
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of CaB6 in the thermal decomposition of the hydrogen storage material Ca(BH4)2†
Christoph J.
Sahle
*a,
Christian
Sternemann
b,
Carlotta
Giacobbe
a,
Yigang
Yan
c,
Christopher
Weis
b,
Manuel
Harder
bd,
Yury
Forov
b,
Georg
Spiekermann
de,
Metin
Tolan
b,
Michael
Krisch
a and
Arndt
Remhof
cf
aEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: christoph.sahle@esrf.fr
bFakultät Physik/DELTA, Technische Universität Dortmund, Dortmund, Germany. E-mail: christian.sternemann@tu-dortmund.de
cMaterials for Energy Conversion, Empa, Ch-8600 Dübendorf, Switzerland
dDeutsches Elektronen-Synchrotron DESY, Notkestrasse 85, 22607 Hamburg, Germany
eDeutsches GeoForschungsZentrum, Section 3.3, Potsdam, Germany
fCrystallography, Institute for Geo- and Environmental Science, Albert-Ludwigs-University Freiburg, D-79098 Freiburg im Breisgau, Germany
First published on 22nd June 2016
Abstract
Using a combination of high resolution X-ray powder diffraction and X-ray Raman scattering spectroscopy at the B K- and Ca L2,3-edges, we analyzed the reaction products of Ca(BH4)2 after annealing at 350 °C and 400 °C under vacuum conditions. We observed the formation of nanocrystalline/amorphous CaB6 mainly and found only small contributions from amorphous B for annealing times larger than 2 h. For short annealing times of 0.5 h at 400 °C we observed neither CaB12H12 nor CaB6. The results indicate a reaction pathway in which Ca(BH4)2 decomposes to B and CaH2 and finally reacts to form CaB6. These findings confirm the potential of using Ca(BH4)2 as a hydrogen storage medium and imply the desired cycling capabilities for achieving high-density hydrogen storage materials.
1 Introduction
Lightweight metal borohydrides are promising candidates for efficient mobile hydrogen storage devices because of their high gravimetric hydrogen density.1–3 For industrial applications such as fuel cell powered cars, a highly cyclable, relatively cheap, and safe hydrogen storage medium is necessary. However, conditions suitable for re-hydrogenation of metal borohydrides are often difficult to achieve in practice and the hydrogen cycling is hindered due to the formation of stable boron phases such as elemental boron or, even more severe, B12H12-species.4 The identification of these (intermediate) reaction products in the decomposition pathway during hydrogen release is difficult as they often form in amorphous phases.4,5 Despite continuous efforts, to date no practical mobile hydrogen storage system has been discovered and therefore new ways to investigate and elucidate the reaction pathways these metal borohydrides follow are essential for the development of potential future applications.6Ca(BH4)2 has a gravimetric hydrogen density of 11.6 mass% (or 124.1 kg m−3)2,7,8 and thus is a promising candidate for use as a reversible hydrogen storage medium. For this material, different desorption pathways under various experimental conditions are vividly discussed and researchers suggest different (amorphous) intermediates and final decomposition products such as CaB2Hx-species,5,8–10 amorphous CaB12H12,9,11–13 and amorphous boron (a-B).8–10,13 Marketable technical applications demand storage media free of these stable phases e.g. by yielding only CaB6 after hydrogen release, which would make complete cycling feasible. Two competing reaction pathways are mainly discussed for Ca(BH4)2, one leading to CaB6 as a favorable end product (1), and the other one resulting in CaB12H12(2):
| Ca(BH4)2 → (amorphous) intermediate |
| → CaB2Hx + (4 − x/2)H2 |
| → 1/3CaB6 + 2/3CaH2 + (x/2 − 2/3)H2 | (1) |
| Ca(BH4)2 → (amorphous) intermediate |
| → 1/6CaB12H12 + 5/6CaH2 + 13/6H2. | (2) |
The formation of CaB2Hx was critically discussed, see e.g.ref. 14, 15 and 16. Riktor et al. assigned this intermediate to be crystalline CaB2H2 with an orthorhombic unit cell and either a Pnna or a Pbna space group using X-ray diffraction (XRD).5 A structure with an orthorhombic unit cell was also found by Aoki et al.17 for this intermediate without reporting the stoichiometry. Recently, based on NMR studies, Yan et al. did not observe CaB12H12 among the decomposition products and identified the intermediate to be CaB2H6 for decomposition between 320 °C and 350 °C.10
In a temperature range close to the melting point of Ca(BH4)2 at 370 °C,18 reaction paths (1) and (2) might compete. Partial and almost complete reversibility of the process was achieved under moderate reaction conditions (ca. 345 °C and 90–185 bar H2).9,10 At decomposition temperatures of 400 °C and 450 °C, amorphous boron has been identified as the major boron containing species, suggesting a third decomposition pathway along19
| Ca(BH4)2 → 2B + CaH2 + 3H2. | (3) |
Here, we report how we used a combination of high resolution powder XRD and X-ray Raman scattering (XRS) spectroscopy at both the K-edge of B and the L2,3-edges of Ca to analyze the thermal decomposition products of Ca(BH4)2. XRD provides accurate information about the crystalline reaction products and yields boundary conditions for a detailed quantitative analysis of the XRS spectra in order to identify the nano-crystalline and/or amorphous decomposition products of the material after hydrogen desorption and thus validate the reaction paths (1), (2), and (3), the suggested decomposition pathways around 400 °C.
