
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of terminal substituents on the electronic properties of rod-shaped [HGaNH]n oligomers†
A. V.
Pomogaeva
 * and
A. Y.
Timoshkin
* and
A. Y.
Timoshkin
Inorganic Chemistry Group, Institute of Chemistry, St. Petersburg State University, Universitetskaya nab. 7/9, St. Petersburg, 199034, Russia. E-mail: avpomogaeva@cc.spbu.ru
First published on 28th June 2016
Abstract
The effect of electron-donating and electron-withdrawing terminal groups on the electronic structure of the rod-shaped X3[HGaNH]nY3 or needle-shaped XGa[HGaNH]nNY oligomers (X, Y = H, CH3, F, CF3; n = 9, 30 and 114) was computationally studied at the B3LYP/SVP level of density functional theory. While the needle-shaped oligomers exhibit moderate variability in the electronic structure upon changing the terminal substituents X and Y, the energy gap of long rod-shaped oligomers varies within 2 eV. For oligomers with n = 114, F3[HGaNH]n(CH3)3 exhibits the largest HOMO–LUMO gap of 2.91 eV, while (CH3)3[HGaNH]nF3 has the smallest gap of 0.94 eV.
Introduction
GaN based nanostructures are among the most popular for nanoengineering due to their high chemical resistance and thermal stability. GaN based semiconducting nanorods can serve as channels for charge carriers with polarization-dependent conductivity in a variety of applications in light emitting diodes, transistors, UV sensors, lasers, piezotronic logical devices and solar cells.1A potentially interesting subclass of GaN based nanostructures is oligomeric clusters composed of [HGaNH]3 rings with the wurtzitic arrangement of the GaN backbone. Such oligomer compounds have been synthesized from solution2 and predicted to be spontaneously formed in the gas phase.3 The stability, structural features and optical properties of the rod-like group 13–15 oligomeric clusters have been the topics of many computational studies.2–12 Thermodynamic analysis3,4 indicates that the spontaneous generation of fairly long oligomers is thermodynamically feasible in a broad temperature range. The rod-shaped oligomers are energetically more favorable than cage-like isomers or 3-dimensional structures.5–8 High polarizability of the rod-shaped oligomers along with tunable band gap makes them promising charge carriers under an external electric field or under strength. Possible pathways for adjusting the band gap in such compounds have been the topic of theoretical studies. The effect of mixing of group 13 and group 15 elements in small oligomers [R(M,M′,M′′)(Y,Y′,Y′′)H]n (M, M′, M′′ = Al, Ga, In; Y, Y′, Y′′ = N, P, As; R = H, CH3; n = 4, 7, 10) was explored computationally.9–12 It was also shown that the termination of the rod-shaped oligomers significantly influences their structural and electronic properties.4,11 A variation of the HOMO–LUMO gap with the increase of the oligomerization degree n for the needle-shaped (“closed”) oligomers RM[RMNH]nNH is significantly smaller than for the rod-like (“open”) oligomers R3[RMNH]nH3. While for [HGaNH]n, HOMO–LUMO gap decreases from 7.1 (n = 7) to 6.5 eV (n = 115), for H3[HGaNH]nH3 it decreases from 6.5 (n = 6) to 1.9 eV (n = 114). For the methyl substituted analogs, the trends are similar, but absolute values are circa 0.62–0.69 eV smaller.4
The strong influence of terminal groups at the oligomer ends on its electronic properties opens a way for fine tuning of the HOMO–LUMO gap by the variation of only terminal substituents. In the present report, we demonstrate that the variation of electron donor/electron acceptor properties of terminal substituents allows us to adjust the HOMO–LUMO gap to cover a wide spectral range. Closed XGa[HGaNH]nNY and open X3[HGaNH]nY3 (X, Y = CH3, H, F, CF3) structures have been considered as the smallest oligomeric clusters (n = 9), as rods of about 10 nm in length (n = 114) and as oligomers of some intermediate lengths (n = 30). Several oligomers with n = 6, 12, and 18 have been considered to evaluate the distribution of partial charges. Schematic structures of open and closed oligomers are shown in Fig. 1a and b, respectively.
