
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of the surface composition of mixed monolayer films on the evaporation coefficient of water
Rachael E. H.
Miles
*a,
James F.
Davies
ab and
Jonathan P.
Reid
a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: rachael.miles@bristol.ac.uk
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 5th July 2016
Abstract
We explore the dependence of the evaporation coefficient of water from aqueous droplets on the composition of a surface film, considering in particular the influence of monolayer mixed component films on the evaporative mass flux. Measurements with binary component films formed from long chain alcohols, specifically tridecanol (C13H27OH) and pentadecanol (C15H31OH), and tetradecanol (C14H29OH) and hexadecanol (C16H33OH), show that the evaporation coefficient is dependent on the mole fractions of the two components forming the monolayer film. Immediately at the point of film formation and commensurate reduction in droplet evaporation rate, the evaporation coefficient is equal to a mole fraction weighted average of the evaporation coefficients through the equivalent single component films. As a droplet continues to diminish in surface area with continued loss of water, the more-soluble, shorter alkyl chain component preferentially partitions into the droplet bulk with the evaporation coefficient tending towards that through a single component film formed simply from the less-soluble, longer chain alcohol. We also show that the addition of a long chain alcohol to an aqueous-sucrose droplet can facilitate control over the degree of dehydration achieved during evaporation. After undergoing rapid gas-phase diffusion limited water evaporation, binary aqueous-sucrose droplets show a continued slow evaporative flux that is limited by slow diffusional mass transport within the particle bulk due to the rapidly increasing particle viscosity and strong concentration gradients that are established. The addition of a long chain alcohol to the droplet is shown to slow the initial rate of water loss, leading to a droplet composition that remains more homogeneous for a longer period of time. When the sucrose concentration has achieved a sufficiently high value, and the diffusion constant of water has decreased accordingly so that bulk phase diffusion arrest occurs in the monolayer coated particle, the droplet is found to have lost a greater proportion of its initial water content. A greater degree of slowing in the evaporative flux can be achieved by increasing the chain length of the surface active alcohol, leading to a greater degree of dehydration.
I. Introduction
Predicting the rates of water evaporation and condensation in an aerosol is important in areas including atmospheric science, drug delivery by inhalation therapies and spray drying. The rate of mass transport of water to and from large droplets (>20 μm) and flat surfaces containing dilute solutes is usually limited by the rate of gas diffusional transport and thermal conduction. However, interfacial transport across the surface and slow diffusional transport within the particle bulk can become increasingly important, especially as the particle size diminishes and/or as solute concentrations surpass their saturation/solubility limit. While it is often not feasible to gain control over the rate of mass transport by modifying thermal conductivity or gas-phase diffusion (through a change in carrier gas or total gas pressure respectively), manipulation of the surface or bulk composition may allow the rate of mass transfer to be controlled via surface transport and bulk diffusion processes. For example, condensed films formed from long-chain alcohols can slow the rate of water evaporation, reducing the evaporative mass flux by many orders of magnitude.1,2 Further, the high solute concentrations reached in evaporating aqueous droplets containing sucrose can lead to particles of high viscosity (>107 Pa s), significantly enhancing water retention within a particle due to slow bulk phase diffusion, and leading to particles that continually lose water over many hours or days.3–7The mass accommodation coefficient of water at a water surface is used to characterise the proportion of condensing molecules that make the transition from the gas phase into the droplet bulk, as opposed to re-entering the gas phase. By microscopic reversibility, the evaporation coefficient for water evaporation is assumed to be equal to the mass accommodation coefficient.8 Although widely debated for many years, the most recent measurements suggest that the mass accommodation coefficient for water accommodating at a pure water surface is close to 1, in agreement with molecular dynamics models.9–15 By contrast, measurements of rates of water evaporation from an aqueous surface with a condensed monolayer film of a long-chain alcohol have shown that the evaporation coefficient can be suppressed by many orders of magnitude, depending on the length of the hydrocarbon chain, falling from a value of 2.4 × 10−3 when the alcohol has a C12 chain to 1.7 × 10−5 for a C17 chain.2 Such suppressions are consistent with measurements of the impact of monolayer films on the reactive uptake coefficients of species such as N2O516–18 and broadly consistent with the structures and permeabilities of films expected from molecular dynamics simulations.14,19–22
Although the transport of water and reactive species through single component monolayers on aerosol droplets has been considered in some detail, systematic studies of the structure and transport through more complex film compositions have not been so extensive. This is despite the fact that it is clear that in most real-world applications, the transport of water or reactive species will be through a complex surface film containing multiple organic components with a composition that depends on surface pressure. Molecular dynamics simulations have investigated the equilibrium ordering of multiple organic species at the surface of an aqueous phase.23,24 For example, Habartová et al. have recently considered the film composition and packing of mixed films of palmitic acid and 1-bromoalkanes at aqueous surfaces, using molecular dynamics simulations to show that stable mixed monolayers are formed between palmitic acid (C15H31COOH) and bromohexadecane (C16H33Br), while stable mixed monolayers of palmitic acid with the much shorter chain bromodecane (C10H21Br) and bromopentane (C5H11Br) do not form.23 Simulations indicated that, prior to film collapse, increasing surface pressure leads to the “squeezing out” of the bromoalkane from the film.