2 Methods
The native Ca(BH4)2 sample (Sigma Aldrich) was decomposed using dynamical vacuum conditions at 350 °C in a stainless steel autoclave for 50 h. A second set of samples was decomposed under the same vacuum conditions at 400 °C for 0.5, 2, and 15 h, respectively. For reference compounds we used partially amorphous B, CaB6, CaB12H12, CaO, Ca(OH)2, and H3BO3. H3BO3, CaB6, CaO, Ca(OH)2, and B were purchased from Sigma Aldrich, whereas CaB12H12 was obtained from Katchem. We used a purified Ar filled glove box (H2O and O2 content below 0.1 ppm) to fill the sample powders into quartz glass capillaries (1 mm diameter/0.01 mm wall thickness) for the measurements.XRD is a well established and accurate structural analysis probe and is routinely used for the study of hydrogen storage materials; for XRD studies on Ca(BH4)2, see e.g.ref. 5, 8, 9, 13, 17 and 20. XRS, on the other hand, is less known, but is an emerging technique to probe soft X-ray absorption edges using hard X-rays.21 XRS enables us to study lightweight elements with bulk sensitivity even when contained in a matrix of heavier elements. Consequently, XRS allows for in situ studies of chemical reactions under realistic reaction conditions, i.e. of samples inside chemical reaction cells under realistic reaction atmospheres. Thus, this inelastic X-ray scattering technique combines the sensitivity of absorption spectroscopy to reveal information about the local atomic and electronic structures with the unique properties of hard X-rays. XRS can be employed for the analysis of low Z-element containing samples, both crystalline and amorphous, under in situ conditions.
XRS was used to study e.g. chemical reactions22–24 or to investigate low-Z materials under extreme pressure and temperature conditions.25,26 This method was applied to analyze the B K-edge e.g. in MgB2, BN, C2B10H12, B2O3, and Li2B4O7.25,27–30 The Ca L2,3-edges were studied so far using only electron energy loss spectroscopy or soft X-ray absorption spectroscopy e.g. to characterize materials of geological and biological relevance31,32 with the exception of a high pressure XRS study of the Ca L-edge in CaSiO3 glass.33 Recently, in situ XRS spectroscopy was used to investigate hydrogen desorption in LiBH4, LiBH4 nano-composites, and Mg(BH4)2.23,34,35
The high resolution powder XRD data were collected at ID22 of the ESRF (Grenoble, France) using a multi-channel detector. We measured the different patterns in two different runs (using incident wavelengths of λ = 0.40 Å and λ = 0.32 Å, respectively). The capillaries were mounted on a rotating support and were spun at approximately 2 Hz to improve the powder average. We collected data to cover a momentum transfer range of 0.0 < |q| < 9.0 Å−1 and binned the data onto a constant step size of 0.002 Å−1.
We performed the measurements of the B K-edge at beamline ID20 of the ESRF using the multi-analyzer-crystal XRS spectrometer. The analyzer energy was fixed at 9.7 keV using the Si(660) Bragg reflection and the incident energy was scanned to obtain energy losses in the vicinity of the B K-edge from 180 eV to 225 eV. The spectrometer is equipped with 6 Maxipix Timepix area detectors, one for each set of 12 spherically bent Si analyzer crystals. We utilized three of these analyzer arrays and set the mean scattering angles of these arrays to 50° for an average momentum transfer of 4.2 ± 0.2 Å−1. With this setup, we achieved an overall energy resolution of 0.6 eV. We measured the Ca L2,3- and B K-edges at beamline P01 of PetraIII (Hamburg, Germany) using 11 Si(660) analyzers focused on 4 avalanche photodiodes. The scattering angle was 32° resulting in an average momentum transfer of 2.9 ± 0.4 Å−1 and an overall energy resolution of 0.9 eV. The incident energy was scanned to measure the Ca L2,3-edges (B K-edge) for the energy loss between 340 eV (173 eV) and 375 eV (223 eV). For the final spectra shown in this report, we averaged over the scattering signals from the different analyzer crystals. Before further processing, we corrected for the valence electrons' response as discussed in ref. 36 and 37. Details of the experimental data processing can be found in ref. 37. After background subtraction, we normalized the B K-edge XRS spectra to their integrated intensity between 183 and 223 eV energy loss and the Ca L2,3-edge spectra to their integrated intensity between 345 and 373 eV energy loss. Both these intergrated intensities relate to the scattering of one B and one Ca atom in each compound, respectively.