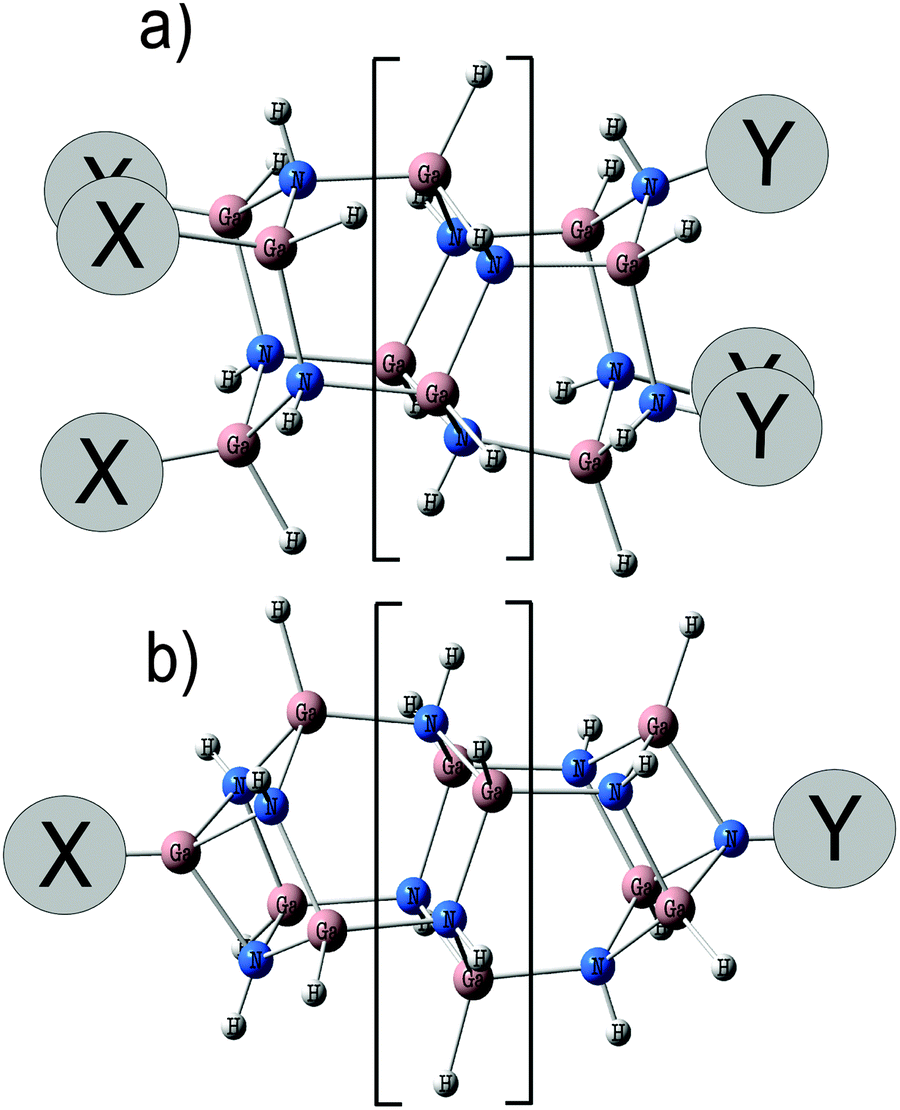 | ||
| Fig. 1 Schematic representation of the molecular structure of open X3[HGaNH]nY3 (a) and closed XGa[HGaNH]nNY (b) oligomers. The fragment in the square brackets is repeated 8 or 36 times for n = 30 and 114, respectively. | ||
Results and discussion
Earlier,2–4 it was shown that the stability of [RGaNH]3n increases linearly with the elongation of the oligomer. The introduction of the substituents on the oligomer ends does not change this trend. Closed oligomers in general are energetically more favourable than the open rods. Reactions of capping of open rods are exothermic for all X, Y pairs with the exception of X = F and Y = H.| X3[HGaNH]nY3 + GaH3 + NH3 = XGa[HGaNH]nNY + 2HX + 2HY + H2 |
The exothermicity of capping reactions increases with the increase of the oligomerization degree (see Table S1, ESI,† for details).
As has been comprehensively shown by Kormos et al.,2 the relative displacement of nitrogen and gallium planes provides a non-zero dipole moment of the cyclotrigallazane ring [H2GaNH2]3 that increases upon the elongation of the rod-like [HGaNH]n oligomer due to the increase of the distance between the oppositely charged ends. The dipole moment is directed from the negative charge accumulated near the Ga saturated end of the oligomer to the positive charge accumulated near the N saturated end. As an example, Fig. 2 shows the optimized structures, dipole moments and charge distributions of CH3Ga[HGaNH]9NCF3 (Fig. 2a), CH3Ga[HGaNH]30NCF3 (Fig. 2b), and CH3Ga[HGaNH]114NCF3 (Fig. 2c) oligomers. The dipole moment values of the longest (n = 114) oligomers are given in Fig. 3.
 | ||
| Fig. 2 Optimized structures and partial charge distribution of closed CH3Ga[HGaNH]9NCF3 (n = 9, 30, 114) oligomers. Dipole moment vectors are also shown for the each structure. | ||
 | ||
| Fig. 3 Dipole moments of open X3[HGaNH]114Y3 (a) and closed XGa[HGaNH]114NY (b) oligomers. | ||
Capping of the [HGaNH]n oligomer by terminal XGa and NY groups significantly (by 317–330 D) reduces the dipole moment (Fig. 3b) not only because the Ga terminated end has only one X-Ga group instead of three in open oligomers, but also because of a different atomic environment around the Ga atom. In closed oligomers terminal Ga atom is bonded with one X group and three N atoms, but in open oligomers each terminal Ga atom is bonded to one X group, one H and two N atoms. It leads to an overall decrease of the charge at the Ga-terminated end in closed oligomers. For example, the ESP charge13 in the outermost Ga atom of CH3Ga[HGaNH]6NCH3 is about 0.478, while the charge of each Ga atom in the outermost plane in (CH3)3[HGaNH]6(CH3)3 is 0.515. The sum of partial charges in the vicinity of Ga and N terminated ends for several open and closed oligomers is presented in Table 1. The graphical distribution of ESP charges over selected oligomers is provided in the ESI† (Fig. S1). Partial charge accumulated on the Ga terminated end in open oligomers (Table 1) is more than three times larger than the one on XGa groups in closed oligomers.
