
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction between ionic liquid cation and water: infrared predissociation study of [bmim]+·(H2O)n clusters†
Jonathan M.
Voss
,
Brett M.
Marsh
,
Jia
Zhou
and
Etienne
Garand
*
Department of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA. E-mail: egarand@wisc.edu
First published on 29th June 2016
Abstract
The infrared predissociation spectra of [bmim]+·(H2O)n, n = 1–8, in the 2800–3800 cm−1 region are presented and analyzed with the help of electronic structure calculations. The results show that the water molecules solvate [bmim]+ by predominately interacting with the imidazolium C2–H moiety for the small n = 1 and 2 clusters. This is characterized by a redshifted and relatively intense C2–H stretch. For n ≥ 4 clusters, hydrogen-bond interactions between the water molecules drive the formation of ring isomers which interact on top of the imidazolium ring without any direct interaction with the C2–H. The water arrangement in [bmim]+·(H2O)n is similar to the low energy isomers of neutral water clusters up to the n = 6 cluster. This is not the case for the n = 8 cluster, which has the imidazolium ring disrupting the otherwise preferred cubic water structure. The evolution of the solvation network around [bmim]+ illustrates the competing [bmim]+–water and water–water interactions.
I. Introduction
The ubiquitous presence of water within ionic liquids, both as an impurity and a co-solvent, makes their interactions with each other topics of active investigations.1–4 While the anionic half of the ionic liquid often plays a major role in determining the extent of water sorption and hydrophobicity, the cations can also contribute to interactions with water molecules.5,6 Salts containing imidazolium cations, such as 1-butyl-3-methylimidazolium ([bmim]+) shown in Fig. 1, are a class of widely used and studied room temperature ionic liquids. Owing to the electron deficiency of the N–C–N moiety, the C2–H group can actively participate in hydrogen-bond like interactions in addition to electrostatic interactions.7–9 Raman, IR, NMR, and crystallography studies have shown that the C2–H of [bmim]+ is capable of forming hydrogen-bonds with various anions.10–17 Although the dominant interaction is likely electrostatic,18 the presence of these hydrogen-bonds can have a significant effect on the properties of these ionic liquids.19 When water is added to [bmim]+[X]− ionic liquids, experimental and theoretical results point to the water molecules individually coordinating with the anions and cations at low concentrations, but forming water clusters and swelling the ionic liquid at higher water concentrations.20–22 These results highlight the competition between water–water, cation–anion, and water–ionic liquid interactions.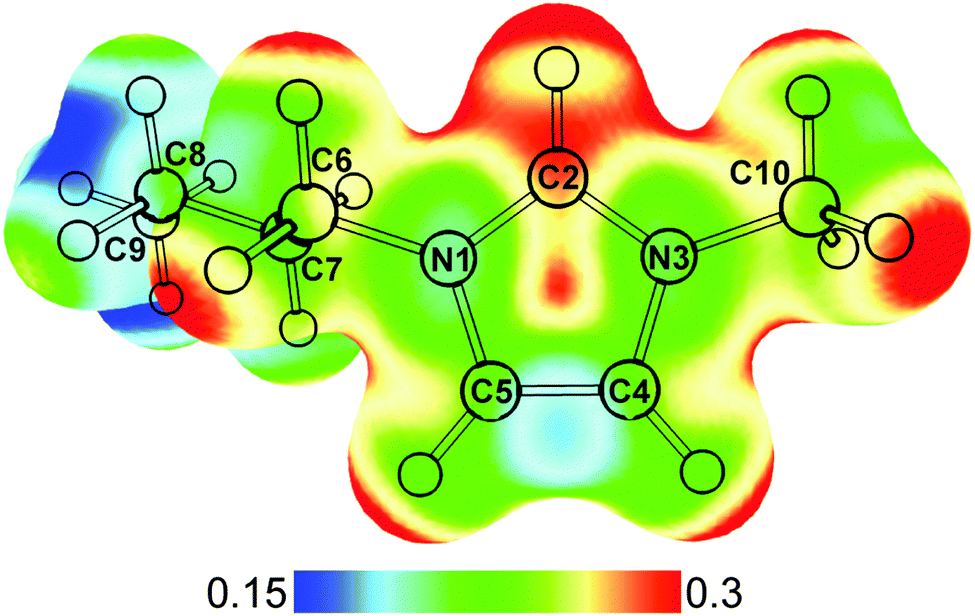 | ||
| Fig. 1 Optimized structure of [bmim]+ calculated at MP2/def2TZVP level. The electrostatic potential of [bmim]+ mapped onto the density surface (iso = 0.02) is shown. Atom labelling is also indicated. | ||
Here, we present the infrared predissociation spectroscopy of mass selected [bmim]+·(H2O)n clusters to provide a molecular level look at the interactions between the imidazolium cation and water molecules. The isolated cluster approach allows us to look at the intermolecular interactions individually, providing a better understanding of the forces responsible for the observed bulk behaviors. Similar gas phase spectroscopic studies of ionic liquid clusters have revealed detailed information on their molecular properties and provided valuable benchmark data for theoretical calculations.23–29 Additionally, the [bmim]+–water interaction itself is of interest because it is unclear how strongly the C2–H group can hydrogen-bond with a water molecule and whether that interaction competes with the overall electrostatic interactions.