3 Results
3.1 X-ray diffraction
The high-resolution powder XRD patterns are shown in Fig. 1 together with the Rietveld refinements (black dashed lines). All refinements were achieved using the TOPAS program package.38 The overall agreement between the refinement and the measured powder diffraction patterns is satisfactory (5.76% < Rwp < 8.55%). The quantitative analysis yields the values summarized in Fig. 4a (and in the ESI† in a tabular form). The diffraction patterns indicate significant contributions from oxides (violet) and hydroxides (gray) in the samples (Fig. 4a). Ca(BH4)2 is highly sensitive to contamination (see e.g.ref. 5, 17 and 39) and the formation of oxides and hydroxides could not be completely avoided. We assume that the formation of the oxides occured during initial sample treatment, and that their contribution does not affect the decomposition process due to their high thermal stability. As we will show later, the independent analysis of the B K-edge supports this assumption.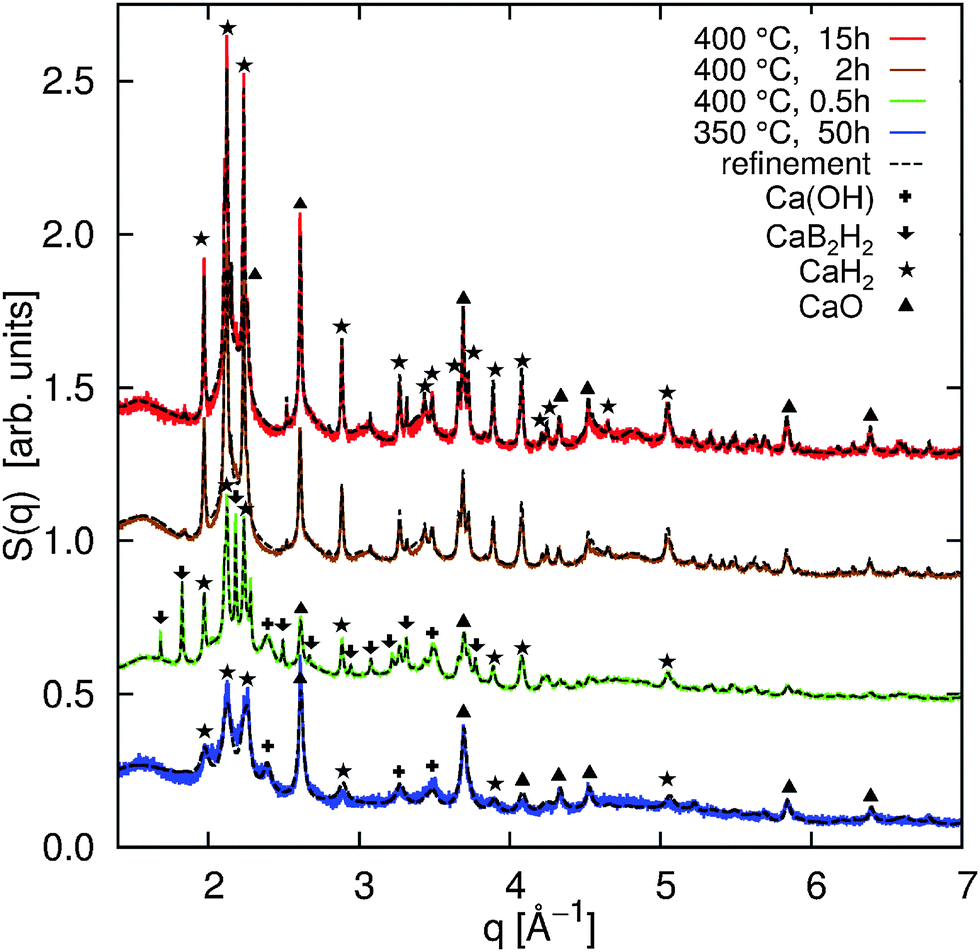 | ||
| Fig. 1 High resolution XRD patterns of annealed samples (colored solid lines) and the corresponding best fits (dashed lines) from Rietveld refinement. The incident X-ray wavelength was λ = 0.40 Å for the sample annealed at 350 °C and for the sample annealed at 400 °C for 0.5 h, the X-ray wavelength used to measure the diffraction pattern of the samples annealed at 400 °C for 2 h and 15 h was λ = 0.32 Å. | ||
For all annealing conditions with the exception of 400 °C and 0.5 h, we find nanocrystalline contributions from CaB6. The averaged size of the CaB6 nanocrystals is the smallest at 350 °C and increases at higher annealing temperatures (compare the table in the ESI†). This supports the improved cycling capabilities when Ca(BH4)2 is decomposed at 350 °C compared to 400 °C due to the increased crystallinity at 400 °C.10 We could not identify crystalline CaB12H12 or crystalline B in any of the samples. CaH2 is found to be a constituent in all samples and its amount increases with annealing time. For the sample annealed at 400 °C for 0.5 h, we can identify a crystalline intermediate with a diffraction pattern that resembles the one observed earlier.5,17
It has to be noted that according to pathway (1), the ratio between CaH2 and CaB6 should be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Instead, based on XRD, we observe a higher ratio in favor of CaH2. This can be ascribed to amorphous contributions from CaB6 or to the formation of amorphous B. Thus, we cannot exclude the competing pathways (2) and (3) because XRD is not sensitive to extract and distinguish possible amorphous contributions from CaB12H12, CaB6, and B in such complex systems. These amorphous contributions can be accessed by XRS spectroscopy.
:1. Instead, based on XRD, we observe a higher ratio in favor of CaH2. This can be ascribed to amorphous contributions from CaB6 or to the formation of amorphous B. Thus, we cannot exclude the competing pathways (2) and (3) because XRD is not sensitive to extract and distinguish possible amorphous contributions from CaB12H12, CaB6, and B in such complex systems. These amorphous contributions can be accessed by XRS spectroscopy.