| Compound | (CH3)3[HGaNH]n(CF3)3 | (CH)3[HGaNH]n(CH3)3 | CH3Ga[HGaNH]nNCF3 | CH3Ga[HGaNH]nNCH3 | ||||
|---|---|---|---|---|---|---|---|---|
| n\group | (CH3GaH)3 | (NHCF3)3 | (CH3GaH)3 | (NHCH3)3 | CH3Ga | NCF3 | CH3Ga | NCH3 |
| 6 | 1.653 | −1.342 | 1.580 | −0.555 | 0.285 | −0.372 | 0.299 | −0.087 |
| 9 | 1.553 | −1.103 | 1.431 | −0.429 | 0.275 | −0.348 | 0.272 | −0.072 |
| 12 | 1.434 | −0.941 | 1.382 | −0.237 | 0.263 | −0.319 | 0.239 | −0.026 |
| 18 | 1.272 | −0.835 | 1.162 | −0.165 | 0.261 | −0.286 | 0.225 | 0.014 |
| 30 | 1.211 | −0.794 | 1.139 | −0.069 | 0.249 | −0.240 | 0.255 | 0.006 |
| 114 | 1.240 | −0.682 | 1.238 | −0.003 | 0.213 | −0.284 | 0.213 | 0.026 |
For the longest considered compounds with n = 144, the dipole moment calculated per HGaNH unit varies from 3.24 (X = CH3, Y = CF3) to 3.44 D (X = CF3, Y = CH3) for open and from 0.44 (X = CH3, Y = CF3) to 0.54 D (X = CF3, Y = CH3) for the closed oligomers.
Substituents X and Y change the dipole moment by donating/withdrawing the electron density to/from the neighboring Ga or N atoms. The electron-withdrawing ability decreases in row CF3 > F > H > CH3.14 Our results for the distribution of dipole moment values over different variations of terminal groups (Fig. 3) are in accord with such a trend. The oligomer with a CF3 group on the gallium atom (X) and a CH3 group on the nitrogen atom (Y) has the maximal dipole moments in the case of both open (Fig. 3a) and closed (Fig. 3b) rods. F or CF3 terminal groups on the gallium, and H or CH3 groups on the nitrogen atoms result in similar values of the dipole moments. In contrast, H or CH3 groups attached to terminal Ga atoms along with F or CF3 groups attached to terminal N atoms provide small dipole moments. The oligomer with X = CH3 and Y = CF3 has the smallest dipole moment. Combinations of X = F/CF3 with Y = F/CF3 or X = H/CH3 with Y = H/CH3 provide intermediate values of the dipole moment.
In the case of the shortest closed oligomers XGa[HGaNH]9NY with X = H or CH3 and Y = F or CF3 the electron-withdrawing power of the F and CF3 groups overcomes the electrostatic field provided by the spatial separation of gallium and nitrogen planes. It results in the change in the dipole moment direction for some compounds (Fig. 2a and Fig. S1, ESI†).
In general, the dipole moments of the oligomers increase linearly with the increase of the oligomer lengths, though some small nonlinearity is notable for open types of oligomers of shorter lengths (see Fig. S2 in the ESI†).
Atomic charge distribution for the combination X = CH3 and Y = CF3, which provides the smallest dipole moments for the fixed n (with the reversal of the dipole moment vector in the case of the shortest oligomers), is compared with the one in structures where both ends have the methyl substituents. Partial charges accumulated at opposite ends (the outermost Ga and N planes with the adjusted substituents) are provided in Table 1 for the oligomers with n = 6, 9, 12, 18, 30 and 114. The CH3 substituted N-terminated end is essentially neutral, while F atoms along with N atoms of the outermost plain accumulate significant negative charge that counterpoises or even overcome the negative charge accumulated at CH3 groups of the Ga-terminated end. In all cases, the partial charges on the negatively and positively charged ends decrease with the elongation of the oligomer. The NCF3 group of closed oligomers loses about 0.09ē when the oligomerization degree increases from n = 6 to 114. For the open structures, three NCF3 groups lose altogether more than 0.5ē when n is increased from 6 to 18 in (CH3)3[HGaNH]n(CF3)3. The respective change in the charge of three GaCH3 groups in (CH3)3[HGaNH]n(CH3)3 is about 0.4ē. Thus, some notable charge transfer between opposite ends occurs in short oligomers. It depends on the particular substituents and affects oligomers with lengths up to about 14Å (n ≤ 18).
