature will remain to serve as a vital inspiration for the research activities in the area of supramolecular protein assemblies.
Acknowledgements
We thank the Netherlands Research School for Chemical Biology (NRSCB, J. M.), the European Research Council (ERC Advanced Grant, ALPROS-290886, R. J. M. N.) and the Netherlands Organisation for Scientific Research (Rubicon fellowship, M. B. v. E.) for financial support.Notes and references
- G. A. Petsko and D. Ringe, Protein Structure and Function, New Sciences Press, 2004 Search PubMed.
- J. A. Marsh and S. A. Teichmann, Annu. Rev. Biochem., 2015, 84, 551–575 CrossRef CAS PubMed.
- C. V. Robinson, A. Sali and W. Baumeister, Nature, 2007, 450, 973–982 CrossRef CAS PubMed.
- R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed.
- N. P. King and Y.-T. Lai, Curr. Opin. Struct. Biol., 2013, 23, 632–638 CrossRef CAS PubMed.
- J. Zhang, F. Zheng and G. Grigoryan, Curr. Opin. Struct. Biol., 2014, 27, 79–86 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- R. Dominguez and K. C. Holmes, Annu. Rev. Biophys., 2011, 40, 169–186 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- M. M. Hingorani and M. O'Donnell, Nat. Rev. Mol. Cell Biol., 2000, 1, 22–30 CrossRef CAS PubMed.
- S. F. M. van Dongen, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2014, 53, 11420–11428 CrossRef CAS PubMed.
- X.-P. Kong, R. Onrust, M. O'Donnell and J. Kuriyan, Cell, 1992, 69, 425–437 CrossRef CAS PubMed.
- M. Hedglin, R. Kumar and S. J. Benkovic, Cold Spring Harbor Perspect. Biol., 2013, 5, a010165 Search PubMed.
- B. Liu, W. K. Eliason and T. A. Steitz, Nat. Commun., 2013, 4, 2495 Search PubMed.
- R. A. Kovall and B. W. Matthews, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7893–7897 CrossRef CAS.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS PubMed.
- J. M. Alonso, M. Ł. Górzny and A. M. Bittner, Trends Biotechnol., 2013, 31, 530–538 CrossRef CAS PubMed.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1866 CrossRef CAS PubMed.
- J. Jiang, B. L. Pentelute, R. J. Collier and Z. H. Zhou, Nature, 2015, 521, 545–549 CrossRef CAS PubMed.
- L. Sun, L. N. Young, X. Zhang, S. P. Boudko, A. Fokine, E. Zbornik, A. P. Roznowski, I. J. Molineux, M. G. Rossmann and B. A. Fane, Nature, 2014, 505, 432–435 CrossRef CAS PubMed.
- E. R. Ballister, A. H. Lai, R. N. Zuckermann, Y. Cheng and J. D. Mougous, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3733–3738 CrossRef CAS PubMed.
- W. R. Wikoff, L. Liljas, R. L. Duda, H. Tsuruta, R. W. Hendrix and J. E. Johnson, Science, 2000, 289, 2129–2133 CrossRef CAS PubMed.
- D. R. Boutz, D. Cascio, J. Whitelegge, L. J. Perry and T. O. Yeates, J. Mol. Biol., 2007, 368, 1332–1344 CrossRef CAS PubMed.
- B. I. Lee, K. H. Kim, S. J. Park, S. H. Eom, H. K. Song and S. W. Suh, EMBO J., 2004, 23, 2029–2038 CrossRef CAS PubMed.
- Z. Cao, A. W. Roszak, L. J. Gourlay, J. G. Lindsay and N. W. Isaacs, Structure, 2005, 13, 1661–1664 CrossRef CAS PubMed.
- C. M. Zimanyi, N. Ando, E. J. Brignole, F. J. Asturias, J. Stubbe and C. L. Drennan, Structure, 2012, 20, 1374–1383 CrossRef CAS PubMed.
- M. J. Smeulders, T. R. M. Barends, A. Pol, A. Scherer, M. H. Zandvoort, A. Udvarhelyi, A. F. Khadem, A. Menzel, J. Hermans, R. L. Shoeman, H. J. C. T. Wessels, L. P. van den Heuvel, L. Russ, I. Schlichting, M. S. M. Jetten and H. J. M. Op den Camp, Nature, 2011, 478, 412–416 CrossRef CAS PubMed.