3.2 X-ray Raman scattering at the Ca L2,3-edges
With the quantitative Rietveld refinement of the crystalline phases of the different decomposed samples, we can analyze the XRS spectroscopy results using spectral fingerprinting. Fig. 2a shows the Ca L2,3-edges of the native Ca(BH4)2 sample and the different spectra of the sample after decomposition at 350 °C and 400 °C. Using spectra of the reference compounds CaB6, CaH2, CaO, and Ca(OH)2 (shown in part (b) of the same figure) and the spectrum of the native sample, we found the best component fits shown as dashed black lines in Fig. 2a. For all linear combination fits, we used the CaO/Ca(OH)2/CaH2 ratio revealed by the Rietveld refinement of the XRD data. References for pure intermediate phases are not available so far. Allowing also for CaB12H12 in the fit routine, we find a contribution of less than 2% in all spectra. Only at 400 °C after 15 h of annealing time a contribution of 6% of CaB12H12 was found. As the formation of CaB12H12 is expected to occur in the first stages of decomposition we conclude that within the limits of the experimental error CaB12H12 is not contained in our samples. Consequently, we have not considered this compound in the final fitting procedure.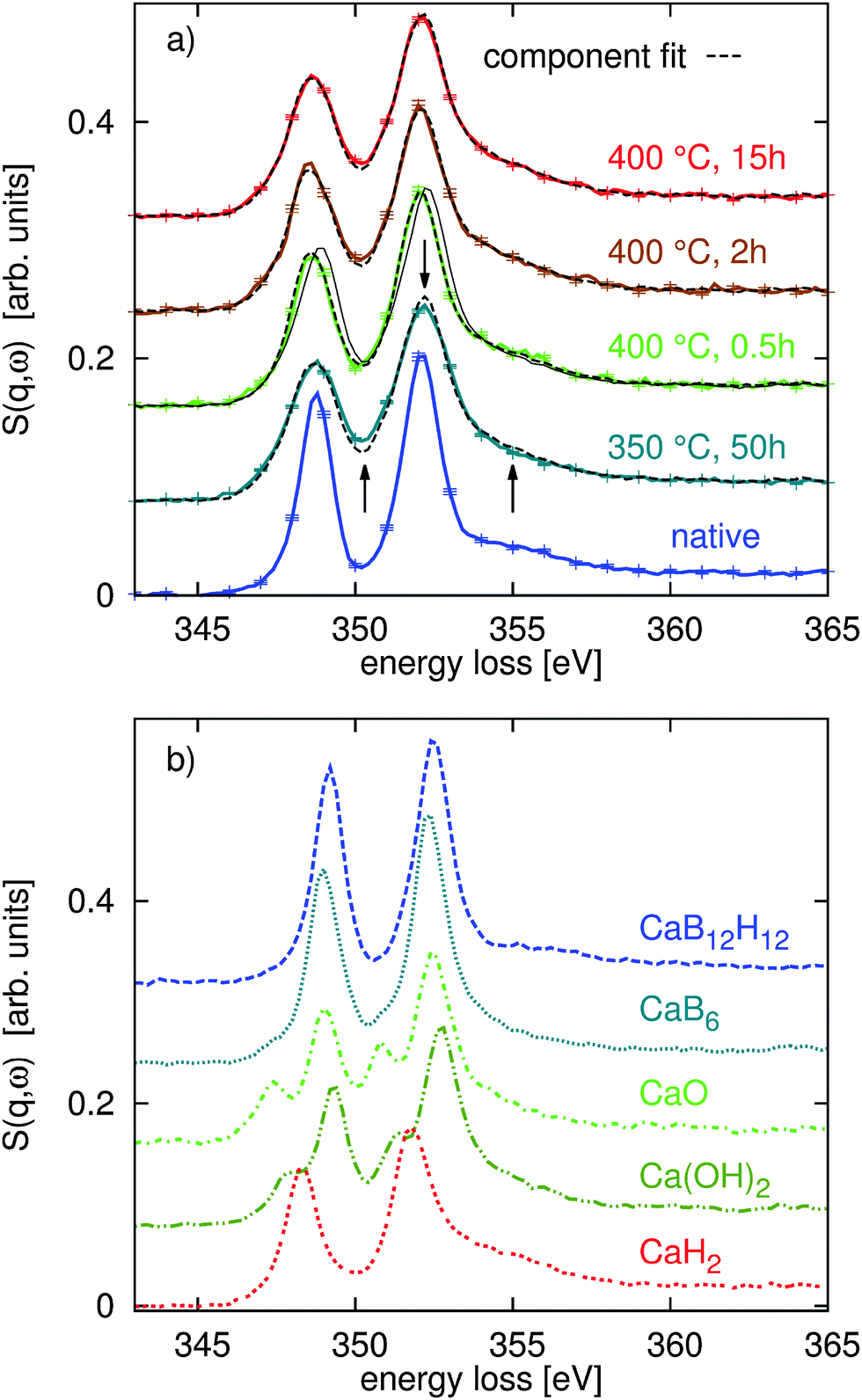 | ||
| Fig. 2 (a) XRS spectra of the Ca L2,3-edges for the native Ca(BH4)2 sample and the sample after thermal decomposition at different temperatures and annealing durations. The component fits are shown as thin dashed black lines, the largest deviations between the best fits and spectra are indicated by arrows. The solid line of the spectrum recorded at 400 °C and 0.5 h indicates a composition as inferred from the B K-edge data (see text for details), which does not resemble the shape of the Ca L-edge spectrum. (b) XRS spectra of the Ca L2,3-edges of the reference compounds CaB12H12, CaB6, CaO, Ca(OH)2, and CaH2. The Ca L2,3-edges show distinctly different shapes and different features for the different reference compounds. | ||
At 400 °C after annealing for only 0.5 h, we find a strong contribution of native Ca(BH4)2 to the Ca L2,3- and B K-edge spectra. For all other annealing conditions this native compound is not observed. As we verified a crystalline intermediate by XRD, it is likely that this compound exhibits a similar spectral shape to the native reference samples. Consequently, the Ca(BH4)2 content at 400 °C after 0.5 h might indicate the amount of intermediate present in the sample. However, we have no access to a pure reference sample of the intermediate so that we cannot draw a final conclusion here. It is also possible that Ca(BH4)2 melts at 370 °C18 and occurs in the amorphous state after partial solidification as no signal of Ca(BH4)2 was observed by XRD. As both, the intermediate and the native sample, have the same Ca:B ratio, we are still able to use the results for a stoichiometric analysis. We would like to note that we cannot identify CaB6 in the first annealing phase at 400 °C (0.5 h) as proposed in reaction path (1). All fit results are summarized in Fig. 4b (see the ESI† for error bars of the fit results).
