
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin–orbit effects on the 119Sn magnetic-shielding tensor in solids: a ZORA/DFT investigation†
Fahri
Alkan
*a,
Sean T.
Holmes
a,
Robbie J.
Iuliucci
b,
Karl T.
Mueller
cd and
Cecil
Dybowski
*a
aDepartment of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: alkan@udel.edu; dybowski@udel.edu
bDepartment of Chemistry, Washington and Jefferson College, Washington, PA 15301, USA
cDepartment of Chemistry, Pennsylvania State University, University Park, PA 16802, USA
dPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 23rd June 2016
Abstract
Periodic-boundary and cluster calculations of the magnetic-shielding tensors of 119Sn sites in various co-ordination and stereochemical environments are reported. The results indicate a significant difference between the predicted NMR chemical shifts for tin(II) sites that exhibit stereochemically-active lone pairs and tin(IV) sites that do not have stereochemically-active lone pairs. The predicted magnetic shieldings determined either with the cluster model treated with the ZORA/Scalar Hamiltonian or with the GIPAW formalism are dependent on the oxidation state and the co-ordination geometry of the tin atom. The inclusion of relativistic effects at the spin–orbit level removes systematic differences in computed magnetic-shielding parameters between tin sites of differing stereochemistries, and brings computed NMR shielding parameters into significant agreement with experimentally-determined chemical-shift principal values. Slight improvement in agreement with experiment is noted in calculations using hybrid exchange–correlation functionals.
1. Introduction
Solid-state nuclear magnetic resonance (NMR) spectroscopy is an important tool for studying 119Sn-containing materials because experimental parameters, particularly the magnetic-shielding tensor, are quite sensitive to structural features.1–3 Experimental 119Sn NMR parameters3–7 depend on the local co-ordination geometry and share important similarities with those of 207Pb.8–10 For instance, tin(II) species generally exhibit hemidirected co-ordination chemistry11,12 with characteristically wide chemical-shift spans (Ω = |δ33 − δ11|) of 600–1200 ppm.5,6,13 In contrast, tin(IV) species generally exhibit holodirected co-ordination chemistry11,14,15 with spans under 400 ppm.4,5 The differences in magnetic-shielding parameters between the two structural motifs reflect the fact that crystal structures of tin(II) compounds usually have large void spaces to accommodate the lone pair of electrons, whereas the co-ordination geometry around tin(IV) sites is more nearly spherically symmetric. A recent computational study of 207Pb NMR parameters illustrates the differences among NMR parameters of lead nuclei at sites of different stereochemistry and how these differences are enhanced by relativistic effects, particularly by spin–orbit (SO) coupling.16 A similar result is expected for 119Sn.Quantum-chemical calculations provide the connection between NMR parameters and structure.17,18 For NMR-active nuclei in period 6, such as 195Pt, 199Hg, and 207Pb, one must use relativistic theory, including SO coupling, for accurate predictions of the principal components of the magnetic-shielding tensors, and thus parameters such as the isotropic chemical shift.19–31 For nuclei in period 5 (119Sn, 125Te, 113Cd), there remains some ambiguity in the literature regarding the impact of relativistic effects on calculated NMR parameters.22,32–37 Benchmark studies are important in determining the efficacy of various computational protocols.
There are suggestions in the literature that relativistic effects, particularly SO coupling, are important in calculating magnetic shielding of 119Sn nuclei.38–40 For example, Malkin et al.41 recently proposed that the 119Sn magnetic-shielding scale should be increased by around 1000 ppm, a result obtained from the four-component (4c) relativistic theory. Strong correlations between experiment and theory have been suggested for various tin-containing molecules at the non-relativistic DFT level as well.33,42,43 Non-relativistic calculations are usually justified by the possible cancellation of relativistic effects when calculated 119Sn magnetic shieldings are converted to the chemical-shift scale (relative to a reference compound such as Sn(CH3)4).22,34 In other studies, it is suggested that SO coupling cannot be ignored for accurate predictions of chemical shifts.37,44 Bagno et al.37 have discussed applying the zeroth-order regular approximation (ZORA) to 119Sn magnetic shielding and spin–spin coupling constants of tin(IV)-containing species. They have shown that inclusion of SO coupling is important when other heavy atoms such as bromine or iodine are directly bonded to tin.37 When tin is coordinated to lighter atoms, chemical shifts and spin–spin couplings calculated with inclusion of SO coupling and without it are quite similar, suggesting that inclusion of such terms is not significant.37 Predicted isotropic 119Sn chemical shifts in tin-containing solids calculated with plane-wave DFT techniques employing relativistic pseudopotentials at the ZORA level (without the SO component) are in good agreement with isotropic chemical shifts, although the calculated values do deviate by up to 200 ppm from experiment in some cases.45
The role of SO coupling on calculated magnetic-shielding tensors is highly system-dependent.16 In this study, we present calculations of the principal components of 119Sn magnetic-shielding tensors for tin sites with different oxidation states and different co-ordination environments. In all cases, we compare the tensor elements, rather than isotropic shifts.29,31 Comparisons of various theoretical approaches are presented, with or without SO coupling, using the ZORA-DFT methodology. Calculations are performed in plane-wave and cluster-based frameworks to demonstrate the differences in these two approaches. The utility of cluster-based calculations for the predictions of magnetic-shielding tensors of nuclei in solids has been established in several studies.29,31,46–55 The effect of hybrid functionals on computed magnetic shielding is also assessed. Our motivation is to understand the impact of various theoretical considerations on the quality of predicted values of magnetic shielding for 119Sn-containing systems.
2. Computational details
Twelve tin-containing solids with known X-ray or neutron diffraction structures and with known principal components of the chemical-shift tensor determined by solid-state NMR spectroscopy have been investigated. The tin(II)-containing solids are SnO,15,56 SnHPO4,13,57 SnHPO3,13,57 SnC2O4,13,58 SnSO413,59 and BaSnF4.13,60 The tin(IV)-containing solids are SnO2,5,12 Ca2SnO4,4,61 SnS2,7,62 Pb2SnO4,63,64 Na6Sn2S77,65 and Sr2SnO4.4,66 The 119Sn magnetic-shielding tensors in these solids were calculated using (1) a cluster-based approach and (2) a periodic approach, as discussed below.Calculations on cluster models were performed using the Amsterdam Density Functional (ADF) program package.67–69 All clusters were expanded around the 119Sn site up to the third co-ordination shell. The all-electron (AE) TZ2P basis set was employed for the NMR-active nucleus (119Sn) and the first co-ordination shell around the NMR-active nucleus, whereas the remainder of the cluster was treated with the smaller AE TZP basis set. Two example clusters, including a schematic of the partitioning of the basis sets, are illustrated in Fig. 1. Calculations were carried out using the PBE70 or PBE071 density functionals. Relativistic effects were incorporated using the ZORA Hamiltonian at the scalar (ZORA/SC) or the spin–orbit (ZORA/SO) level.72–75 Magnetic-shielding tensors were calculated using the GIAO76,77 formalism as implemented in ADF2014.19,78–80 A linear-dependence threshold of 10−4 was applied for the cluster calculations employing the PBE functional. For calculations employing the PBE0 functional, a more stringent threshold parameter was necessary for numerical problems associated with the linear dependence of the basis functions; therefore the threshold parameter was increased to 5 × 10−3 in these calculations. Effects of this procedure on the calculated magnetic-shielding tensor are given in the ESI† in Table S1 and Fig. S1.