In this study, we use the OH and CH stretch vibrations, with the aid of electronic structure calculations, to follow the evolution of both the [bmim]+–water interaction and structure of the water network as a function of cluster size. We find that the solvation geometry is driven mostly by water–water hydrogen-bonds as well as a subtle balance between the C2–H–water interaction and [bmim]+–water electrostatic and dispersion interactions. The small clusters consisting of one to three water molecules do exhibit a weak hydrogen-bond like interaction between the C2–H and water, redshifting the frequency and increasing the intensity of the C2–H stretching vibration. However, for larger clusters, hydrogen-bond interactions between the water molecules drive the formation of ring isomers which interact primarily with the delocalized charge over the top of the imidazolium ring, similar to those found in solvated hydrophobic cations.30
II. Experimental and theoretical details
The infrared predissociation spectra of the [bmim]+·(H2O)n complexes were acquired using a homebuilt dual trap cryogenic ion vibrational spectrometer, described in detail previously.31 Briefly, [bmim]+ ions were generated via electrospray ionization of ∼1 mM aqueous solution of [bmim][Cl], and were brought into a cryogenic linear octopole ion trap cooled to 80 K by liquid nitrogen. Inside this trap, the [bmim]+ ions were exposed to helium buffer gas seeded with water vapor, resulting in the formation of [bmim]+·(H2O)n clusters.31 These clusters were then gently transferred into a cryogenic 3D quadrupole ion trap held at 10 K. Collisions with buffer gas consisting of 10% D2 in He further cooled the ions. The clusters were size-selected inside the mass spectrometer prior to being mass gated and intersected with the output of a Nd:YAG pumped tunable OPO/OPA laser system (Laservision). When the IR photon energy was resonant with a vibrational transition, the absorption of a single photon was sufficient to induce the evaporation of one water molecule. Photofragment ions were then separated from the parent ions by a two-stage reflectron. Infrared predissociation spectra were generated by integrating the intensity of the water loss photofragment peak as a function of laser wavelength. The use of water as messenger tag means that the clusters studied here were likely to have higher internal energies compared to D2 tagging due to higher binding energies. Unfortunately, the delocalized charge on [bmim]+ made it difficult to form significant amount of D2 adducts.To aid the analysis of the experimental spectra, electronic structure calculations were performed using the Gaussian 09 computational package.32 Geometry optimizations and harmonic frequency calculations were carried out at the MP2/def2TZVP level for n = 1–3 clusters and wB97XD/def2TZVP level for the larger clusters. The def2TZVP33,34 basis is used for its relatively low computational cost and relatively high accuracy. We have previously used this basis set to reproduce the experimental IR spectra of solvated metal clusters.35 Comparisons between MP2 and wB97XD results were made for the n = 3 and 4 clusters to ensure that both methods arrived at similar structures and IR spectra. For the MP2 harmonic frequencies, a scaling factor of 0.957 was applied for comparison to experimental spectra. For the DFT harmonic frequencies, a constant 0.96 scaling factor was applied to the CH stretches while a linear scaling factor36 of 0.688x + 989 was applied to the OH stretches (for further detail see Fig. S1 in ESI†). Such a linear scaling factor is useful for the larger clusters because their OH stretches span a wider frequency range where a constant scaling factor is no longer sufficient.
III. Results and analysis
An overview of the experimental IR predissociation spectra of [bmim]+·(H2O)n, n = 1–8, in the 2800–3800 cm−1 region is shown in Fig. 2. These spectra are dominated by features in the OH stretch region, and general assignments can be made based on characteristic frequencies. The vibrational features above 3600 cm−1 are comparably narrow and correspond to the stretches of free OH groups, i.e. those not involved in a donor hydrogen-bond interaction. These features can be separated into two distinct categories. First, peaks appearing around 3640 cm−1 and 3730 cm−1 can be assigned to the symmetric and antisymmetric OH stretches of water with two free OH groups. Second, peaks appearing near 3700 cm−1 can be assigned to the free OH stretch of a water involved in a single donor hydrogen-bond interaction. The vibrational features in the 3200–3600 cm−1 region are generally broader, more intense, and gradually redshift with increasing cluster size. These features correspond to the stretches of hydrogen-bond donating OH groups with the interaction strength proportional to the size of the redshift. Note that additional features at 3500–3600 cm−1 appear for n ≥ 6 clusters, indicating a change in the hydrogen-bond network that leads to weaker hydrogen-bonds. Lastly, there is a distinct feature at a fairly constant frequency around 3225 cm−1 for the n = 5–8 clusters. | ||
| Fig. 2 Experimental IR predissociation spectra of [bmim]+·(H2O)n, n = 1–8. | ||
The CH stretches of the [bmim]+ ion, appearing in the 2900–3200 cm−1 region, are very weak compared to the OH stretches. These features have been studied in detail for the isolated [emim]+ ion,24,25 as well as in neat [bmim]+[X]− liquid.14,15 The three imidazolium ring C–H stretches generally appears around 3150 cm−1 while the aliphatic C–H stretches of the methyl and butyl groups appear around 2950 cm−1. In the spectrum of the n = 1 cluster, there is a notable broad feature at 3080 cm−1, which gradually redshifts to 3045 cm−1 and 3020 cm−1 in the spectra of the n = 2 and 3 clusters. Besides this feature, the C–H stretch region shows no solvation-dependent changes.
(a) [bmim]+·(H2O)1
The experimental and calculated IR spectra of [bmim]+·(H2O)1 are shown in Fig. 3. The peaks at 3638 cm−1 and 3723 cm−1 are readily assigned to the symmetric and antisymmetric stretches of a water molecule having two free OH groups. These values are slightly redshifted (19 cm−1 and 33 cm−1 respectively) compared to the OH stretches of an isolated water molecule, but are typical of a water molecule interacting with a positively charged ion.30,37,38 The positive charge on [bmim]+ is delocalized over the imidazolium ring, as shown in Fig. 1, giving rise to several possible interaction sites for the water molecule. The two isoenergetic lowest energy isomers have the water molecule mainly interacting with the imidazolium C2–H, on either the butyl C6–H (Fig. 3B) or the methyl C10–H (Fig. 3C) side. The transition state between these two structures, shown in Fig. S2 (ESI†), has a linear C2–H⋯OH2 geometry and is calculated to lie only 15 cm−1 above the minima. Therefore these geometries likely represent a single structure in which the water undergoes large amplitude motion between C6–H and C10–H. The calculated IR spectra of these two isomers are essentially the same and have an intense and redshifted C2–H at 3105 and 3108 cm−1. This calculated frequency agrees well with the experimentally observed broad feature at 3080 cm−1, but the calculated intensity is much larger than the experimental observation. The interaction with the C6–H/C10–H is too weak to induce any noticeable change in their vibrational frequencies. It should be noted that the C2–H stretch is calculated to be further redshifted to 3074 cm−1 at the transition state between the two minima. Therefore, the motion of the water from one minima to the other likely explains the broad appearance to the C2–H feature in the experimental spectrum. | ||
| Fig. 3 Experimental (A) and MP2/def2TZVP calculated (B–G) IR spectra of [bmim]+·(H2O)1. The relative energies of each isomer, including ZPE correction, are listed. The imidazolium C–H stretches and symmetric and antisymmetric H2O stretches are labelled in (B). | ||
Two other isomers in which the water is mainly interacting with C4–H or C5–H are found to lie 290 cm−1 and 405 cm−1 higher in energy, and are shown in Fig. 3D and E. The weaker water interaction leads to a slightly redshifted C4–H/C5–H at 3130 cm−1. Additionally, an isomer in which the water is located on top of the imidazolium ring (Fig. 3F) is found to be 637 cm−1 higher in energy. This isomer has no redshifted C–H and all the imidazolium C–H stretches appear near 3160 cm−1. Finally, an isomer with the water situated between C4–H and C5–H is found to lie 669 cm−1 higher (Fig. 3G), with minimally perturbed C–H stretches. The presence of small amount of these higher lying isomers, particularly those shown in Fig. 3D and E, may contribute to the broad appearance of the spectrum between 3100 cm−1 and 3200 cm−1.