At this point, we can conclude that the fit results obtained at 400 °C for 2 h and 15 h point toward reaction path (1) but the ratio of CaH2:CaB6 indicates that a minor part of the reaction follows pathway (3). Consequently, we expect some contribution from amorphous boron. We can exclude reaction pathway (2) as we find no formation of CaB12H12. The situation changes remarkably when the composition of the sample annealed at 400 °C for only 0.5 h is considered. Here, we clearly observe an intermediate state. Surprisingly, no CaB6 is observed and we have identified CaH2 together with CaB2Hx/Ca(BH4)2. The sample annealed for 50 h at 350 °C resembles closely the composition of the sample annealed for 2 h at 400 °C due to the lower activation temperature.
We have no direct evidence for B from the fits of the Ca L2,3-edges, but based on the amount of CaH2 from the Ca L2,3-edges and the reactions (1) and (3), we can estimate the amount of B contained in the samples in the form of CaB6 and elemental B, respectively, by assigning all excess CaH2 from pathway (1) to the partial decomposition occurring via pathway (3). These values are reported in Fig. 4d and we can confront them with the values derived from a multi-component fit of the B K-edge as follows.
3.3 X-ray Raman scattering at the B K-edge
Fig. 3 shows the XRS spectra of the measured B K-edges of the native and decomposed samples in part (a) and the spectra of the reference compounds CaB12H12, CaB6, B, and H3BO3 in part (b). Again, the dashed black lines in Fig. 3a represent the best fits of the multi-component fit. Based on the fitting results of the Ca L2,3-edges, we used B and CaB6 as components of the fit and considered contributions from the native sample only for annealing conditions of 400 °C and 0.5 h. We found no evidence of boron oxidation using the H3BO3 as a reference for oxides. | ||
| Fig. 3 (a) XRS spectra of the B K-edge for the native Ca(BH4)2 sample and the sample after decomposition at different temperatures and annealing durations. The component fits are shown as dashed black lines (combination of CaB6, B, and Ca(BH4)2), a thin grey line represents the results from a linear combination of reference spectra (B and Ca(BH4)2) according to the fit results of the Ca L2,3-edges and the subsequent estimation of the boron containing phases. (b) XRS spectra of the B K-edge of the reference compounds CaB12H12, CaB6, B, and H3BO3. As the Ca L2,3-edges, the B K-edge shows distinctly different shapes and different features for the different reference compounds. | ||
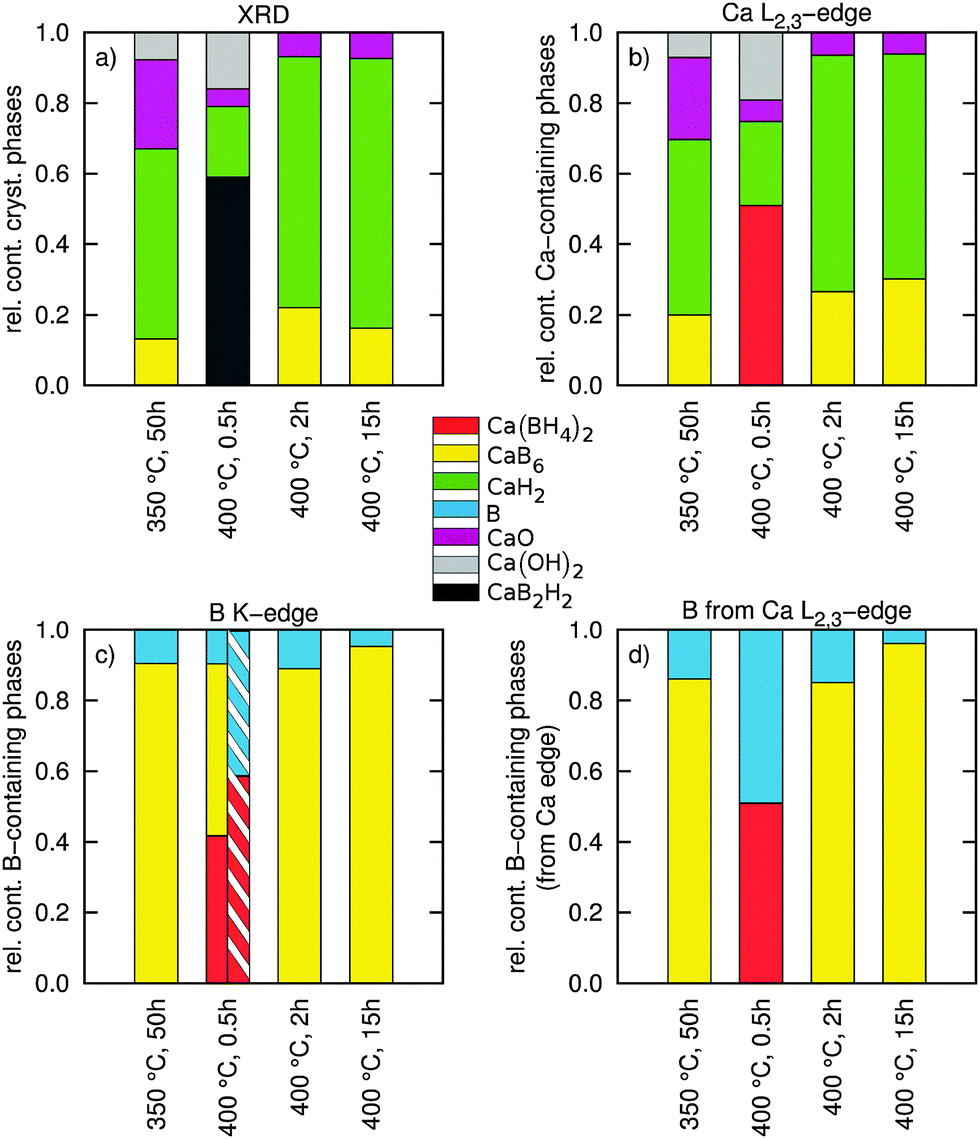 | ||