The HOMO and LUMO of the rod-shaped open oligomers are strictly localized at the ends of the molecules. Fig. 4a and b show the HOMO and LUMO of (CH3)3[HGaNH]30F3 as an example. The HOMO is almost entirely localized at the gallium terminated end in long oligomers and the LUMO is almost entirely localized at the nitrogen terminated end. It should be noted that all other occupied orbitals with energy within at least 2 eV below the HOMO are also localized at the Ga-terminated end. Likewise, all unoccupied orbitals with energy within at least 2 eV higher than the LUMO are localized at the N-terminated end. It is expected that the HOMO energy will be sensitive to the X substituent while the LUMO energy will depend on the Y substituent.
 | ||
| Fig. 4 HOMO (a and c), HOMO−2 (e) and LUMO (b and d) of (CH3)3[HGaNH]30F3 (a and b) and CH3Ga[HGaNH]30NF (c–e) oligomers. Blue color refers to N, pink to Ga, white to H, gray to C and light blue to F atoms. | ||
In closed oligomers, the LUMO of A1 symmetry is strongly localized at the N-terminated end (Fig. 4c), but the HOMO of E symmetry is more delocalized and does not extend over the terminal group at the Ga-terminated end (Fig. 4d). In the most of the considered closed structures with n = 10, the HOMO is found to be moderately localized in the vicinity of Ga-terminated end as shown for CH3Ga[HGaNH]30NF3 in Fig. 4d. However, there are MOs in the vicinity of the HOMO which are localized at the N-terminated end. For example, in Fig. 4e the p-type HOMO−2 of CH3Ga[HGaNH]30NF3 is shown which is localized at the terminal F and adjacent N atoms. The energy difference between HOMO and HOMO−2 is below 0.1 eV. Thus, the HOMO−2–LUMO energy difference is similar to the HOMO–LUMO energy difference, while HOMO−2–LUMO transition does not involve an end-to-end intramolecular charge transfer. A similar situation is observed for other closed oligomers with intermediate length (n = 30). In some cases (X = CF3, Y = F; X = CF3, Y = CH3; X = H, Y = CH3; X = F, Y = CH3; X = F, Y = F) the occupied molecular orbital localized at the N-terminated end shifts to somewhat higher energy than the one localized at the Ga-terminated end and becomes the HOMO (see example in Fig. S3 in the ESI†). Fig. 5a and b show the energies of LUMOs for open and closed oligomers. The estimation of electronegativities of functional groups based on core-ionization energies15 implies that the CF3 group is significantly less electronegative than F. It correlates with the trend in the LUMO energies. The lowest energy of the LUMO corresponds to Y = F for both closed and open oligomers with n = 30 or n = 114; for open oligomers Y = CF3 yields much higher LUMO energies (Fig. 5a). The difference in LUMO energies between Y = F and Y = CF3 is circa 0.9 eV in open oligomers with n = 114 or n = 30. The difference in LUMO energies between Y = CH3 and Y = H for analogues is circa 0.5 eV.
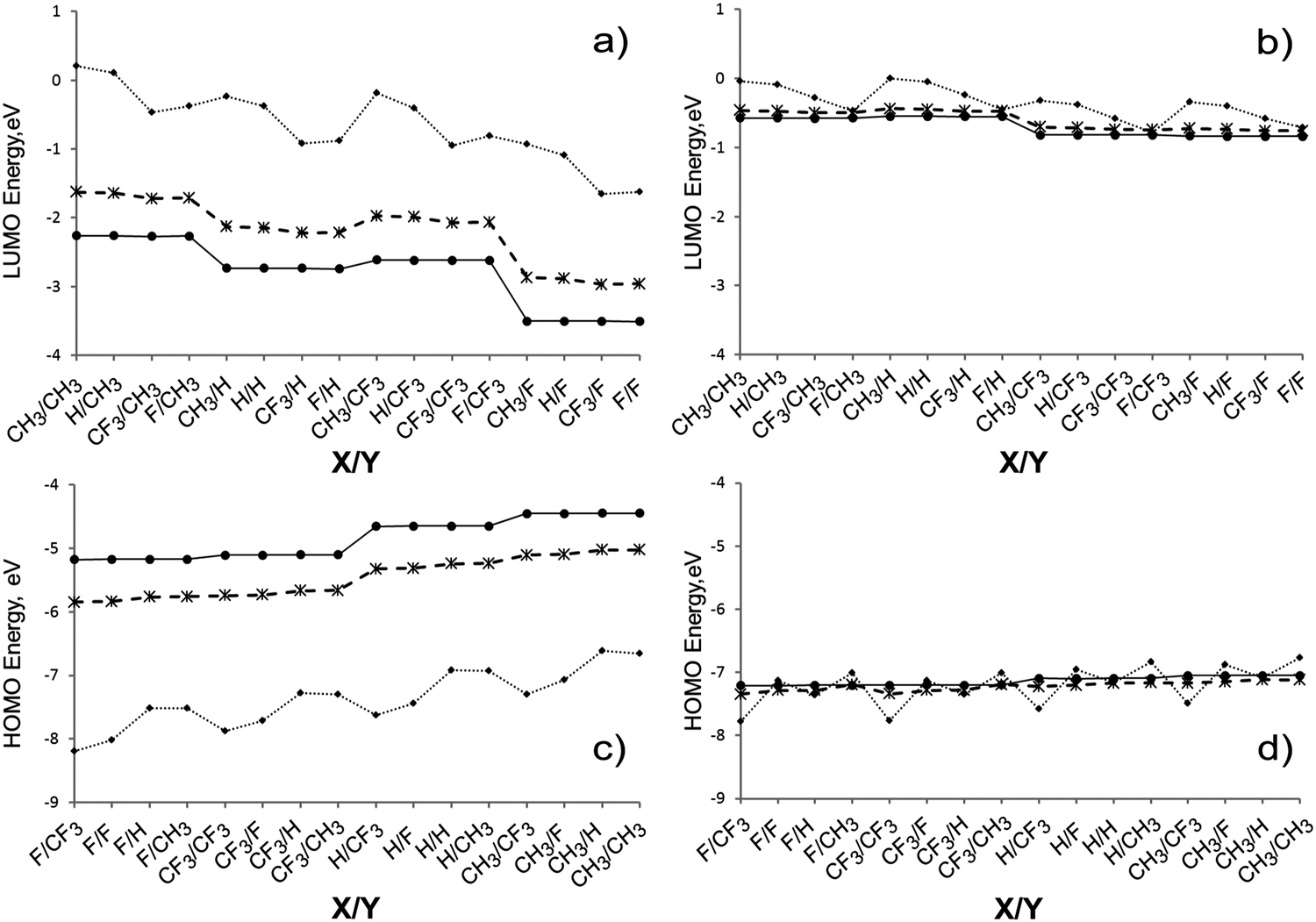 | ||