(b) [bmim]+·(H2O)2
The experimental and calculated IR spectra of [bmim]+·(H2O)2 are shown in Fig. 4. Compared to the n = 1 spectrum, the addition of the second water molecule gives rise to two new peaks at 3700 cm−1 and 3476 cm−1, characteristic of a water having one free OH and donating one hydrogen-bond. Features corresponding to the symmetric and antisymmetric stretch of a water having two free OH are still present but slightly blueshifted to 3647 cm−1 and 3735 cm−1. This peak pattern points to a structure in which the two water molecules are hydrogen-bonded to each other. The two isoenergetic lowest energy isomers are found with such a structure, in which the first water is interacting with C2–H on either the C6–H or C10–H side. Both isomers are calculated to have a slightly more redshifted C2–H stretch than the n = 1 clusters at 3066 and 3069 cm−1, in agreement with the observed feature at 3045 cm−1. This indicates that the water–water hydrogen-bonding leads to a stronger C2–H–water interaction. Again, both isomers show a strong relative intensity for the calculated C2–H stretch that is not reflected in the experimental spectrum. Rotation of the water chain into the imidazolium ring plane leads to low lying isomers (29 cm−1 and 129 cm−1) in which the water molecules are also interacting with the alkyl groups. The energies of these isomers point to a very loose and flexible binding motif between the water dimer and [bmim]+, which likely contributes to the observed broadening of the C2–H and OH features.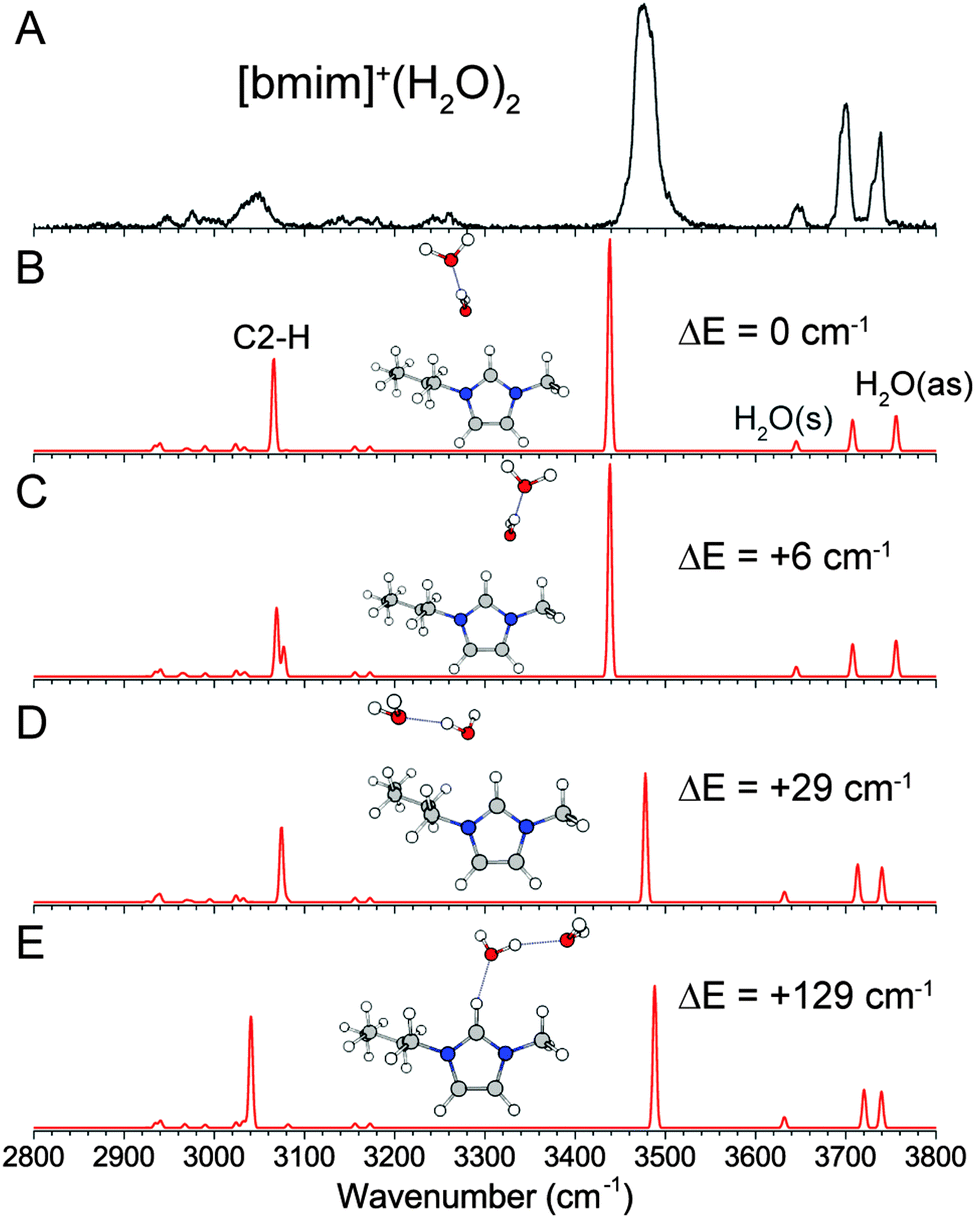 | ||
| Fig. 4 Experimental (A) and MP2/def2TZVP calculated (B–E) IR spectra of [bmim]+·(H2O)2. The relative energies of each isomer, including ZPE correction, are listed. The imidazolium C2–H and symmetric and antisymmetric H2O stretches are labelled in (B). | ||
(c) [bmim]+·(H2O)3
The number of features in the OH stretch region in the experimental IR spectrum of [bmim]+·(H2O)3, shown in Fig. 5A, suggests that there are more than one distinct isomer present. The lowest energy isomer found has a linear chain of water molecules interacting with C2–H and C6–H (Fig. 5B), a continuation of the structure of the n = 2 cluster. This isomer gives rise to the observed H2O symmetric and antisymmetric features at 3646 cm−1 and 3736 cm−1, as well as the hydrogen-bonded features around 3390 and 3430 cm−1. The cooperative hydrogen-bonding interaction in the water chain leads to an intense and redshifted C2–H stretch calculated at 3046 cm−1, in agreement with the observed broad feature around 3020 cm−1. Similarly to the n = 2 cluster, the linear water chain is fairly free to rotate, and additional isomers, such as that shown in Fig. 5D, are energetically accessible. The presence of these isomers also likely contributes to the broad appearance of the C2–H and associated OH stretches. | ||
| Fig. 5 Experimental (A) and MP2/def2TZVP calculated (B–D) IR spectra of [bmim]+·(H2O)3. The relative energies of each isomer, including ZPE correction, are listed. The imidazolium C2–H and symmetric and antisymmetric H2O stretches are labelled in (B). | ||