| Fig. 4 (a) Results of a quantitative phase analysis using the Rietveld method of the high resolution powder XRD patterns showing the relative abundence of (nano-)crystalline constituents. (b) Results of a fingerprinting analysis of the Ca L2,3-edge showing the relative content of the used Ca containing reference compounds (both amorphous and crystalline). (c) Results of a fingerprinting analysis of the B K-edge showing the relative abundance of B containing phases in different samples (both amorphous and crystalline). The hatched area shows the relative content of B containing phases from a two-component fit. (d) Extraction of the B content from the fit results of the Ca L2,3-edge via the reaction pathways (1) and (3) (both amorphous and crystalline). | ||
The results of this fit procedure are shown in Fig. 4c and are compiled in a tabular form in the ESI.† In general, all experimental spectra are well reproduced by the results of the component fits. Larger differences can be observed around 200 eV where the structure related to CaB6 is less pronounced and for the energy loss above 205 eV where the spectra are less structured than the respective best fits. This can be related to the fact that all boron containing decomposition products occur in amorphous or nanocrystalline form, whereas the reference spectra are recorded from crystalline or partly amorphous samples.
Except for the sample annealed at 400 °C for 0.5 h we find very good agreement between the fit results with respect to the content B and CaB6 and the compositions derived from the Ca L2,3-edges via reaction pathways (1) and (3) (compare Fig. 4c and d). The composition estimated for the sample in the intermediate state (400 °C, 0.5 h) disagrees with the fit results of the B K-edge as the estimate based on the Ca L2,3-edges suggests no contribution from CaB6. We used these B K-edge fit results (marked with an asterisk in the ESI†) to model the shape of the corresponding Ca L-edge shown as a solid line in Fig. 3a. It deviates significantly from the shape found experimentally. As the fit of the boron K-edge might be less sensitive to distinguish between elemental B and CaB6 due to the similar spectral shape, as can be inferred from the error bars of the fit (see the table in the ESI†), we repeated the fit of this spectrum by neglecting the CaB6 contribution, i.e. considering only B and Ca(BH4)2. The results of this fit (see the hatched bar in Fig. 4c and ESI† tables) are in line with the estimated boron content using stoichiometric considerations (Fig. 4d and ESI† tables) and the shape of the fitted spectrum deviates from the measured spectrum around the edge onset but agrees well with the experiment at a slightly higher energy loss.