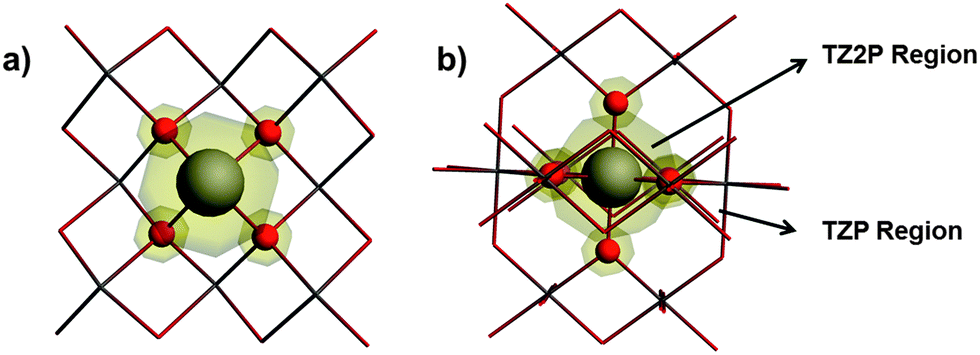 | ||
| Fig. 1 Cluster models for (a) SnO and (b) SnO2. The central ball-and-stick region, representing the NMR-active 119Sn center and the first-co-ordination shell, are treated with the TZ2P basis set. The outer co-ordination shells are treated with the smaller TZP basis set. | ||
For materials containing hydrogen atoms, a preliminary geometry optimization was run at the scalar (SC) level where the positions of the light atoms were allowed to relax while the heavy atoms remained fixed at their experimental coordinates. See the ESI† for details of the coordinates for each material. The terminal atoms of the clusters were treated with valence modification of terminal atoms using bond valence theory81–84 or VMTA/BV.31 In this scheme, the valence of a terminal atom is modified by altering the nuclear charge, Zmod, by the following relation:
| Zmod = Znuc + ΔS | (1) |
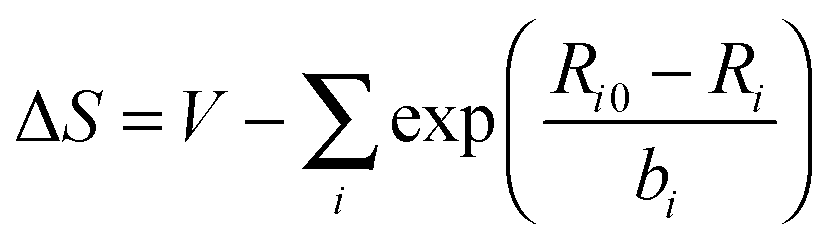 | (2) |
Calculations employing periodic-boundary conditions (PBCs) were performed using the CASTEP module of Materials Studio 7.0.84,85 These calculations were performed at the PBE level with core orbitals replaced by ultrasoft pseudopotentials generated on the fly and with a plane-wave cutoff energy of 600 eV.85 Convergence of the computed magnetic-shielding parameters was tested by running several additional calculations with higher cutoff energies and finer k-point grids for integration over the Brillouin zone. Calculations of the 119Sn magnetic-shielding tensors employed the GIPAW method of Pickard and Mauri.86 Relativistic effects were included at the ZORA/SC level through the pseudopotential approximation of Yates and co-workers.87
3. Results and discussion
3.1. Spin–orbit effects on calculated 119Sn magnetic-shielding tensors
All calculations discussed in this subsection were performed with the PBE density functional. Fig. 2 shows the correlations between the principal components of calculated magnetic-shielding tensors and the principal components of experimental chemical-shift tensors. Calculations were performed with the periodic PBE/GIPAW method (Fig. 2a), at the PBE/ZORA/SC level (Fig. 2b), and at the PBE/ZORA/SO level (Fig. 2c). Table 1 presents the parameters of the linear best-fit lines for tin(II) and tin(IV)-containing solids.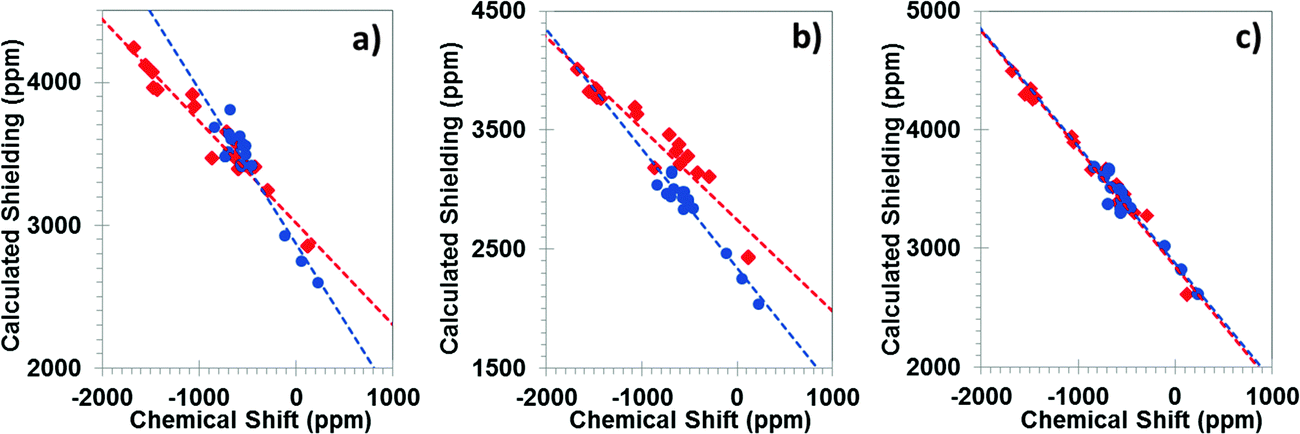 | ||
| Fig. 2 Correlations between calculated principal components of 119Sn magnetic-shielding tensors and experimental 119Sn chemical-shift tensors for twelve tin-containing solids, as determined with different methodologies. Computed shielding constants were obtained using (a) the PBE/GIPAW method, (b) the PBE/ZORA/SC method, and (c) the PBE/ZORA/SO method. Tin(II) sites are shown in red; tin(IV) sites are shown in blue. | ||
| Method | Slope | σ ref (ppm) | R 2 |
|---|---|---|---|
| Tin(ii)-containing solids | |||
| PBE/GIPAW | −0.71 ± 0.04 | 3019 ± 38 | 0.95 |
| PBE/ZORA/SC | −0.77 ± 0.06 | 2745 ± 60 | 0.91 |
| PBE/ZORA/SO | −0.99 ± 0.03 | 2849 ± 34 | 0.98 |
| Tin(iv)-containing solids | |||
| PBE/GIPAW | −1.08 ± 0.10 | 2869 ± 55 | 0.89 |
| PBE/ZORA/SC | −1.00 ± 0.07 | 2338 ± 41 | 0.92 |
| PBE/ZORA/SO | −0.99 ± 0.06 | 2875 ± 37 | 0.94 |
| All Systems | |||
| PBE/GIPAW | −0.77 ± 0.04 | 3001 ± 36 | 0.90 |
| PBE/ZORA/SC | −0.92 ± 0.07 | 2499 ± 52 | 0.85 |
| PBE/ZORA/SO | −0.98 ± 0.03 | 2867 ± 22 | 0.97 |
For tin(II)-containing solids analyzed as a separate subset (Table 1), the correlation between calculated magnetic shielding at the SC level and experimental chemical-shift values deviates significantly from ideal agreement. The deviation from the ideal case (slope = −1.00) is 29% and 23% for the linear best-fit lines obtained using periodic PBE/GIPAW and PBE/ZORA/SC methods, respectively. The extrapolated shielding of the reference compound, σref, given by the intercept of the best-fit line, is 3019 ppm by the PBE/GIPAW method. The PBE/ZORA/SC value of σref is 2745 ppm. For the subset of tin(IV)-containing solids, the PBE/GIPAW and PBE/ZORA/SC methods are much closer to the ideal value of −1.00. However, the predicted reference shieldings predicted by the two methods differ by 531 ppm. These results indicate that the predicted magnetic shieldings determined either with the cluster model treated with the ZORA/SC Hamiltonian or with the GIPAW formalism with scalar-relativistic pseudopotentials are dependent on the oxidation state and the co-ordination geometry of the tin atom in the solid system.