A different isomer, with a three-membered water ring situated on top of the imidazolium ring (Fig. 5C), is found to be nearly isoenergetic with the lowest energy structure, lying only 38 cm−1 higher in energy. The water network in this isomer corresponds to the lowest energy structure of the neutral water cluster,39–41 where all three water molecules are involved in a similar single-donor–single-acceptor hydrogen-bonding environment, leading to a single peak at 3700 cm−1 in the free OH region. Additionally, there is no direct C2–H–water interaction, and all the imidazolium CH have frequencies near 3160 cm−1. This isomer can account for the strong peak at 3700 cm−1 as well as the two higher frequency hydrogen-bonded feature at 3480 cm−1 and 3540 cm−1. It should be noted that no stable structures were found for a 3-membered water ring interacting more directly with C2–H at the MP2 level of theory. This differs from DFT calculations, as shown in Fig. S3 (ESI†). The wB97XD functional yielded a similar structure as MP2, but the water ring is tilted more towards the C2–H, resulting in a shorter C2–H⋯O distance. The cam-B3LYP functional, on the other hand, gives a structure in which the water ring is mostly interacting with the C2–H and is almost perpendicular to the plane of the imidazolium ring. Upon inclusion of the GD3BJ empirical dispersion, the structure is again similar to the MP2 result. These results indicate that the exact location of the water ring involves subtle competition between the two binding sites and requires accurate treatment of long-range interactions. The importance of including dispersion correction in DFT is similar to theoretical treatment of pure ionic liquids.42,43
(d) [bmim]+·(H2O)4 and [bmim]+·(H2O)5
The experimental IR spectra of the [bmim]+·(H2O)4 and [bmim]+·(H2O)5 clusters, shown in Fig. 6, are simpler than the smaller clusters. The presence of a single free OH feature at 3700 cm−1 indicates that all water molecules are involved in a hydrogen-bond donor interaction. Additionally, a single dominant broad feature centered at 3430 cm−1 indicates that all the water molecules are in a fairly similar hydrogen-bonding environment. These observations are consistent with the 4- and 5-membered ring structures of the isolated neutral water clusters.39–41 We find that the lowest energy isomer for both clusters has such a water ring located on top of the imidazolium ring. The resulting calculated IR spectra show a good agreement with the experiment for both species. It should be noted that there are multiple isomers with this general structure, i.e. the water ring can sit nearer to C2–H or C4–H/C5–H. In particular, MP2 calculations give the isomer shown in Fig. 6B as the lowest energy isomer for n = 4 cluster, while wB97XD calculations show the isomer with the water ring sitting closer to C2–H to be 17 cm−1 lower. This points to a very flexible interaction between the water network and the top of the imidazolium ring, similar to the interaction with the C2–H. For the n = 5 cluster, we also considered isomers with a more cage like water network involving a 4-membered ring, but the calculated IR spectrum contained additional vibrational features near 3600 cm−1, which is not observed experimentally. The 3220 cm−1 feature in the experimental spectrum of the n = 5 cluster is not reproduced in the calculation. We attribute this feature to the Fermi resonance between the overtone of the H2O bend and the most redshifted OH stretch. This coupling, in which the overtone band borrows intensity from the OH stretch, has been described in detail in the case of benzene–water clusters.36,44 Lastly, the larger and more flexible water ring in the n = 5 cluster allows for some C2–H–water interaction, as indicated by a redshifted calculated C2–H stretch. However, with increasing number of water molecules, the OH stretches dominate the experimental spectrum and redshift into the CH spectral region, making C–H stretch assignments difficult.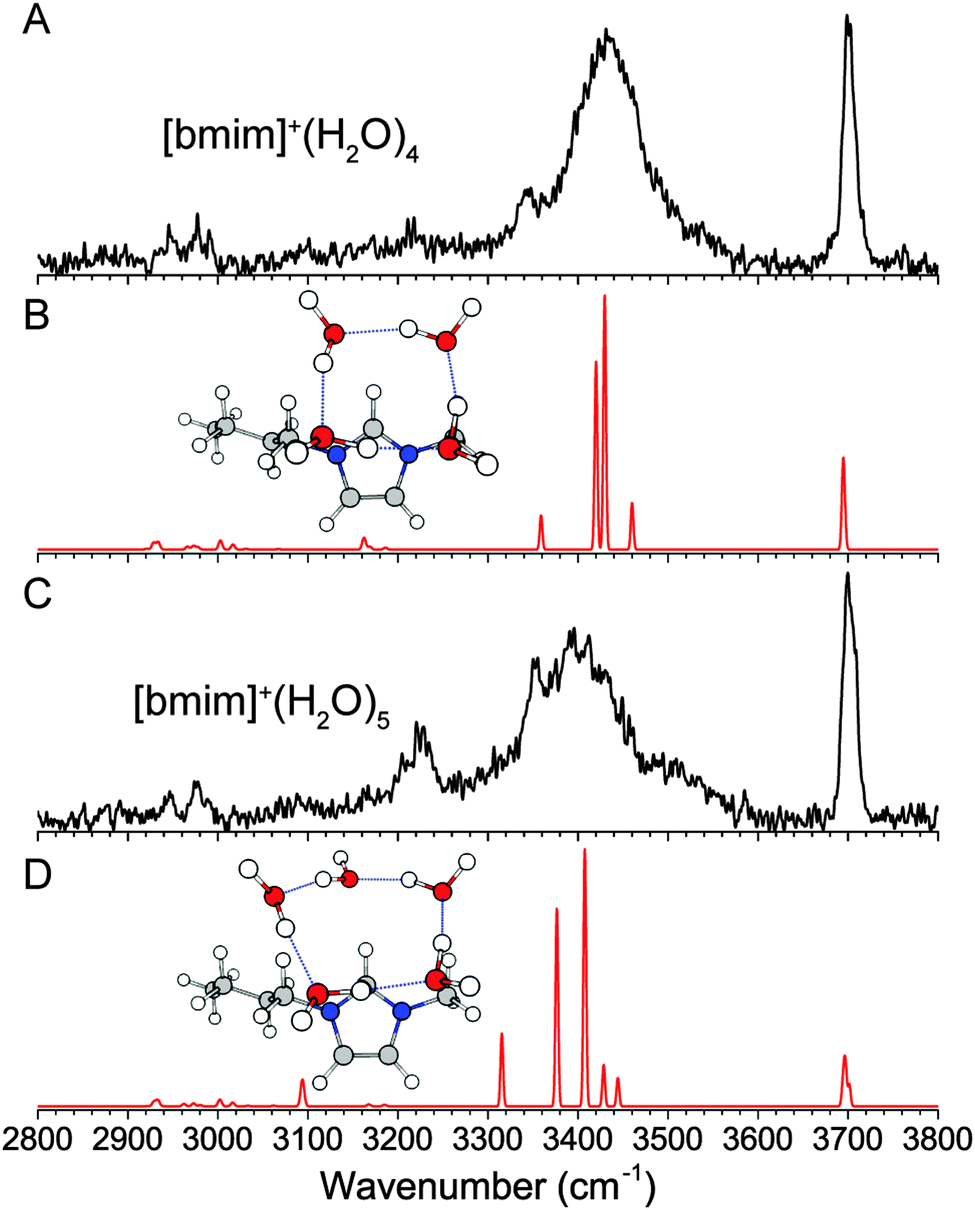 | ||