4 Discussion
Using a combination of high resolution X-ray powder diffraction and X-ray Raman scattering spectroscopy at the B K-edge and Ca L2,3-edges, we analysed the thermally induced decomposition of Ca(BH4)2. The reaction conditions (dynamic vacuum, 350 °C and 400 °C) were chosen to compare with ref. 10. The long annealing time of 50 h at 350 °C was chosen to guarantee complete decomposition. Under these conditions the decomposition into CaB6 and CaH2 as solid reaction products could be verified. The annealing series at 400 °C was intended to clarify the observation of amorphous boron. Here, the combined XRD and XRS results, together with the findings of ref. 10, suggest that at temperatures of 400 °C and short annealing times (0.5 h) there is a competition between the decomposition along pathways (1) and (3). The observation of the crystalline intermediate CaB2Hx indicates that the decomposition partially proceeds along pathway (1). The XRS spectra, on the other hand, indicate that Ca(BH4)2 also decomposes along pathway (3), resulting in amorphous B as the major boron containing species.Longer annealing times of up to 15 h initiate the formation of CaB6 from CaH2 and B. This is supported by the increase of the CaB6 proportion at the expense of CaH2 and the B content as observed after longer thermal treatment. Comparing the standard enthalpy of formation of CaH2 (ΔH0 = 186 kJ mol−1) and CaB6 (ΔH0 = 121 kJ mol−1), CaH2 is found to be more stable. However, with increasing temperature the entropy gain from the release of gaseous H2 (TΔS ≈ 90 kJ mol−1, at T = 400 °C and ΔS(H2) = 135 J mol−1 K−1) supports the formation of CaB6 in line with our observations. The degree of crystallinity of CaB6, as observed by XRD, could be the key for the reversibility of the hydrogen release reaction, i.e. the rehydrogenation of the decomposition products. Yan et al. observed that the rehydrogenation conditions degrade with respect to the initial decomposition temperature.10 Here, we see that the crystallite size together with the amorphous contribution of CaB6 may be an indicator to tune the material for improved reversibility at lower temperatures.
5 Summary and outlook
The presented results show that XRS, especially in combination with complementary techniques such as XRD, is a powerful tool for the spectroscopic identification and quantification of solid phases, thereby unveiling the chemical reaction pathways and the role of intermediates. Once fully established, this method will be a useful tool beyond the hydride research. In the case of the thermal hydrogen release and decomposition of Ca(BH4)2 we could show that the formation of unwanted intermediates and products that restrict the re-hydrogenation and thereby the application as a hydrogen storage material, can be avoided (e.g. CaB12H12) or can be converted (e.g. elemental B) by appropriate choice of the dehydrogenation conditions. Decomposition under dynamic vaccum at 350 °C results in CaH2 and nano-crystalline CaB6 as residual solid phases that favor rehydrogenation. At higher temperatures, elemental B, resulting from the decomposition, can be further converted into CaB6 by a prolonged heating process. The resulting grain size of CaB6 seems to be unfavorably large for decomposition at 400 °C compared to the grain size obtained at 350 °C. The discussion so far had not involved any additives or catalysts that may increase the kinetics of the reaction or act as grain refiners, leaving vast space for further improvement in order to develop a technically usable borohydride-based hydrogen storage material.Acknowledgements
We thank the European Synchrotron Radiation Facility for providing synchrotron radiation and the technical and scientific staff of ESRF beamlines ID20 and ID22 for support and valuable discussions. We are further grateful for synchrotron radiation provided by PetraIII. We thank H. Yavas and the P01 beamline staff for support during the measurements. This work was supported by RESOLV (EXC 1069) and BMBF (Project No. 05K13PE2) within FSP-302. Y. Yan and A. Remhof acknowledge financial support granted by Switzerland through the Swiss contribution to the enlarged European Union.References
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- H.-W. Li, Y. Yan, S. Orimo, A. Züttel and C. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS.
- E. Rönnebro, Curr. Opin. Solid State Mater. Sci., 2011, 15, 44–51 CrossRef.
- M. Riktor, M. Sørby, K. Chłopek, M. Fichtner and B. Hauback, J. Mater. Chem., 2009, 19, 2754–2759 RSC.
- M. Ley, L. Jepsen, Y.-S. Lee, Y. Cho, J. Von Colbe, M. Dornheim, M. Rokni, J. Jensen, M. Sloth and Y. Filinchuk, et al. , Mater. Today, 2014, 17, 122–128 CrossRef CAS.
- K. Miwa, M. Aoki, T. Noritake, N. Ohba, Y. Nakamori, S. Towata, A. Züttel and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 155122 CrossRef.
- J.-H. Kim, S.-A. Jin, J.-H. Shim and Y. Cho, J. Alloys Compd., 2008, 461, L20–L22 CrossRef CAS.
- Y. Kim, S.-J. Hwang, J.-H. Shim, Y.-S. Lee, H. Han and Y. Cho, J. Phys. Chem. C, 2012, 116, 4330–4334 CAS.
- Y. Yan, A. Remhof, D. Rentsch, A. Züttel, S. Giri and P. Jena, Chem. Commun., 2015, 51, 11008–11011 RSC.
- L.-L. Wang, D. Graham, I. Robertson and D. Johnson, J. Phys. Chem. C, 2009, 113, 20088–20096 CAS.
- A. Kulkarni, L.-L. Wang, D. Johnson, D. Sholl and J. Johnson, J. Phys. Chem. C, 2010, 114, 14601–14605 CAS.
- C. Bonatto Minella, S. Garroni, C. Pistidda, R. Gosalawit-Utke, G. Barkhordarian, C. Rongeat, I. Lindemann, O. Gutfleisch, T. Jensen, Y. Cerenius and J. Christensen, J. Phys. Chem. C, 2011, 115, 2497–2504 Search PubMed.
- T. Frankcombe, J. Phys. Chem. C, 2010, 114, 9503–9509 CAS.