In Table 2, we present the predicted chemical-shift parameters resulting from each method, along with reported experimental values for the twelve tin-containing solids. Calculated magnetic-shielding parameters have been converted to the chemical-shift scale using the predicted σref from the linear best-fit correlations for all systems given in Table 1. The residuals between the experimental and calculated principal components of the chemical-shift tensors are given in Table 1, which is a measure of the overall quality of performance of each computational methodology (Table 2).
| Compounds | δ 11 (ppm) | δ 22 (ppm) | δ 33 (ppm) | δ iso (ppm) | Ω (ppm) | Residuala (ppm) |
|---|---|---|---|---|---|---|
a
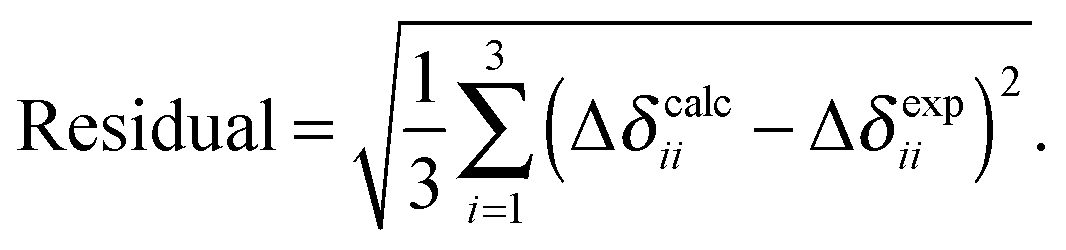
|
||||||
| Tin(II)-containing solids | ||||||
| SnO | 121 | 121 | −867 | −208 | 988 | — |
| PBE/GIPAW | 145 | 145 | −472 | −61 | 617 | 229 |
| PBEZORA/SC | 45 | 45 | −677 | −196 | 722 | 126 |
| PBE/ZORA/SO | 256 | 253 | −793 | −94 | 1049 | 117 |
| SnHPO4 | −606 | −712 | −1553 | −957 | 947 | — |
| PBE/GIPAW | −564 | −655 | −1119 | −779 | 555 | 254 |
| PBE/ZORA/SC | −874 | −954 | −1323 | −1050 | 449 | 247 |
| PBE/ZORA/SO | −669 | −808 | −1429 | −969 | 760 | 97 |
| SnHPO3 | −290 | −420 | −1435 | −715 | 1145 | — |
| PBE/GIPAW | −247 | −409 | −949 | −535 | 702 | 282 |
| PBE/ZORA/SC | −602 | −638 | −1262 | −834 | 660 | 241 |
| PBE/ZORA/SO | −405 | −430 | −1402 | −745 | 996 | 69 |
| SnC2O4 | −523 | −639 | −1474 | −879 | 951 | — |
| PBE/GIPAW | −421 | −479 | −965 | −622 | 544 | 314 |
| PBE/ZORA/SC | −778 | −816 | −1266 | −953 | 488 | 216 |
| PBE/ZORA/SO | −587 | −651 | −1392 | −877 | 805 | 61 |
| SnSO4 | −1047 | −1070 | −1679 | −1265 | 632 | — |
| PBE/GIPAW | −834 | −912 | −1239 | −995 | 405 | 297 |
| PBE/ZORA/SC | −1130 | −1183 | −1510 | −1274 | 380 | 127 |
| PBE/ZORA/SO | −1028 | −1075 | −1630 | −1245 | 602 | 30 |
| BaSnF4 | −596 | −596 | −1486 | −893 | 890 | — |
| PBE/GIPAW | −394 | −394 | −1073 | −620 | 679 | 290 |
| PBE/ZORA/SC | −708 | −708 | −1340 | −919 | 632 | 124 |
| PBE/ZORA/SO | −520 | −520 | −1478 | −839 | 958 | 62 |
| Tin(IV)-containing solids | ||||||
| SnO2 | −550 | −573 | −686 | −603 | 136 | — |
| PBE/GIPAW | −564 | −617 | −633 | −605 | 69 | 41 |
| PBE/ZORA/SC | −471 | −475 | −631 | −526 | 160 | 79 |
| PBE/ZORA/SO | −605 | −630 | −785 | −673 | 180 | 73 |
| Ca2SnO4 | −459 | −512 | −664 | −545 | 205 | — |
| PBE/GIPAW | −415 | −491 | −597 | −501 | 182 | 48 |
| PBE/ZORA/SC | −334 | −389 | −495 | −406 | 161 | 141 |
| PBE/ZORA/SO | −474 | −529 | −647 | −550 | 173 | 16 |
| SnS2 | −730 | −730 | −835 | −765 | 105 | — |
| PBE/GIPAW | −474 | −475 | −684 | −544 | 211 | 226 |
| PBE/ZORA/SC | −456 | −456 | −527 | −479 | 71 | 286 |
| PBE/ZORA/SO | −741 | −742 | −819 | −767 | 77 | 13 |
| Pb2SnO4 | −558 | −566 | −692 | −605 | 134 | — |
| PBE/GIPAW | −410 | −419 | −512 | −447 | 101 | 159 |
| PBE/ZORA/SC | −328 | −421 | −436 | −395 | 108 | 216 |
| PBE/ZORA/SO | −436 | −468 | −509 | −471 | 73 | 139 |
| Na6Sn2S7 | 232 | 60 | −107 | 62 | 339 | — |
| PBE/GIPAW | 409 | 255 | 78 | 247 | 331 | 186 |
| PBE/ZORA/SC | 468 | 257 | 46 | 257 | 423 | 199 |
| PBE/ZORA/SO | 249 | 41 | −150 | 47 | 399 | 29 |
| Sr2SnO4 | −510 | −548 | −681 | −580 | 171 | — |
| PBE/GIPAW | −551 | −551 | −805 | −636 | 253 | 75 |
| PBE/ZORA/SC | −407 | −412 | −645 | −488 | 238 | 100 |
| PBE/ZORA/SO | −536 | −539 | −801 | −625 | 265 | 71 |