| Fig. 6 Experimental (A and C) and wB97XD/def2TZVP calculated (B and D) IR spectra of [bmim]+·(H2O)4 and [bmim]+·(H2O)5. | ||
(e) [BMIM]+·(H2O)6
The experimental IR spectrum of [bmim]+·(H2O)6, shown in Fig. 7A, has a broad distribution of peaks in the hydrogen-bonded region, but again only one feature in the free OH region, indicating that all water molecules are hydrogen-bond donors. The corresponding isolated neutral water cluster has five energetically close-lying isomers, namely the “ring”, “bag”, “book”, “cage”, and “prism” isomers, which have been the subject of numerous studies.40,45,46 We considered solvation geometries where these water clusters are interacting with the imidazolium ring in various configurations. The lowest energy isomer found, shown in Fig. 7B, has the water molecules arranged in the “book” geometry, draped on top and onto the C2–H side of the imidazolium ring. This structure involves a stable water cluster geometry that overlaps with the two strongest binding sites on the imidazolium. The calculated IR spectrum shows that it is sufficient to account for all the observed experimental features if the peak at 3225 cm−1 is again assigned to a Fermi resonance involving the H2O bend overtone. Note that the transition away from a ring structure results in a higher frequency OH stretch, at around 3585 cm−1, corresponding to one of the OH stretch of the water molecule that is a double hydrogen-bond donor. The next isomer, calculated to be 206 cm−1 higher in energy, involves a 6-membered water ring (Fig. 7C), and may also contribute to the experimental spectrum. These two isomers have water network that maximizes direct interaction with the charged imidazolium, in contrast to the more three-dimensional structures such the bag (Fig. 7D), cage or prism isomers (Fig. S4, ESI†), which all lie higher in energy. | ||
| Fig. 7 Experimental (A) and wB97XD/def2TZVP calculated (B–D) IR spectra of [bmim]+·(H2O)6. The relative energies of each isomer, including ZPE correction, are listed. | ||
(f) [BMIM]+·(H2O)7 and [BMIM]+·(H2O)8
A comprehensive isomer search for the n = 7 and 8 clusters is not feasible here, but the geometries determined so far can be used to extrapolate plausible structures. We know a more open arrangement of the water molecules that can wrap around the imidazolium ring is energetically favorable, and the water molecules favor four-membered and five-membered rings. Additionally, the spectrum for the n = 7 and 8 clusters are very similar to that of the n = 6 cluster, supporting the assignment to similar structures. Therefore, we considered the extension of the n = 6 “book” water network, leading to the 5 + 2 “book” arrangement for the n = 7 cluster and the 4 + 4 “open-u” arrangement for the n = 8 cluster, as shown in Fig. 8B and D. The calculated IR spectra show similar features as the experimental spectra, suggesting that these isomers are reasonable candidates. The arrangement of the water molecules in the n = 7 cluster is similar to one of the low energy neutral water isomers.40 However, the n = 8 isomer is different than the corresponding isolated water cluster which distinctly favors a cubic geometry. We do not think such an isomer has a major presence in our experiment because a cubic water arrangement situated on top of the imidazolium ring would result in half of the water molecules not interacting with [bmim]+. Additionally, this type of water clusters would lead to stronger hydrogen-bonds and lower frequency OH stretches,47 which are not observed in the n = 8 spectrum.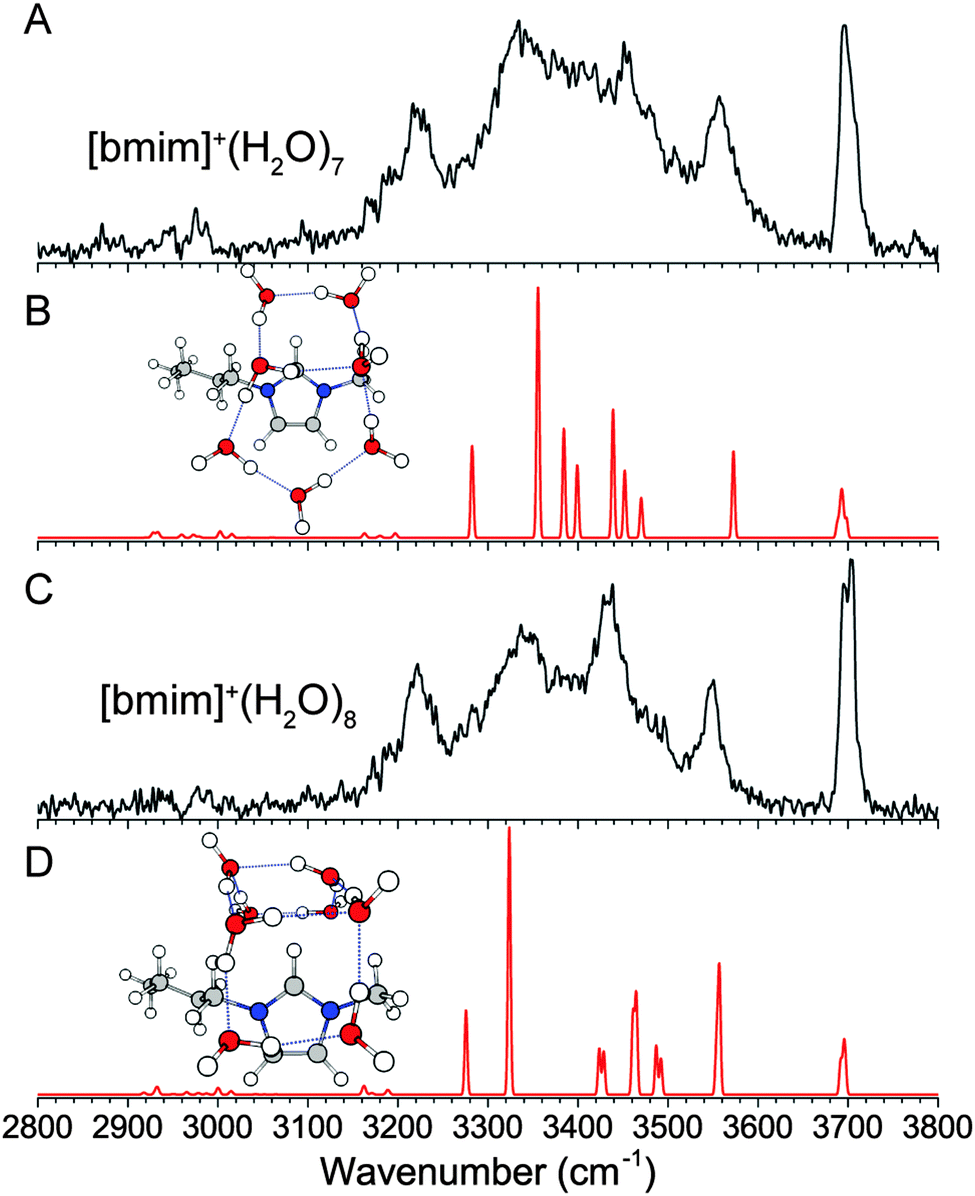 | ||