| Fig. 5 Energies of LUMO (a and b) and HOMO (c and d) for open X3[HGaNH]nY3 (a and c) and closed XGa[HGaNH]nNY (b and d) oligomers with n = 9 (dotted line), n = 30 (dashed line) and n = 114 (solid line) for different combinations of X/Y substituents. | ||
In the case of closed oligomers (Fig. 5b), if they are long enough to neglect the influence of terminal groups at opposite ends (n = 30 or 114), there is no difference between Y = F and Y = CF3, or Y = H and Y = CH3. The only factor lowering the LUMO energy (for ∼0.3 eV compared with Y = H or Y = CH3) is the presence of fluorine atoms (Y = F or Y = CF3) in substituents at the nitrogen terminated end.
The HOMO energies of long oligomers (Fig. 5c and d) increase in the order X = F < CF3 < H < CH3. The effect of terminal substituents on the HOMO energies is less pronounced than on the LUMO energies. The largest difference in the energies between structures with X = F and X = CH3 of long open oligomers (n = 30 or 114) is about 0.65 eV (Fig. 5c). For long needle-shaped oligomers (Fig. 5d) the difference does not exceed 0.16 eV. It correlates with the observation that terminal groups in the Ga-terminated end of long closed oligomers do not contribute to the HOMO (see Fig. 4d).
The elongation of open oligomers has a drastic effect on the HOMO and LUMO energies (see Fig. 5). The average downshift of LUMO energies of open rods is circa 0.58 eV (Fig. 5a) with elongation from n = 30 to 114. The respective upshift for the HOMO energies is circa 0.62 eV (Fig. 4b).
As expected, X substituents do not influence the energy of LUMO and Y substituents do not influence the HOMO energies for oligomers with n = 30 or 114. However, the substituents play a significant role in molecules with n = 9 where the opposite ends of the molecule are close enough to interact with each other. For any particular substituents at the nitrogen atom, LUMO energies decrease in the order of substituents at the gallium atom (X) as CH3 > H > CF3 > F for closed and as CH3 > H > F > CF3 for open oligomers with n = 9. On the other hand, for any particular substituent at the gallium atom, HOMO energies of oligomers with n = 9 increase with Y substitutions as CF3 < F < H ≈ CH3 for open, and as CF3 < H ≈ F < CH3 for closed oligomers. On the whole, for short oligomers the substituents Y at the nitrogen terminated end have a larger effect on the energies of HOMO and LUMO and, consequently, the HOMO–LUMO gap values than the substituents X at the gallium terminated end.
Electronic energies and partial density of states (PDOS) in the vicinity of HOMO–LUMO gaps of F3[HGaNH]114F3 and (CH3)3[HGaNH]114(CH3)3 oligomers are given in Fig. 6. At such a length (circa 10 nm) terminal groups do not affect the opposite ends of the rod. Thus, the PDOS in the vicinity of the HOMO and LUMO of the oligomers with the smallest (X = CH3 and Y = F) (see Fig. 4) and the largest (X = F and Y = CH3) energy gaps are expected to be the same as the respective local PDOS shown in Fig. 6a and b.