For tin(II)-containing solids, PBE/GIPAW calculations consistently give large deviations from experimental values, with residuals ranging between 229 and 314 ppm. For all tin(II)-containing solids, the δ33 component has the largest deviation between experiment and theory, when using the PBE/GIPAW approach. The performance of PBE/ZORA/SC calculations is somewhat better, as the residuals range from 124 to 250 ppm. With both computational protocols, the calculated spans (Ω = |δ33 − δ11|) are 200–500 ppm smaller than the experimental values. The agreement between experiment and theory is considerably stronger with the PBE/ZORA/SO method for tin(II)-containing solids, with residuals under 100 ppm for five of the six systems. Spans predicted by the PBE/ZORA/SO calculations are in better agreement with experiment than spans obtained by PBE/GIPAW and PBE/ZORA/SC calculations.
For tin(IV)-containing solids, the performance of PBE/GIPAW shows some improvement over its performance in calculations of the chemical shifts in tin(II) systems. For example, the accuracies of calculated principal components for SnO2, Ca2SnO4, Pb2SnO4 and Sr2SnO4 by the PBE/GIPAW method are comparable to results obtained with PBE/ZORA/SO methods. However, the deviation of PBE/GIPAW results from experimental values is not as good for materials like SnS2 and Na6Sn2S7. In these latter systems, the first co-ordination shell around tin consists of sulfur atoms rather than oxygen atoms. The magnitude of SO effects on 119Sn magnetic shielding is probably increased by the presence of the heavier sulfur atom in the co-ordination environment. In these two cases, the residuals determined with the PBE/ZORA/SO calculations are 13 and 29 ppm, respectively, whereas residuals by the PBE/ZORA/SC are 286 and 199 ppm.
To understand the effect of SO coupling on the 119Sn magnetic-shielding tensor, we present the differences (Δσii) between principal components of magnetic-shielding tensors calculated at the PBE/ZORA/SO level and those calculated at the PBE/ZORA/SC level (Fig. 3). It is evident that SO effects on magnetic-shielding tensors exhibit a strong dependence on the oxidation state of tin. For tin(II)-containing systems, the contribution of SO effects on magnetic shielding is largest for the σ33 component where Δσ33 values are around 500 ppm. The SO effects are less for σ11 and σ22, with Δσii ranging between 154 and 260 ppm. In comparison, the contribution of SO effects on each principal component of the magnetic-shielding tensor is more uniform for tin(IV)-containing systems, with Δσii varying between 435 and 654 ppm. The largest change in any magnetic-shielding tensor between PBE/ZORA/SO and PBE/ZORA/SC results is observed for SnS2 where Δσii ∼ 650 ppm. Indeed, among the tin(IV)-containing materials, the residuals of PBE/GIPAW and cluster-based PBE/ZORA/SC results in Table 2 are highest for this compound, due to the presence of significant spin–orbit effects relative to oxygen-co-ordinated tin sites.
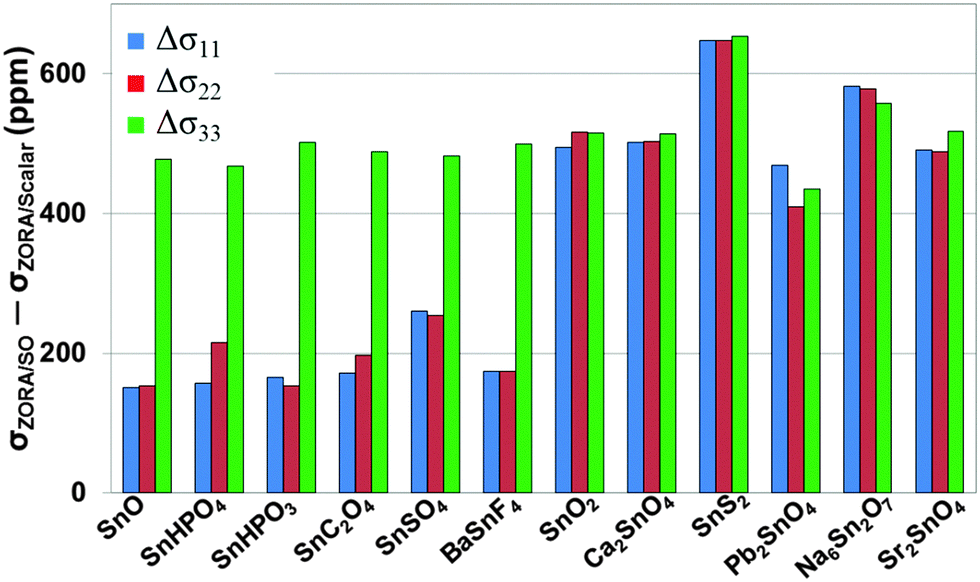 | ||
| Fig. 3 The differences (Δσii) in principal components of 119Sn magnetic-shielding tensors calculated with the ZORA/SO method and the ZORA/SC method. All calculations model the solid-state environment with the cluster-based VMTA/BV approach. Magnetic-shielding calculations use the PBE functional. | ||
The results in Fig. 3 for SO effects on the 119Sn magnetic-shielding tensor show a striking resemblance to the recently-investigated SO effects on co-ordination compounds of lead.16 In the case of 207Pb-containing solids, SO effects show a similar dependence on the oxidation state (+2 or +4) and co-ordination geometry (hemidirected or holodirected) around the 207Pb nuclei. The magnitudes of the SO effects for 119Sn and 207Pb are quite different, as expected. Overall, the magnitude of SO effects (Δσii) for the 119Sn-containing systems varies between 154 and 654 ppm. In comparison, the SO effects on 207Pb magnetic-shielding tensor are generally 2000–3000 ppm. This difference is likely due to the larger nuclear charge on 207Pb, resulting in stronger SO coupling effects on magnetic shielding.