| Fig. 8 Experimental (A and C) and wB97XD/def2TZVP calculated (B and D) IR spectra of [bmim]+·(H2O)7 and [bmim]+·(H2O)8. | ||
IV. Discussions
IR predissociation spectroscopy of the isolated [bmim]+·(H2O)n clusters allow us to examine in detail the competing long range interactions present here. The calculated structures show that the water arrangement in [bmim]+·(H2O)n is similar to those found for neutral water clusters for n = 1–5. For the smallest n = 1 and n = 2 clusters, the preferred water binding site is centered on the C2–H group. The n = 3 cluster represents the tipping point in which the water molecules form a ring and the interaction site switches to the top of the imidazolium ring. For clusters with n ≥ 6, the water network evolves in such a manner that the interaction with the delocalized charge on the imidazolium is maximized. This leads to water arrangements with a more open structure than the isolated water clusters.The interaction between water and C2–H has been described as a hydrogen-bonding interaction.7,21 Our experimental spectra and calculation results show that this interaction have some hydrogen-bonding character. Notably, interaction with the first water molecule redshifts the C2–H frequency from 3160 cm−1 to 3080 cm−1. The formation of a water chain in n = 2 and 3 clusters further redshifts the C2–H frequency to 3045 cm−1 and 3020 cm−1, respectively. The calculated structures show that the C2–H bond lengthens slightly from 1.078 Å in the bare [bmim]+ ion to 1.082 Å in n = 1, 1.085 Å in n = 2 and 1.086 Å for the n = 3 water chain isomer. This relatively small bond length change is the only notable change in the geometry of [bmim]+ upon solvation, and points to a weak hydrogen-bonding interaction. A weak hydrogen-bond is also consistent with the lack of directionality observed for the C2–H–water interaction. Hydrogen-bonds usually have a linear X–H⋯A geometry while the C2–H⋯O angle is calculated to be about 150° with a very flat double-well potential. However, these weak hydrogen-bond characteristics are not consistent with the substantial 9.8 kcal mol−1 binding energy calculated for the water in the n = 1 clusters. Therefore, electrostatic and dispersion interactions with the overall positively charged imidazolium are the main contributions to the overall binding energy. This is evidenced by the n = 1 isomer shown in Fig. 3F, in which the water is located on top the imidazolium ring and not interacting with any C–H group. The binding energy for this isomer is only reduced by 1.8 kcal mol−1. Additionally, the binding energy of a water molecule to the hydrophobic tetramethylammonium cation is calculated to be at a similar value of 10.5 kcal mol−1.30
A typical mass spectrum showing the distribution of solvated [bmim]+ clusters formed in the cold ion trap was published in ref. 31. It shows a distinct intensity distribution which indicates that the sixth water should have a comparably lower binding energy while the fourth water should have a distinctively higher binding energy. This is confirmed by the calculated binding energy of each subsequent additional water molecule, listed in Table 1. The wB97XD/def2TZVP calculations predict a 9.9 kcal mol−1 binding energy for the sixth water molecule. This is noticeably smaller than the 12.5 kcal mol−1 binding energy calculated for the fourth water, as well as the consistent 10.4–10.5 kcal mol−1 binding energies of the fifth, seventh, and eighth water molecule.