For mixed films formed from organic components that are considered to be good surrogates for organic constituents of atmospheric aerosol or biological membranes, measurements of phase behaviour and surface tension have been made using conventional techniques such as surface tensiometry and Brewster angle microscopy.25–28 Such measurements have shown the considerable complexity in film properties that can arise for mixed component films, particularly following repeated compression and expansion cycles, exactly what would be expected to occur for atmospheric aerosol when progressing through repeated evaporation and condensation cycles.26 Surface compositions inferred from sum-frequency generation measurements have shown the greater propensity for the less-soluble component to reside at the surface. For example, Voss et al. investigated the competition between atmospherically relevant fatty acid monolayers at the air/water interface, showing that oleic acid preferentially displaced palmitic acid from the surface.29 On exposure to ozone, the greater solubility and volatility of the products from the oleic acid ozonolysis led to an increase in partitioning of palmitic acid to the surface over time.
Gilman and Vaida studied the influence of multicomponent films on the mass transport of acetic acid across an aqueous interface.30 In particular, they concluded that the permeabilities of mixed films containing equimolar 1-triacontanol/nonacosane and equimolar 1-triacontanol/cis-9-octadecen-1-ol were in between their single component values, demonstrating that the mass transfer rate is not governed solely by either the most or least permeable species. By contrast, Burden et al. examined the uptake of HCl into sulfuric acid doped with pentanoic acid and hexanol, concluding that the greater surface activity of hexanol led to uptake that was reminiscent of uptake onto a surface coated in hexanol alone.31 In studies of the reactive uptake of N2O5 by aqueous sulfuric acid, Cosman and Bertram found that the addition of a small amount of a branched surfactant (phytanic acid) to a monolayer largely composed of a straight chain surfactant (octadecanol) led to a significant increase in the permeability of the film.16
In this study, we extend our earlier work on the kinetics of water evaporation from aqueous droplets containing long-chain alcohols of varying carbon chain length to studies of the evaporation of aqueous droplets containing multiple components. In addition, we consider for the first time the interplay of slowing the mass transport rate across the interface through reducing the evaporation coefficient, and a reduction in rate of bulk diffusional transport of water within an evaporating particle that becomes increasingly viscous during drying. In Section II we present a description of the experiment and the method for simulating the evaporation kinetics of water from droplets. This is followed by a discussion of the temperature dependence of the kinetics of water evaporation from droplets coated in a single-component film, measurements of evaporation from droplets coated in binary component films, and then the evaporation of droplets that include both a long-chain alcohol and sucrose, a viscosity modifying constituent.