Fig. 4 shows the orientations of the axes of the 119Sn magnetic-shielding tensors obtained at the ZORA/SO level of theory for two tin(II) systems in their local frames of reference. For both SnO and BaSnF4, the principal axis of the most-shielded component is aligned with the symmetry axis of the molecular orbital (MO) which results mostly from the mixing of the 5s and 5p atomic orbitals of the tin nuclei. Such MOs are often associated with the ‘lone-pair’ on an atom. In comparison, the σ11 and σ22 axes are in the plane formed by the tin atoms for both systems. The relationship between the principal axes and the lone-pair of the tin(II) nuclei are analogous to recent findings for hemidirected lead(II) systems.16
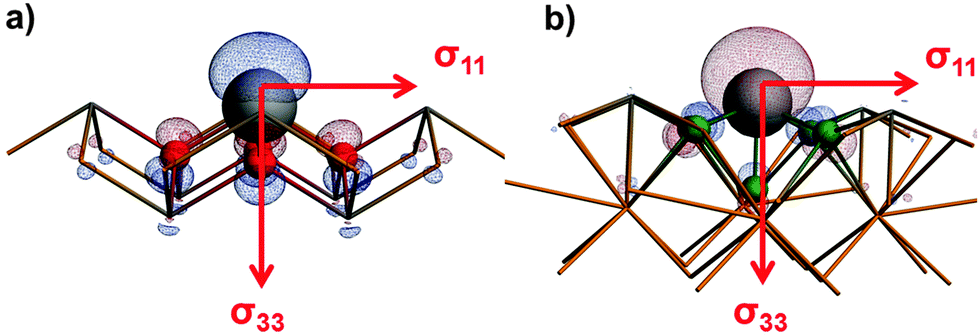 | ||
| Fig. 4 The orientations of magnetic-shielding tensor axes along with MOs associated with the ‘lone pair’ on tin(II) for (a) SnO and (b) BaSnF4. | ||
The accuracy of calculated 119Sn NMR parameters has been systematically investigated for a series of isolated tin(IV) molecules by Bagno et al.,37 using the ZORA/SC and ZORA/SO methods. The results indicate that the both ZORA/SC and ZORA/SO methods work quite well for predicting chemical shifts when no other heavy atom is bound to tin. In such systems, the predicted SO effects on the isotropic magnetic shielding vary by around 500 ppm and mostly cancel out when magnetic shieldings are converted to the chemical-shift scale. These findings partially agree with the PBE/GIPAW and PBEZORA/SC results for tin(IV)-containing solids. However, for tin(II)-containing solids, the assumption that there are negligible SO effects on the chemical shift is incorrect.
The magnetic shielding (or absolute shielding) of tetramethyltin, σref, can be estimated from the intersection of the best-fit correlation lines in Table 1. From the PBE/ZORA/SO method, σref is predicted to be 2867 ppm from the correlation obtained for all tin-containing systems. By comparison, a single calculation on tetramethyltin at the same level of theory gives 2852 ppm for σref, a discrepancy of only 15 ppm. With the 4c relativistic DFT (with the BP86 functional), σref is computed as 3199 ppm.41 It is clear that PBE/ZORA/SO underestimates σref by ∼12% compared to the 4c method. The underestimation of absolute shieldings predicted by ZORA calculations has been discussed previously.27,88,89 Nevertheless, the current results, as well as previous studies on other heavy nuclei such as 207Pb16,31 and 199Hg,29 demonstrate that ZORA/SO predictions for the chemical-shift tensor agree with the experimental values within ∼2%, possibly due to the cancellation of higher-order relativistic effects beyond SO coupling.
In the previous investigations of lighter nuclei such as 13C, 19F or 29Si, the performances of the GIPAW method and cluster models for the predictions of magnetic-shielding tensors in solids are similar, provided that sufficiently large clusters are used for the comparison.53–55,90 In contrast, the current results show that although PBE/GIPAW and cluster-based PBE/ZORA/SC methods yield similar trends for tin(II)-containing and tin(IV)-containing solids, the two methods yield quite different results for σref (Table 1).
To compare the two methods in the absence of solid-state effects, we performed NMR calculations on isolated molecules (SnF2, Sn(CN)2, Sn(OH)2, SnF4, Sn(CH3)4, and SnH4) containing 119Sn in oxidation states of +2 or +4. The results in Table 3 show that for the tin(II) species (SnF2, Sn(CN)2, and Sn(OH)2), the calculated magnetic shieldings determined by the PBE/ZORA/SC and PBE/GIPAW methods are quite similar, with the PBE/GIPAW approach yielding results that are more shielded by 26–58 ppm. On the other hand, the calculated magnetic shieldings of the tin(IV) species (SnF4, Sn(CH3)4, and SnH4) are predicted by the PBE/GIPAW approach to be 362–460 ppm more shielded. Therefore, one should expect that the calculated magnetic shieldings with the PBE/ZORA/SC Hamiltonian and the PBE/GIPAW method deviate from one another for 119Sn nuclei. Moreover, the difference in the calculated magnetic shieldings depends on the electronic structure of the system investigated.