| n | [bmim]+·(H2O)n | |
|---|---|---|
| BE (MP2) | BE (wB97XD) | |
| 1 | 9.76 | 9.89 |
| 2 | 9.07 | 9.36 |
| 3 | 9.23 | 9.67 |
| 4 | 13.14 | 12.54 |
| 5 | 10.43 | |
| 6 | 9.92 | |
| 7 | 10.48 | |
| 8 | 10.42 | |
The [bmim]+·(H2O)n results presented here can be compared to those of a similar solvated cluster, benzene·(H2O)n, studied by Zwier and coworkers.44,47,48 For the n = 1–6 clusters, we find that the water network adopts very similar structures in both cases and the vibrational spectra in the OH stretch region are remarkably similar. The main difference is that in the benzene·(H2O)n clusters, one water always has an OH pointed towards the electron density above the benzene ring, leading to an OH stretch feature at ∼3650 cm−1 for all clusters. In the case of [bmim]+·(H2O)n, the positive charge on the [bmim]+ forces the water to interact via the oxygen end with the OH pointing outward. Interestingly, the IR spectra of the two systems differ significantly for the n = 8 cluster. The weak interaction between benzene and water leads to the formation of a cubic water network situated on top of benzene, which gives rise to a group of OH stretches below 3200 cm−1 and a group of OH stretches above 3400 cm−1. The [bmim]+·(H2O)8 cluster, on the other hand, has a water network wrapped around [bmim]+, giving rise to OH stretches extending the range between 3200 cm−1 and 3600 cm−1. This difference highlights the effect of the delocalized charge on the solvation structure. We also note that the IR predissociation spectra of [bmim]+·(H2O)n, especially the smaller clusters, are also very similar to those of [tetramethylammonium]+(H2O)n, a solvated hydrophobic system.30
Finally, it is of interest to compare our results to what is known about the interaction of water in bulk [bmim]+[BF4]− ionic liquid. The cation–anion, cation–water and anion–water interactions have been studied by NMR using nuclear Overhauser enhancement measurements20 and analyzed by molecular dynamics simulations.21 The NMR data showed two distinct hydration regimes as the water concentration increased. At low concentrations, the water molecules were found to disrupt the short range network of the neat ionic liquid by interacting mainly with the imidazolium C–H groups, with preferential interaction with the C2–H. Additionally, the H2 NMR shift was found to be linearly correlated with those of the H6 and H10. This is consistent with our results for the n = 1 and n = 2 clusters, which show that the water interaction is centered on C2–H but is loose and flexible enough to also interact with C6–H and C10–H. At higher water concentrations, the ionic liquid network is perturbed by the insertion of whole water clusters, and the interaction between [bmim]+ and water becomes non-selective with respect to the various C–H groups. This is again consistent with the picture emerging from the work presented here, in which the dominant electrostatic and dispersion interactions and comparably stronger water–water hydrogen-bonds lead to water clusters located on top of the imidazolium ring for the n ≥ 3 clusters.
V. Conclusions
The intermolecular interactions between water molecules and imidazolium cation are revealed by the IR predissociation spectra of [bmim]+·(H2O)1–8 in the CH and OH stretch region. The experimental and calculated results show that the imidazolium C2–H can form a weak hydrogen-bond type interaction with a water molecule, but the solvation is energetically dominated by electrostatic and dispersion interactions. For the small n = 1–3 clusters, water molecules solvate the [bmim]+ by predominately interacting with the C2–H moiety, and the linear chain water arrangement strengthens the C2–H–water interaction. For the larger clusters, water–water hydrogen-bond dominates and drives the formation of water ring isomers, which preferentially interact with the delocalized positive charge by situating on top of the imidazolium ring. The geometry of the water arrangement in [bmim]+·(H2O)n mirrors the low energy isomers of neutral water clusters for the n = 1–6 cluster sizes, while the larger clusters adopt a more open arrangement that envelops the positively charged imidazolium ring rather than developing a tight water–water network. The observed solvation behavior reported here is in agreement with solution phase studies that showed concentration-dependent solvation effects in ionic liquid–water mixtures.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award no. DE-SC0010326. The computational resources used in this work are supported by National Science Foundation Grant CHE-0840494.References
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- L. M. Varela, T. Mendez-Morales, J. Carrete, V. Gomez-Gonzalez, B. Docampo-Alvarez, L. J. Gallego, O. Cabeza and O. Russina, J. Mol. Liq., 2015, 210, 178–188 CrossRef CAS.
- H. Weingaertner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef CAS PubMed.
- Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119–7130 RSC.
- Y. Cao, Y. Chen, X. Sun, Z. Zhang and T. Mu, Phys. Chem. Chem. Phys., 2012, 14, 12252–12262 RSC.
- M. G. Freire, C. M. S. S. Neves, P. J. Carvalho, R. L. Gardas, A. M. Fernandes, I. M. Marrucho, L. M. N. B. F. Santos and J. A. P. Coutinho, J. Phys. Chem. B, 2007, 111, 13082–13089 CrossRef CAS PubMed.
- M. Brehm, H. Weber, A. S. Pensado, A. Stark and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 5030–5044 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- K. Fumino, A. Wulf and R. Ludwig, Phys. Chem. Chem. Phys., 2009, 11, 8790–8794 RSC.
- E. R. Talaty, S. Raja, V. J. Storhaug, A. Dolle and W. R. Carper, J. Phys. Chem. B, 2004, 108, 13177–13184 CrossRef CAS.
- S. Saha, S. Hayashi, A. Kobayashi and H. Hamaguchi, Chem. Lett., 2003, 32, 740–741 CrossRef CAS.