- P. Vajeeston, P. Ravindran, B. Hauback and H. Fjellvåg, Int. J. Hydrogen Energy, 2011, 36, 10149–10158 CrossRef CAS.
- I. Llamas-Jansa, O. Friedrichs, M. Fichtner, E. Bardaji, A. Züttel and B. Hauback, J. Phys. Chem. C, 2012, 116, 13472–13479 CAS.
- M. Aoki, K. Miwa, T. Noritake, N. Ohba, M. Matsumoto, H.-W. Li, Y. Nakamori, S. Towata and S. Orimo, Appl. Phys. A: Mater. Sci. Process., 2008, 92, 601–605 CrossRef CAS.
- M. Paskevicius, M. Ley, D. Sheppard, T. Jensen and C. Buckley, Phys. Chem. Chem. Phys., 2013, 15, 19774–19789 RSC.
- Y. Yan, A. Remhof, D. Rentsch and A. Züttel, Chem. Commun., 2015, 51, 700–702 RSC.
- Y. Kim, S.-J. Hwang, Y.-S. Lee, J.-Y. Suh, H. Han and Y. Cho, J. Phys. Chem. C, 2012, 116, 25715–25720 CAS.
- W. Schülke, Electron dynamics by inelastic X-ray scattering, OUP Oxford, 2007 Search PubMed.
- W. Mao, H.-k. Mao, Y. Meng, P. Eng, M. Hu, P. Chow, Y. Cai, J. Shu and R. Hemley, Science, 2006, 314, 636–638 CrossRef CAS PubMed.
- P. Miedema, P. Ngene, A. van der Eerden, T.-C. Weng, D. Nordlund, D. Sokaras, R. Alonso-Mori, A. Juhin, P. de Jongh and F. de Groot, Phys. Chem. Chem. Phys., 2012, 14, 5581–5587 RSC.
- J. Inkinen, J. Niskanen, T. Talka, C. Sahle, H. Müller, L. Khriachtchev, J. Hashemi, A. Akbari, M. Hakala and S. Huotari, Sci. Rep., 2015, 5, 15851 CrossRef CAS PubMed.
- S. Lee, J.-F. Lin, Y. Cai, N. Hiraoka, P. Eng, T. Okuchi, H.-k. Mao, Y. Meng, M. Hu and P. e. a. Chow, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7925–7929 CrossRef CAS PubMed.
- C. Sahle, C. Sternemann, C. Schmidt, S. Lehtola, S. Jahn, L. Simonelli, S. Huotari, M. Hakala, T. Pylkkänen and A. e. a. Nyrow, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6301–6306 CrossRef CAS PubMed.
- A. Mattila, J. Soininen, S. Galambosi, S. Huotari, G. Vankó, N. Zhigadlo, J. Karpinski and K. Hämäläinen, Phys. Rev. Lett., 2005, 94, 247003 CrossRef.
- S. Lee, P. Eng, H.-k. Mao, Y. Meng, M. Newville, M. Hu and J. Shu, Nat. Mater., 2005, 4, 851–854 CrossRef CAS.
- Y. Feng, J. Soininen, A. Ankudinov, J. Cross, G. Seidler, A. Macrander, J. Rehr and E. Shirley, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 165202 CrossRef.
- T. Fister, F. Vila, G. Seidler, L. Svec, J. Linehan and J. Cross, J. Am. Chem. Soc., 2008, 130, 925–932 CrossRef CAS PubMed.
- S. Naftel, T. Sham, Y. Yiu and B. Yates, J. Synchrotron Radiat., 2001, 8, 255–257 CrossRef CAS PubMed.
- Y. Politi, R. Metzler, M. Abrecht, B. Gilbert, F. Wilt, I. Sagi, L. Addadi, S. Weiner and P. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17362–17366 CrossRef CAS PubMed.
- S. Lee, P. Eng and H.-k. Mao, Rev. Mineral. Geochem., 2014, 78, 139–174 CrossRef CAS.
- P. Miedema, P. Ngene, A. Van Der Eerden, D. Sokaras, T.-C. Weng, D. Nordlund, Y. Au and F. De Groot, Phys. Chem. Chem. Phys., 2014, 16, 22651–22658 RSC.
- C. Sahle, S. Kujawski, A. Remhof, Y. Yan, N. Stadie, A. Al-Zein, M. Tolan, S. Huotari, M. Krisch and C. Sternemann, Phys. Chem. Chem. Phys., 2016, 18, 5397 RSC.
- H. Sternemann, C. Sternemann, G. Seidler, T. Fister, A. Sakko and M. Tolan, J. Synchrotron Radiat., 2008, 15, 162–169 CrossRef CAS PubMed.
- C. Sahle, A. Mirone, J. Niskanen, J. Inkinen, M. Krisch and S. Huotari, J. Synchrotron Radiat., 2015, 22, 400–409 CrossRef CAS PubMed.
- A. Coelho, General Profile and Structure Analysis Software for Powder Diffraction Data, version, 2007, vol. 4 Search PubMed.
- Y. Kim, D. Reed, Y.-S. Lee, J. Lee, J.-H. Shim, D. Book and Y. Cho, J. Phys. Chem. C, 2009, 113, 5865–5871 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02495e |
| This journal is © the Owner Societies 2016 |