| Moleculeb | σ iso (ppm) | |
|---|---|---|
| PBE/ZORA/SC | PBE/GIPAW | |
| a For GIPAW calculations the isolated molecular state is approximated by employing large cubic unit cells. (a = 20 Å). b Geometries are optimized at PBE/ZORA/SC level of theory. | ||
| SnF2 | 2854 | 2880 |
| Sn(CN)2 | 2053 | 2111 |
| Sn(OH)2 | 2465 | 2516 |
| SnF4 | 3002 | 3364 |
| Sn(CH3)4 | 2370 | 2829 |
| SnH4 | 2982 | 3354 |
3.2. The performance of hybrid DFT methods when combined with ZORA/SO
In general, hybrid density functionals improve the accuracy of prediction of NMR parameters. A recent study of 207Pb-containing solids indicates that the inclusion of Hartree–Fock (HF) exchange via hybrid functionals can have significant effects on calculated principal components of the magnetic-shielding tensor.16 Therefore, we have investigated the performance of the PBE0 functional (with 25% HF exchange), using cluster models and the ZORA/SO Hamiltonian.In Fig. 5, we show the correlation between the principal components of the calculated magnetic-shielding tensor at the PBE0/ZORA/SO level of theory and the principal components of the experimental chemical-shift tensor. The slope of the correlation line when all systems are considered is −1.03 ± 0.02. There is less scatter about the best-fit line (R2 = 0.99) than was obtained at the PBE/ZORA/SO level. In general, the calculated principal components obtained with the PBE0/ZORA/SO method are 100–200 ppm more shielded than the calculated principal components obtained with the PBE/ZORA/SO method. The predicted shielding of the reference compound is found to be 3003 ± 16 ppm, indicating a slightly more shielded value obtained at PBE0/ZORA/SO level of theory than at the PBE/ZORA/SO level.
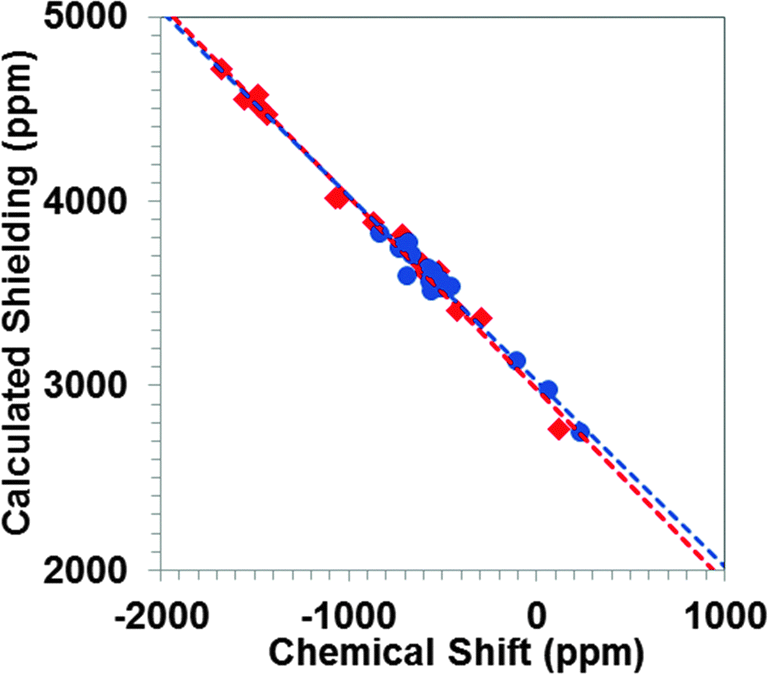 | ||
| Fig. 5 Correlation between calculated principal components of 119Sn magnetic-shielding tensors and experimental 119Sn chemical-shift tensors for twelve tin-containing solids. Calculations were performed at the PBE0/ZORA/SO level of theory. Tin(II) sites are shown in red; tin(IV) sites are shown in blue. | ||
In Table 4, the predicted principal components of the chemical-shift tensors at the PBE0/ZORA/SO level of theory are tabulated, along with the experimental values. For all tin-containing solids, the calculated residuals between theory and experiment are below 100 ppm and the largest residual (94 ppm) is seen for SnO. In general, the agreement between experiment and theory improves when PBE0 is employed instead of PBE. However, this improvement, for most cases, is quite small. As the NMR calculations employing hybrid functionals are considerably larger than for GGA functionals, the latter may be more cost-effective for calculations of 119Sn magnetic-shielding tensor in similar systems.
| Compounds | δ 11 (ppm) | δ 22 (ppm) | δ 33 (ppm) | δ iso (ppm) | Ω (ppm) | Residual (ppm) |
|---|---|---|---|---|---|---|
| Tin(II)-containing solids | ||||||
| SnO | 121 | 121 | −867 | −208 | 988 | — |
| PBE0/ZORA/SO | 236 | 236 | −885 | −138 | 1121 | 94 |
| SnHPO4 | −606 | −712 | −1553 | −957 | 947 | — |
| PBE0/ZORA/SO | −657 | −813 | −1547 | −1006 | 890 | 66 |
| SnHPO3 | −290 | −420 | −1435 | −715 | 1145 | — |
| PBE0/ZORA/SO | −363 | −404 | −1465 | −744 | 1102 | 46 |
| SnC2O4 | −523 | −639 | −1474 | −879 | 951 | — |
| PBE0/ZORA/SO | −618 | −683 | −1512 | −938 | 894 | 65 |
| SnSO4 | −1047 | −1070 | −1679 | −1265 | 632 | — |
| PBE0/ZORA/SO | −1011 | −1015 | −1715 | −1247 | 704 | 43 |
| BaSnF4 | −596 | −596 | −1486 | −893 | 890 | — |
| PBE0/ZORA/SO | −613 | −613 | −1571 | −932 | 958 | 51 |
| Tin(IV)-containing solids | ||||||
| SnO2 | −550 | −573 | −686 | −603 | 136 | — |
| PBE0/ZORA/SO | −617 | −639 | −770 | −675 | 153 | 73 |
| Ca2SnO4 | −459 | −512 | −664 | −545 | 205 | — |
| PBE0/ZORA/SO | −536 | −569 | −713 | −606 | 177 | 62 |
| SnS2 | −730 | −730 | −835 | −765 | 105 | — |
| PBE0/ZORA/SO | −747 | −747 | −821 | −772 | 75 | 16 |
| Pb2SnO4 | −558 | −566 | −692 | −605 | 134 | — |
| PBE0/ZORA/SO | −513 | −558 | −597 | −556 | 85 | 61 |
| Na6Sn2S7 | 232 | 60 | −107 | 62 | 339 | — |
| PBE0/ZORA/SO | 252 | 21 | −134 | 46 | 387 | 30 |
| Sr2SnO4 | −510 | −548 | −681 | −580 | 171 | — |
| PBE0/ZORA/SO | −526 | −531 | −772 | −610 | 246 | 54 |
4. Conclusion
The effects of SO coupling on the magnetic-shielding or chemical-shift tensors of 119Sn nuclei in various tin compounds are significant. Neglecting the SO coupling in calculations (GIPAW or ZORA/SC) usually results in calculated values that deviate by 20–30% from the experimental values for tin(II) compounds. The deviation is clearly seen for the calculated chemical shifts and spans.When the ZORA/SO Hamiltonian and cluster models are employed, one obtains correlations between calculated and experimental principal components of chemical-shift tensors that are very close to the ideal relationship. In fact, using this level of theory provides agreement between results for tin(II)- and tin(IV)-containing solids. The residuals between calculated and experimental principal components are below 100 ppm for the majority of tin-containing solids. These results are in contrast to the GIPAW and ZORA/SC results, where large deviations from experiment are present. At the moment, calculations with the SO Hamiltonian are not available in the GIPAW formalism. Inclusion of the SO Hamiltonian in the GIPAW formalism may improve the systematic deviations of 119Sn magnetic-shielding tensors from experiment.