- N. E. Heimer, R. E. Del Sesto, Z. Z. Meng, J. S. Wilkes and W. R. Carper, J. Mol. Liq., 2006, 124, 84–95 CrossRef CAS.
- J. H. Antony, D. Mertens, A. Dolle, P. Wasserscheid and W. R. Carper, ChemPhysChem, 2003, 4, 588–594 CrossRef CAS PubMed.
- S. Cha, M. Ao, W. Sung, B. Moon, B. Ahlstrom, P. Johansson, Y. Ouchi and D. Kim, Phys. Chem. Chem. Phys., 2014, 16, 9591–9601 RSC.
- A. Yokozeki, D. J. Kasprzak and M. B. Shiflett, Phys. Chem. Chem. Phys., 2007, 9, 5018–5026 RSC.
- E. I. Izgorodina and D. R. MacFarlane, J. Phys. Chem. B, 2011, 115, 14659–14667 CrossRef CAS PubMed.
- Y. Gao, L. Zhang, Y. Wang and H. Li, J. Phys. Chem. B, 2010, 114, 2828–2833 CrossRef CAS PubMed.
- S. Tsuzuki, H. Tokuda and M. Mikami, Phys. Chem. Chem. Phys., 2007, 9, 4780–4784 RSC.
- K. Fumino, S. Reimann and R. Ludwig, Phys. Chem. Chem. Phys., 2014, 16, 21903–21929 RSC.
- A. Mele, C. D. Tran and S. H. D. Lacerda, Angew. Chem., Int. Ed., 2003, 42, 4364–4366 CrossRef CAS PubMed.
- M. Moreno, F. Castiglione, A. Mele, C. Pasqui and G. Raos, J. Phys. Chem. B, 2008, 112, 7826–7836 CrossRef CAS PubMed.
- Q.-G. Zhang, N.-N. Wang, S.-L. Wang and Z.-W. Yu, J. Phys. Chem. B, 2011, 115, 11127–11136 CrossRef CAS PubMed.
- D. Strasser, F. Goulay, M. S. Kelkar, E. J. Maginn and S. R. Leone, J. Phys. Chem. A, 2007, 111, 3191–3195 CrossRef CAS PubMed.
- J. A. Fournier, C. T. Wolke, C. J. Johnson, A. B. McCoy and M. A. Johnson, J. Chem. Phys., 2015, 142, 064306 CrossRef PubMed.
- C. J. Johnson, J. A. Fournier, C. T. Wolke and M. A. Johnson, J. Chem. Phys., 2013, 139, 224305 CrossRef PubMed.
- E. I. Obi, C. M. Leavitt, P. L. Raston, C. P. Moradi, S. D. Flynn, G. L. Vaghjiani, J. A. Boatz, S. D. Chambreau and G. E. Douberly, J. Phys. Chem. A, 2013, 117, 9047–9056 CrossRef CAS PubMed.
- R. Cooper, A. M. Zolot, J. A. Boatz, D. P. Sporleder and J. A. Stearns, J. Phys. Chem. A, 2013, 117, 12419–12428 CrossRef CAS PubMed.
- A. J. A. Harvey, A. Sen, N. Yoshikawa and C. E. H. Dessent, Chem. Phys. Lett., 2015, 634, 216–220 CrossRef CAS.
- K. Hanke, M. Kaufmann, G. Schwaab, M. Havenith, C. T. Wolke, O. Gorlova, M. A. Johnson, B. P. Kar, W. Sander and E. Sanchez-Garcia, Phys. Chem. Chem. Phys., 2015, 17, 8518–8529 RSC.
- J. S. Prell and E. R. Williams, J. Am. Chem. Soc., 2009, 131, 4110–4119 CrossRef CAS PubMed.
- B. M. Marsh, J. M. Voss and E. Garand, J. Chem. Phys., 2015, 143, 204201 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- B. M. Marsh, J. M. Voss, J. Zhou and E. Garand, Phys. Chem. Chem. Phys., 2015, 17, 23195–23206 RSC.
- D. P. Tabor, R. Kusaka, P. S. Walsh, E. L. Sibert III and T. S. Zwier, J. Phys. Chem. Lett., 2015, 6, 1989–1995 CrossRef CAS PubMed.
- T. D. Vaden, J. M. Lisy, P. D. Carnegie, E. D. Pillai and M. A. Duncan, Phys. Chem. Chem. Phys., 2006, 8, 3078–3082 RSC.
- T. D. Vaden, C. J. Weinheimer and J. M. Lisy, J. Chem. Phys., 2004, 121, 3102–3107 CrossRef CAS PubMed.
- R. Ludwig, Angew. Chem., Int. Ed., 2001, 40, 1808–1827 CrossRef CAS.
- B. Temelso, K. A. Archer and G. C. Shields, J. Phys. Chem. A, 2011, 115, 12034–12046 CrossRef CAS PubMed.
- M. F. Vernon, J. M. Lisy, D. J. Krajnovich, A. Tramer, H. S. Kwok, Y. R. Shen and Y. T. Lee, Faraday Discuss., 1982, 73, 387–397 RSC.
- S. Zahn, D. R. MacFarlane and E. I. Izgorodina, Phys. Chem. Chem. Phys., 2013, 15, 13664–13675 RSC.
- S. Grimme, W. Hujo and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 4875–4883 RSC.
- D. P. Tabor, R. Kusaka, P. S. Walsh, T. S. Zwier and E. L. Sibert III, J. Phys. Chem. A, 2015, 119, 9917–9930 CrossRef CAS PubMed.
- J. Kim and K. S. Kim, J. Chem. Phys., 1998, 109, 5886–5895 CrossRef CAS.
- D. M. Bates and G. S. Tschumper, J. Phys. Chem. A, 2009, 113, 3555–3559 CrossRef CAS PubMed.
- C. J. Gruenloh, J. R. Carney, C. A. Arrington, T. S. Zwier, S. Y. Fredericks and K. D. Jordan, Science, 1997, 276, 1678–1681 CrossRef CAS.
- R. N. Pribble, A. W. Garrett, K. Haber and T. S. Zwier, J. Chem. Phys., 1995, 103, 531–544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02730j |
| This journal is © the Owner Societies 2016 |