II. Experimental description and analysis
Water evaporation from single droplets was studied using a concentric cylindrical electrodynamic balance (C-EDB). This technique has been described in detail in previous publications and will only be briefly recounted here.2,9,32–35 Individual droplets with radii ∼25 μm (±100 nm) were produced from the solution of interest using a droplet-on-demand generator (MicroFab, MJ-ABP-01; 30 μm orifice) and charged by an induction electrode. Aqueous solutions (50/50 v/v water/ethanol) containing a single fatty alcohol component, a mixture of two fatty alcohols or a fatty alcohol with sucrose were prepared with starting fatty alcohol concentrations of 0.6 mM. Ethanol was used to solubilise the fatty alcohols due to their very limited solubility in water, as described in our previous publication.2 The ethanol component evaporates from the droplets more rapidly than water leaving a droplet that contains water alone as the solvent after <1 s following droplet generation. The very low concentration of solute in the starting solution and the miscibility of ethanol and water ensures that the starting droplet composition can be assumed to be homogeneous. The charged droplets were injected in to the C-EDB cell, where they became trapped in the electric field generated between the upper and lower pairs of concentric cylindrical electrodes. A constant DC voltage applied to the lower electrode offset the effect of gravity and the Stokes drag force from the gas flow within the trap.The environmental conditions in the gas phase surrounding the droplet were controlled by regulating the relative humidity (RH) and temperature of a 50 sccm nitrogen gas flow which entered the trapping cell through the lower outer electrode. The RH could be varied between fully dry conditions and >95% RH by altering the ratio of humidified to dry nitrogen contributing to the total flow. The temperature of the gas phase was kept constant at 293.15 K by a chiller unit (JULABO F-32) which circulated a 50/50 v/v water/ethylene glycol mixture within the top and bottom plates of the C-EDB trapping cell, through which the gas flow passed. The temperature within the trapping region was confirmed using a hand held temperature probe (Tenma 72-2060).
The angular variation in the intensity of the elastic light scattering (the phase function) was used to infer the change in droplet radius with time. Trapped droplets were illuminated with a Gaussian 532 nm laser beam (Laser Quantum, Ventus) and the phase function, which has the appearance of a pattern of light and dark fringes, was collected over a 24° angular range by a camera (THORLABS DCC1545M) centred at 45° to the direction of laser propagation. For a droplet of known refractive index, and with estimates of camera position and angular range, the fringe spacing can be used to estimate the droplet radius with an accuracy of +100 nm/−50 nm using the geometrical optics approximation.36 For measurements with surfactant doped aqueous droplets, the refractive index was assumed constant at 1.335 during evaporation, the value of pure water at a wavelength of 532 nm.37 For measurements with solute-containing aqueous droplets, the mass fraction of solute (MFS) increases as the droplet evaporates and a correction must be applied to the data to account for the resulting variation in refractive index.33 Measurements of droplet size were recorded every 0.01 s. Droplets were trapped within the C-EDB within ∼0.1 s of generation and the initial droplet size determined to within ±50 nm by extrapolating the measurements of size at early time back to t = 0 s. The low level of charge on the trapped droplets had no measureable effect on the evaporation rate.
The mass flux of water from a trapped aqueous droplet evaporating within the C-EDB can be predicted using the semi analytical treatment of Kulmala et al.38
 | (1) |
The Kulmala et al. treatment uses the Fuchs–Sutugin transition correction factors for mass and heat transport, which have the form given in eqn (2).39
 | (2) |
For water accommodating on/evaporating from a water surface, molecular dynamics simulations have consistently reported values of αM and γ = 111–15 and recent experimental studies have concluded that αM and γ > 0.5, although the available techniques are unable to discriminate more precisely above this value.9,10 Thus, for water evaporation from pure aqueous surfaces, the mass flux is controlled only by the rate of gas phase diffusion, as surface processes are not limiting. In measurements of water evaporation from aqueous droplets coated with fatty alcohol monolayer films, Davies et al. reported values of γ as low as 1.65 × 10−5 for mass transport through heptadecanol films, leading to droplet evaporation rates controlled by interfacial transfer kinetics rather than gas phase diffusion.2 This demonstrates the impact that the presence of a surface film can have on mass transfer rates. The thermal accommodation coefficient for water is widely accepted to be unity, and this is the value used in the current study.40–42