The agreement between calculated and experimental principal components of chemical-shift tensors is improved further by use of the hybrid PBE0 functional. However, this improvement is rather small for most of the cases we have examined.
Direct comparison of the principal components of calculated magnetic-shielding tensors to the principal components of chemical-shift tensors provides a more stringent test of relativistic effects than does the comparison of isotropic values. For example, the effects of SO coupling on the σ33 component of tin(II)-containing materials are significantly larger than observed for the other two principal components. This observation suggests that SO effects may be present for the principal components of the magnetic-shielding tensors of other period 5 nuclei such as 113Cd and 125Te.
Acknowledgements
C. D. acknowledges the support of the National Science Foundation under Grant CHE-0956006 and K. T. M. acknowledges the support of the National Science Foundation under Grant CHE-1213451. Authors acknowledge the Pennsylvania State University Center for Nanoscale Science for access to Accelrys MATERIALS STUDIO and use of the Lionxj cluster.References
- E. D. Becker, High Resolution NMR: Theory and Chemical Applications, Academic Press, San Diego, 3rd edn, 2000 Search PubMed.
- M. J. Duer, Introduction to Solid-State NMR Spectroscopy, Wiley-Blackwell, Oxford, 2nd edn, 2005 Search PubMed.
- B. Wrackmeyer, Annu. Rep. NMR Spectrosc., 1999, 38, 203–264 CrossRef CAS.
- N. J. Clayden, C. M. Dobson and A. Fern, J. Chem. Soc., Dalton Trans., 1989, 843–847 RSC.
- C. Cossement, J. Darville, J. M. Gilles, J. B. Nagy, C. Fernandez and J. P. Amoureux, Magn. Reson. Chem., 1992, 30, 263–270 CrossRef CAS.
- R. K. Harris, S. E. Lawrence, S. W. Oh and V. G. K. Das, J. Mol. Struct., 1995, 347, 309–319 CrossRef CAS.
- C. Mundus, G. Taillades, A. Pradel and M. Ribes, Solid State Nucl. Magn. Reson., 1996, 7, 141–146 CrossRef CAS PubMed.
- C. Dybowski and G. Neue, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 153–170 CrossRef CAS.
- F. Fayon, I. Farnan, C. Bessada, J. Coutures, D. Massiot and J. P. Coutures, J. Am. Chem. Soc., 1997, 119, 6837–6843 CrossRef CAS.
- G. Neue, C. Dybowski, M. L. Smith, M. A. Hepp and D. L. Perry, Solid State Nucl. Magn. Reson., 1996, 6, 241–250 CrossRef CAS PubMed.
- P. G. Harrison, Orgaometallic Compounds of Germanium, Tin, and Lead, Chapman and Hall, London, 1985 Search PubMed.
- W. H. Baur, Acta Crystallogr., 1956, 9, 515–520 CrossRef CAS.
- P. Amornsakchai, D. C. Apperley, R. K. Harris, P. Hodgkinson and P. C. Waterfield, Solid State Nucl. Magn. Reson., 2004, 26, 160–171 CrossRef CAS PubMed.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65–115 CrossRef CAS.
- J. Pannetier and G. Denes, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2763–2765 CrossRef.
- F. Alkan and C. Dybowski, J. Phys. Chem. A, 2016, 120, 161–168 CrossRef CAS PubMed.
- J. C. Facelli, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 58, 176–201 CrossRef PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- A. Rodriguez-Fortea, P. Alemany and T. Ziegler, J. Phys. Chem. A, 1999, 103, 8288–8294 CrossRef CAS.
- S. K. Wolff, T. Ziegler, E. van Lenthe and E. J. Baerends, J. Chem. Phys., 1999, 110, 7689–7698 CrossRef CAS.
- J. Jokisaari, S. Jarvinen, J. Autschbach and T. Ziegler, J. Phys. Chem. A, 2002, 106, 9313–9318 CrossRef CAS.
- J. Autschbach and S. Zheng, Annu. Rep. NMR Spectrosc., 2009, 67, 1–95 CrossRef CAS.
- L. A. Truflandier and J. Autschbach, J. Am. Chem. Soc., 2010, 132, 3472–3483 CrossRef CAS PubMed.
- B. J. Greer, V. K. Michaelis, M. J. Katz, D. B. Leznoff, G. Schreckenbach and S. Kroeker, Chem. – Eur. J., 2011, 17, 3609–3618 CrossRef CAS PubMed.
- J. Roukala, A. F. Maldonado, J. Vaara, G. A. Aucar and P. Lantto, Phys. Chem. Chem. Phys., 2011, 13, 21016–21025 RSC.
- J. Autschbach, J. Chem. Phys., 2012, 136, 15 CrossRef PubMed.
- A. Wodynski, M. Repisky and M. Pecul, J. Chem. Phys., 2012, 137, 014311 CrossRef PubMed.
- J. Vicha, M. Patzschke and R. Marek, Phys. Chem. Chem. Phys., 2013, 15, 7740–7754 RSC.
- F. Alkan and C. Dybowski, Phys. Chem. Chem. Phys., 2014, 16, 14298–14308 RSC.
- J. Autschbach, Philos. Trans. R. Soc., A, 2014, 372, 20120489 CrossRef PubMed.
- F. Alkan and C. Dybowski, Phys. Chem. Chem. Phys., 2015, 17, 25014–25026 RSC.
- Y. Ruiz-Morales, G. Schreckenbach and T. Ziegler, J. Phys. Chem. A, 1997, 101, 4121–4127 CrossRef CAS.