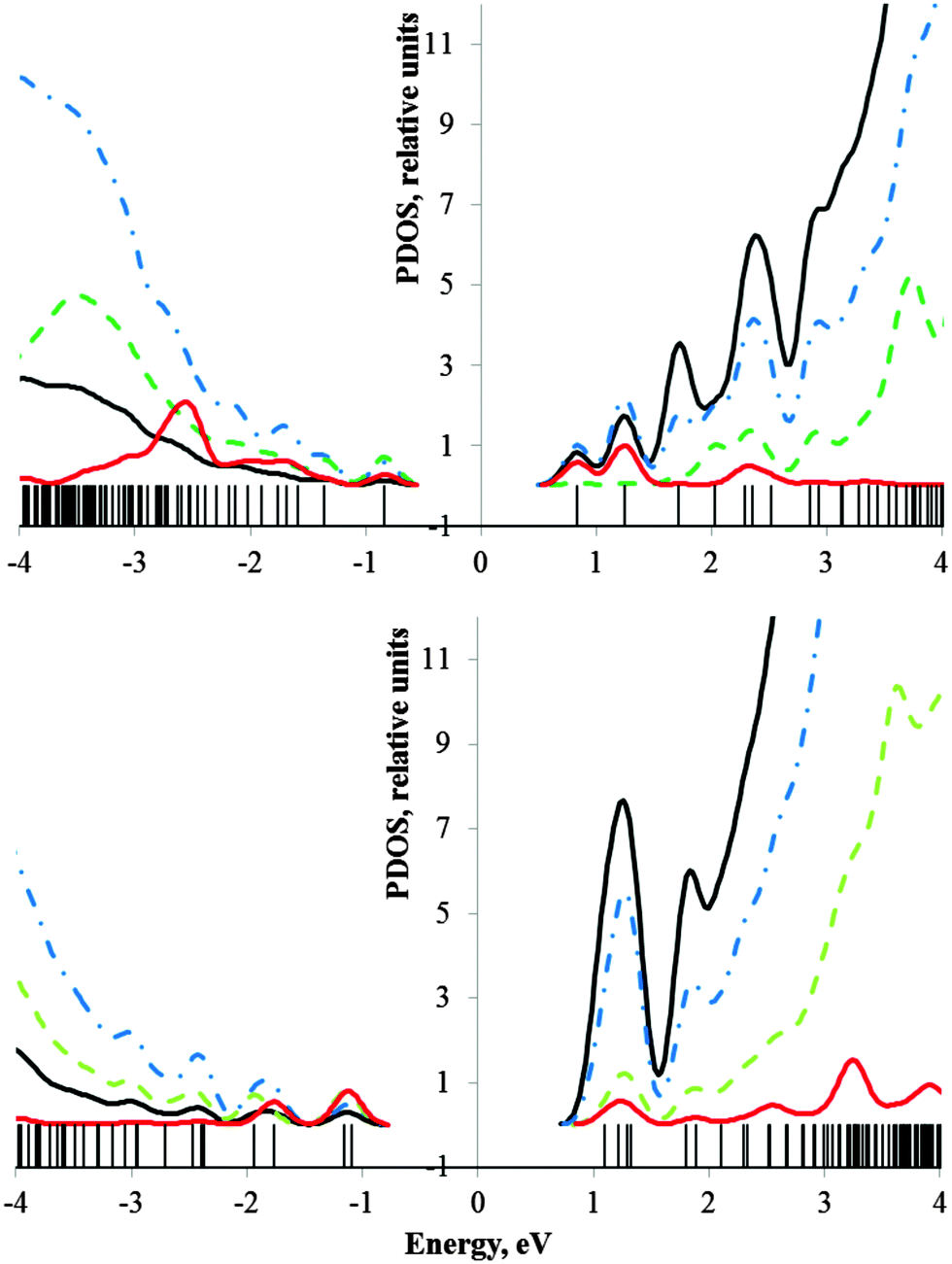 | ||
| Fig. 6 PDOS of F3[HGaNH]114F3 (a) and (CH3)3[HGaNH]114(CH3)3 (b). Contributions from Ga atoms are shown by black lines, from N atoms by blue dash-dotted lines, from backbone H atoms by green dashed lines and contributions from terminal groups (F or CH3) are presented by red lines. For drawing this figure, the middle of the energy gap is chosen as the zero energy. | ||
The estimated band gap of the [HGaNH]∞ polymer, extrapolated from the oligomer computations, is circa 7.0 eV.4 Finite open oligomers have a number of localized states within the band gap of the parental polymer. These states are clearly seen in the energy spectra shown in the Fig. 6 as spatially separated levels in the vicinity of the HOMO–LUMO gap compared with nearly continuous spectra in valence and conductivity bands. Most of the HOMOs exhibit density distribution over the N atoms near the Ga-terminated end, while lower unoccupied states have the main contribution from the Ga atoms near the N-terminated end. The contribution of terminal groups is noticeable not only in HOMO and LUMO states, but also deeper in the spectrum. F substituents produce a relatively sparse spectrum of lower unoccupied localized states which results in the maximal downshift of the LUMO energy (Fig. 6a).
By withdrawing electrons from N atoms, F substituents induce vacant orbitals of the lowest energy which result in lower LUMO energies. In contrast, methyl substituents produce a relatively sparse spectrum of the highest occupied molecular orbitals (Fig. 6b).