- M. B. van Eldijk, I. van Leeuwen, V. A. Mikhailov, L. Neijenhuis, H. R. Harhangi, J. C. M. van Hest, M. S. M. Jetten, H. J. M. Op den Camp, C. V. Robinson and J. Mecinović, Chem. Commun., 2013, 49, 7770–7772 RSC.
- J. C.-Y. Wang, A. Zlotnick and J. Mecinović, Chem. Commun., 2014, 50, 10281–10283 RSC.
- M. B. van Eldijk, B. J. Pieters, V. A. Mikhailov, C. V. Robinson, J. C. M. van Hest and J. Mecinović, Chem. Sci., 2014, 5, 2879–2884 RSC.
- P. Virnau, A. Mallam and S. Jackson, J. Phys.: Condens. Matter, 2011, 23, 033101 CrossRef PubMed.
- T. O. Yeates, T. S. Norcross and N. P. King, Curr. Opin. Chem. Biol., 2007, 11, 595–603 CrossRef CAS PubMed.
- M. Jamroz, W. Niemyska, E. J. Rawdon, A. Stasiak, K. C. Millett, P. Sułkowski and J. I. Sulkowska, Nucleic Acids Res., 2015, 43, D306–D314 CrossRef PubMed.
- A. L. Mallam, J. M. Rogers and S. E. Jackson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8189–8194 CrossRef CAS PubMed.
- G. Kolesov, P. Virnau, M. Kardar and L. A. Mirny, Nucleic Acids Res., 2007, 35, W425–W428 CrossRef PubMed.
- A. L. Mallam and S. E. Jackson, Nat. Chem. Biol., 2012, 8, 147–153 CrossRef CAS PubMed.
- E. W. W. Leung and L. W. Guddat, J. Mol. Biol., 2009, 389, 167–182 CrossRef CAS PubMed.
- F. I. Andersson, D. G. Pina, A. L. Mallam, G. Blaser and S. E. Jackson, FEBS J., 2009, 276, 2625–2635 CrossRef CAS PubMed.
- D. Bölinger, J. I. Sułkowska, H.-P. Hsu, L. A. Mirny, M. Kardar, J. N. Onuchic and P. Virnau, PLoS Comput. Biol., 2010, 6, e1000731 Search PubMed.
- X. Liu and E. C. Theil, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- Y. Zhang and B. P. Orner, Int. J. Mol. Sci., 2011, 12, 5406 CrossRef CAS PubMed.
- A. Fotin, Y. Cheng, P. Sliz, N. Grigorieff, S. C. Harrison, T. Kirchhausen and T. Walz, Nature, 2004, 432, 573–579 CrossRef CAS PubMed.
- H. Tanaka, K. Kato, E. Yamashita, T. Sumizawa, Y. Zhou, M. Yao, K. Iwasaki, M. Yoshimura and T. Tsukihara, Science, 2009, 323, 384–388 CrossRef CAS PubMed.
- J. Mrazek, D. Toso, S. Ryazantsev, X. Zhang, Z. H. Zhou, B. C. Fernandez, V. A. Kickhoefer and L. H. Rome, ACS Nano, 2014, 8, 11552–11559 CrossRef CAS PubMed.
- H. Saibil, Nat. Rev. Mol. Cell Biol., 2013, 14, 630–642 CrossRef CAS PubMed.
- A. Zlotnick, S. Francis, L. S. Lee and J. C.-Y. Wang, in Viral Nanotechnology, ed. Y. Khudyakov and P. Pumpens, CRC Press, 2015, ch. 2, pp. 13–26 Search PubMed.
- R. Ladenstein, M. Fischer and A. Bacher, FEBS J., 2013, 280, 2537–2563 CrossRef CAS PubMed.
- C. A. McHugh, J. Fontana, D. Nemecek, N. Cheng, A. A. Aksyuk, J. B. Heymann, D. C. Winkler, A. S. Lam, J. S. Wall, A. C. Steven and E. Hoiczyk, EMBO J., 2014, 33, 1896–1911 CrossRef CAS PubMed.
- M. Sutter, D. Boehringer, S. Gutmann, S. Gunther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban and N. Ban, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CAS.
- T. O. Yeates, M. C. Thompson and T. A. Bobik, Curr. Opin. Struct. Biol., 2011, 21, 223–231 CrossRef CAS PubMed.
- S. Tanaka, M. R. Sawaya and T. O. Yeates, Science, 2010, 327, 81–84 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2016 |