- P. Avalle, R. K. Harris, P. B. Karadakov and P. J. Wilson, Phys. Chem. Chem. Phys., 2002, 4, 5925–5932 RSC.
- M. Kaupp, M. Buhl and V. G. Malkin, Calculation of NMR and EPR Parameters: Theory and Applications, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- R. Fukuda and H. Nakatsuji, J. Chem. Phys., 2005, 123, 044101 CrossRef PubMed.
- M. Hada, J. Wan, R. Fukuda and H. Nakatsuji, J. Comput. Chem., 2001, 22, 1502–1508 CrossRef CAS.
- A. Bagno, G. Casella and G. Saielli, J. Chem. Theory Comput., 2006, 2, 37–46 CrossRef CAS PubMed.
- A. F. Maldonado and G. A. Aucar, J. Phys. Chem. A, 2014, 118, 7863–7875 CrossRef CAS PubMed.
- J. I. Melo, A. Maldonado and G. A. Aucar, Theor. Chem. Acc., 2011, 129, 483–494 Search PubMed.
- H. Kaneko, M. Hada, T. Nakajima and H. Nakatsuji, Chem. Phys. Lett., 1996, 261, 1–6 CrossRef CAS.
- E. Malkin, S. Komorovsky, M. Repisky, T. B. Demissie and K. Ruud, J. Phys. Chem. Lett., 2013, 4, 459–463 CrossRef CAS PubMed.
- L. F. Wang, C. E. Kefalidis, T. Roisnel, S. Sinbandhit, L. Maron, J. F. Carpentier and Y. Sarazin, Organometallics, 2014, 34, 2139–2150 CrossRef.
- R. Vivas-Reyes, F. De Proft, M. Biesemans, R. Willem and P. Geerlings, J. Phys. Chem. A, 2002, 106, 2753–2759 CrossRef CAS.
- L. Broeckaert, J. Turek, R. Olejnik, A. Ruzicka, M. Biesemans, P. Geerlings, R. Willem and F. De Proft, Organometallics, 2013, 32, 2121–2134 CrossRef CAS.
- M. R. Mitchell, S. W. Reader, K. E. Johnston, C. J. Pickard, K. R. Whittle and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2011, 13, 488–497 RSC.
- J. A. Tossell, J. Magn. Reson., 1997, 127, 49–53 CrossRef CAS.
- G. Valerio, A. Goursot, R. Vetrivel, O. Malkina, V. Malkin and D. R. Salahub, J. Am. Chem. Soc., 1998, 120, 11426–11431 CrossRef CAS.
- G. Valerio and A. Goursot, J. Phys. Chem. B, 1999, 103, 51–58 CrossRef CAS.
- Y. Zhang and E. Oldfield, J. Phys. Chem. B, 2004, 108, 19533–19540 CrossRef CAS.
- D. Stueber, Concepts Magn. Reson., Part A, 2006, 28, 347–368 CrossRef.
- D. H. Brouwer and G. D. Enright, J. Am. Chem. Soc., 2008, 130, 3095–3105 CrossRef CAS PubMed.
- J. Weber and J. Gunne, Phys. Chem. Chem. Phys., 2010, 12, 583–603 RSC.
- S. T. Holmes, R. J. Iuliucci, K. T. Mueller and C. Dybowski, J. Chem. Theory Comput., 2015, 11, 5229–5241 CrossRef CAS PubMed.
- S. T. Holmes, R. J. Iuliucci, K. T. Mueller and C. Dybowski, J. Chem. Phys., 2014, 141, 164121 CrossRef PubMed.
- S. T. Holmes, F. Alkan, R. J. Iuliucci, K. T. Mueller and C. Dybowski, J. Comput. Chem., 2016, 37, 1704–1710 CrossRef CAS PubMed.
- A. W. MacGregor, L. A. O’Dell and R. W. Schurko, J. Magn. Reson., 2011, 208, 103–113 CrossRef CAS PubMed.
- R. C. McDonald and K. Eriks, Inorg. Chem., 1980, 19, 1237–1241 CrossRef CAS.
- A. Gleizes and J. Galy, J. Solid State Chem., 1979, 30, 23–33 CrossRef CAS.
- J. D. Donaldson and D. C. Puxley, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 864–867 CrossRef CAS.
- M. M. Ahmad, Y. Yamane and K. Yamada, J. Appl. Phys., 2009, 106, 074106 CrossRef.
- H. Yamane, Y. Kaminaga, S. Abe and T. Yamada, J. Solid State Chem., 2008, 181, 2559–2564 CrossRef CAS.
- R. M. Hazen and L. W. Finger, Am. Mineral., 1978, 63, 289–292 CAS.
- J. Catalano, A. Murphy, Y. Yao, F. Alkan, N. Zurnbulyadis, S. A. Centeno and C. Dybowski, J. Phys. Chem. A, 2014, 118, 7952–7958 CrossRef CAS PubMed.
- J. R. Gavarri, J. P. Vigouroux, G. Calvarin and A. W. Hewat, J. Solid State Chem., 1981, 36, 81–90 CrossRef CAS.
- J. C. Jumas, J. Olivierf, F. Vermotga, M. Ribes, E. Philippo and M. Maurin, Rev. Chim. Miner., 1974, 11, 13–26 CAS.
- W. T. Fu, D. Visser and D. J. W. Ijdo, J. Solid State Chem., 2002, 169, 208–213 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com. (accessed September 2, 2014).
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Van lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- M. Krykunov, T. Ziegler and E. van Lenthe, J. Phys. Chem. A, 2009, 113, 11495–11500 CrossRef CAS PubMed.
- M. Krykunov, T. Ziegler and E. van Lenthe, Int. J. Quantum Chem., 2009, 109, 1676–1683 CrossRef CAS.
- J. Autschbach, Mol. Phys., 2013, 111, 2544–2554 CrossRef CAS.
- I. D. Brown and R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 266–282 CrossRef CAS.
- I. D. Brown and K. K. Wu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1957–1959 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- J. R. Yates, C. J. Pickard, M. C. Payne and F. Mauri, J. Chem. Phys., 2003, 118, 5746–5753 CrossRef CAS.
- J. Autschbach, Theor. Chem. Acc., 2004, 112, 52–57 Search PubMed.
- V. Arcisauskaite, J. I. Melo, L. Hemmingsen and S. P. A. Sauer, J. Chem. Phys., 2011, 135, 044306 CrossRef PubMed.
- A. Sadoc, M. Body, C. Legein, M. Biswal, F. Fayon, X. Rocquefelte and F. Boucher, Phys. Chem. Chem. Phys., 2011, 13, 18539–18550 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp03807g |
| This journal is © the Owner Societies 2016 |