The energy gaps for all considered oligomers with n = 114 are shown in Fig. 7. In all cases, the smallest HOMO–LUMO gap corresponds to the oligomer with a methyl group at the Ga atom and fluorine at the nitrogen atom. The largest values of the HOMO–LUMO gap correspond to strong electron-withdrawing substituents (F, CF3) at the terminal Ga atoms and electron-donating groups (CH3) at the terminal N atoms. Thus, substituents which reduce the dipole moment of the system (Fig. 3) reduce the energy gap as well (Fig. 7). More details can be found in the ESI† (Fig. S3)
 | ||
| Fig. 7 HOMO–LUMO gaps of open X3[HGaNH]114Y3 (a) and closed XGa[HGaNH]114NY (b) oligomers with different substituents X and Y. | ||
The HOMO–LUMO gap values of closed oligomers vary within circa 0.4 eV depending on terminal groups (Fig. 7b). The change is much more pronounced in the case of open oligomers (Fig. 7a); the HOMO–LUMO gap varies from 0.94 to 2.91 eV. It should be noted that the change in the HOMO–LUMO gap value is smaller in the case of shorter oligomers. Depending on substituents, the HOMO–LUMO gap of open oligomers changes within 1.83 eV (from 2.22 to 4.05 eV) for n = 30 and within 1.33 eV (from 5.23 to 6.56 eV) for n = 9.
Computational details
The DFT B3LYP/SVP16 level of theory was used throughout. The geometries of all compounds were fully optimized. The vibrational frequency calculations were performed to verify that the obtained structures are true minima on their respective potential energy surfaces. Open compounds containing CH3 or CF3 groups turned out to be C3 symmetric since these terminal groups are turned around the main axis of the molecules to minimize the steric strain. All other studied compounds are C3v symmetric. The optimized xyz coordinates for all considered compounds are provided in the ESI.†Selected compounds were optimized using a larger basis set to verify the reliability of the chosen computational level. It was found that the TZVP bases set provides slightly longer bond lengths compared to the SVP basis set. The average difference in Ga–N bond lengths computed using TZVP and SVP basis sets (circa 0.002Å) is the same for H3[HGaNH]9H3 and H3[HGaNH]30H3 compounds. Thus, we conclude that the error does not increase with the elongation of the oligomer. A difference in the reaction energies of formation of H3[HGaNH]nH3 calculated using TZVP and SVP basis sets is about 1% of the absolute value obtained using the TZVP basis set, irrespective of the oligomerization degree. The HOMO–LUMO gaps of H3[HGaNH]nH3 obtained using the TZVP basis set are by 0.27 eV (n = 9) and by 0.20 eV (n = 30) smaller than the values obtained using the SVP basis set.
In general, HOMO–LUMO energies are sensitive to the particular choice of DFT method. B3LYP was found to be slightly more preferable in the case of small GaN based compounds.11 The PBE0 DFT functional was tested for H3[HGaNH]nH3 oligomers with some intermediate lengths.4 It was found that a choice of PBE0 instead of B3LYP causes an increase of the HOMO–LUMO gap value of about 0.55 eV, while the overall tendencies in the changes in the HOMO–LUMO gap upon the rod elongation are similar.
Calculated ESP charges fit to the electrostatic potential13 constraining them to reproduce the dipole moment.
The partial density of states (PDOS) for choosing fragment A was obtained as Gaussian convolution of the discrete PDOS distribution
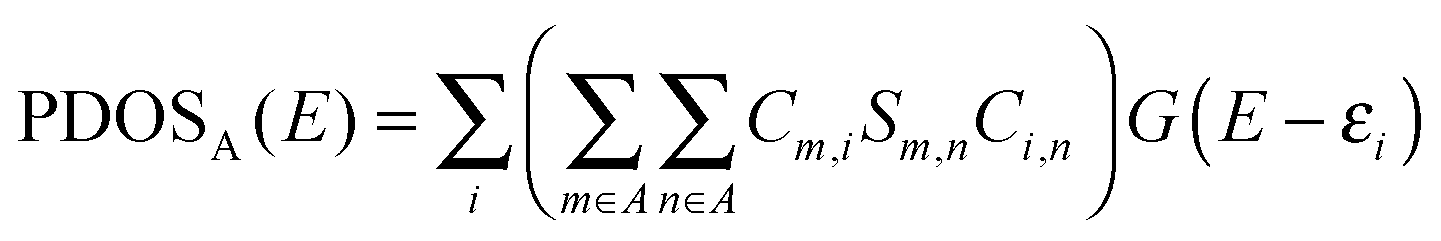
Conclusions
The influence of electron-donating/electron-withdrawing terminal groups on the electronic properties of the rod-shaped GaN-based oligomers was computationally explored. It was found that for the open (rod-like) oligomers, the molecular orbital energy values of end-localized states are highly sensitive to a particular terminal group. This, along with a variation of the length of the oligomer, allows adjusting the HOMO–LUMO gap to cover a wide spectral range. For example, the elongation of F3[HGaNH]n(CH3)3 from n = 9 to 114 reduces the HOMO–LUMO gap from 6.5 to 2.9 eV, the elongation of (CH3)3[HGaNH]nF3 from n = 9 to 114 reduces the HOMO–LUMO gap from 5.2 to 0.9 eV, and the change from F3[HGaNH]n(CH3)3 to (CH3)3[HGaNH]nF3 reduces the HOMO–LUMO gap from 2.9 to 0.9 eV in the case of n = 114 and from 6.5 to 5.2 eV in the case of n = 9.For the long oligomers, where the influence of the substituents at the opposite end of the rod is negligible, HOMO–LUMO gap values decrease in the order F > CF3 > H > Me for the substituents on the Ga atom. The order of substituents for the analogues at the N atom is F < CF3 < H < CH3.
Qualitatively, this trend is observed both for open (rod-like) and closed (needle-shaped) oligomers, but in the latter case the influence of the substituents on energy gap values is much less pronounced.
The HOMO and LUMO of the rod-shaped open oligomers are strictly localized at the opposite ends of the molecules. Moreover, all other occupied orbitals with energy within at least 2 eV below the HOMO are also localized at the Ga-terminated end. Likewise, all unoccupied orbitals with energy within at least 2 eV higher than the LUMO are localized at the N-terminated end. Unlike the open oligomers, in closed ones there are upper occupied molecular orbitals which are localized at the same end of the oligomer as the LUMO.
The HOMO–LUMO transition in long open oligomers requires a relatively small energy change (which could be as low as 0.9 eV for X = CH3, Y = F in oligomers of circa 10 nm of length), but involves an intermolecular end-to-end charge transfer. In contrast, in closed oligomers the transitions occur only at the nitrogen terminated end of the oligomer and require a large energy change (at least 6.3 eV for n = 114, X = CH3, Y = F).
Overall, we conclude that the type of termination (open or closed) has the largest effect on the electronic properties of [HGaNH]n oligomers. The elongation and variation of substituents on the terminal group have comparable effects on the HOMO–LUMO gap and can be effectively combined for turning the energy gap to a desirable value.
Acknowledgements
This work was supported by SPbSU grants 12.38.255.2014 and 12.50.1563.2013. Research was carried out using computational resources provided by Resource Center “Computer Center of SPbU”.References
- (a) S. Han, W. Jin, D. Zhang, T. Tang, C. Li, X. Liu, Z. Liu, B. Lei and C. Zhou, Chem. Phys. Lett., 2004, 389, 176–180 CrossRef CAS; (b) M. Tchernycheva, P. Lavenus, H. Zhang, A. V. Babichev, G. Jacopin, M. Shahmohammadi, F. H. Julien, R. Ciechonski, G. Vescovi and O. Kryliouk, Nano Lett., 2014, 14, 2456–2465 CrossRef CAS PubMed; (c) J. C. Johnson, H.-J. Choi, K. P. Knutsen, R. D. Schaller, P. Yang and R. J. Saykally, Nat. Mater., 2002, 1, 106–110 CrossRef CAS PubMed; (d) R. Yu, W. Wu, Y. Ding and Z. L. Wang, ACS Nano, 2013, 7, 6403–6409 CrossRef CAS PubMed; (e) Y. Dong, B. Tian, T. J. Kempa and C. M. Lieber, Nano Lett., 2009, 9, 2183–2187 CrossRef CAS PubMed.
- B. L. Kormos, J. A. Jegier, P. C. Ewbank, U. Pernisz, V. G. Young, Jr., C. J. Cramer and W. L. Gladfelter, J. Am. Chem. Soc., 2005, 127, 1493–1503 CrossRef CAS PubMed.
- A. Y. Timoshkin and H. F. Schaefer, J. Am. Chem. Soc., 2004, 126, 12141–12154 CrossRef CAS PubMed.
- A. V. Pomogaeva and A. Y. Thimoshkin, J. Phys. Chem. C, 2015, 119, 16475–16482 CAS.
- A. Y. Timoshkin and H. F. Schaefer, J. Phys. Chem. C, 2008, 112, 13816–13836 CAS.
- A. J. Karttunen, M. Linnolahti and T. A. Pakkanen, J. Phys. Chem. C, 2008, 112, 10032–10037 CAS.
- H. Wang, H. S. Wu and J. F. Jia, Chin. J. Chem., 2006, 24, 731–738 CrossRef CAS.
- A. Schaumlöffel, M. Linnolahti, A. J. Karttunen and T. A. Pakkanen, ChemPhysChem, 2007, 8, 62–63 CrossRef PubMed.
- A. Y. Timoshkin, Solid-State Electron., 2003, 47, 543–548 CrossRef CAS.
- A. Mohajeri and M. Ebadi, J. Phys. Chem. A, 2012, 116, 4678–4686 CrossRef CAS PubMed.
- A. V. Pomogaeva and A. Y. Timoshkin, Theor. Chem. Acc., 2014, 133, 1572–1581 CrossRef.
- E. Simon and P. G. Mezey, Theor. Chem. Acc., 2012, 131, 1097–1198 CrossRef.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- W. A. Sheppard, J. Am. Chem. Soc., 1962, 84, 3072–3076 CrossRef CAS.
- J. E. True, T. D. Thomas, R. W. Winter and G. L. Gard, Inorg. Chem., 2003, 42, 4437–4441 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision C.01, Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures of all oligomers. See DOI: 10.1039/c6cp02576e |
| This journal is © the Owner Societies 2016 |