By comparison with predictions from the Kulmala et al. treatment in eqn (1), measurements of water mass flux from a trapped droplet can be used to determine the value of the evaporation coefficient. To do this, the gas phase relative humidity must be known with high accuracy. In this work, probe droplets containing either pure water or aqueous NaCl solutions of known concentration were injected into the C-EDB prior to each sample droplet to determine the trap RH. For water probe droplets, the Kulmala et al. treatment was used to extract the gas phase RH from the water mass flux, taking γ = 1. For aqueous NaCl droplets, the diameter growth factor of the equilibrated droplet was compared with a thermodynamic model (E-AIM) to infer the water activity, and thus determine the gas phase RH.43,44 The uncertainties in the RH retrieved from these two fitting methods were taken as reported in Davies et al. and are on the order of 0.2% at 95% RH, increasing to 1.5% at 50% RH.34
III. Results and discussion
Recently, Langridge et al. studied water mass accommodation on size-selected 200 nm dry diameter nigrosin aerosol at 298.15 K and 284.15 K using photoacoustic spectroscopy (PAS).45 After correcting for RH biases inherent in the PAS technique, the authors observed a marked change in the inferred value of the mass accommodation coefficient with RH for the wet aerosol, with experimental values of αM of approximately unity at 90% RH but falling to 0.05 at RHs below 70% RH. A similar trend was observed for 200 nm dry diameter internally mixed nigrosin–ammonium sulphate aerosol, with the value of αM approaching 1 for all RHs greater than ∼50%, owing to the increased particle hygroscopicity. The authors attributed this change in the value of αM with RH to a change in the surface composition of the aerosol with humidity, with droplets at low RH containing a higher proportion of solute at the surface and those at higher RH being water rich. To our knowledge, the study by Langridge et al. is the only one to have reported a systematic variation in the value of αM with RH, in effect reporting a dependence of αM on the surface composition of the aerosol. Here, we consider systematically the dependence of αM on the droplet surface composition by, firstly, investigating the dependence on RH of the evaporation coefficient of water through a single component monolayer film and, secondly, by exploring the influence of multicomponent films on droplet evaporation.III.i. Impact of experimental RH on γ: single component monolayer films
The evaporation of water from fatty alcohol coated droplets was measured as a function of RH to determine the effect on the evaporation coefficient, γ. This extends the scope of our earlier measurements of the γ value for water droplets coated with single component monolayer films of fatty alcohols in the homologous series of primary alcohols, from dodecanol (C12H25OH) to heptadecanol (C17H35OH) at 80% RH and 293.15 K.2 This previous work also reported measurements of the evaporation coefficient of water from hexadecanol doped droplets at constant RH but varying trap temperature between 284 and 328 K, which revealed a dependence on the fatty alcohol molecular footprint and droplet surface temperature.The evaporation of droplets formed from aqueous solutions (50/50 v/v water/ethanol) containing a single fatty alcohol, either tridecanol (C13H27OH) or tetradecanol (C14H29OH), were investigated with starting fatty alcohol concentrations of 0.6 mM. Droplets of each solution were injected into the C-EDB at RHs ranging from 45 to 95% RH at 293.15 K. The change in droplet radius with time during evaporation was recorded (inset, Fig. 1). Initial rapid loss of ethanol (at t < 0.5 s) from the droplet was followed by swift water loss (0.5 < t/s < 2.3) until a condensed fatty alcohol monolayer film formed around the droplet surface (at ∼12 μm in radius and t = 2.3 s), slowing the rate of further evaporation. The droplet evaporation rate following film formation was used to determine the evaporation coefficient, γ, by comparing the experimental data with predictions using the semi-analytical treatment of Kulmala et al., described in Section II, and varying the value for γ.
 | ||
| Fig. 1 (a) Evaporation coefficient for tridecanol coated droplets, plotted as a function of trap RH. Inset: Radius vs. time profile for a tridecanol doped droplet evaporating in to 52% RH. The Kulmala et al. model prediction for γ = 7.05 × 10−4 is shown as dashed red line. (b) Tridecanol molecular footprint at the point of film formation, plotted as a function of trap RH. (c) Correlation between the measured evaporation coefficient and the calculated droplet surface temperature. | ||
The γ values determined for monolayer coated aqueous droplets evaporating at different RHs are shown for the tridecanol and tetradecanol systems in Fig. 1(a) and 2(a) respectively. Each data point corresponds to a single droplet measurement, with the error bars corresponding to the uncertainty in RH as determined from the probe droplet and the consequent impact on the estimated value of γ. Interestingly, it is clear that a qualitatively similar trend to that observed by Langridge et al. for the mass accommodation coefficient of water on nigrosin is seen, with the measured evaporation coefficients smallest at low relative humidities and increasing rapidly as the RH increases. While the variation in the evaporation coefficient with RH is small, it is clearly resolvable using the C-EDB technique. For both fatty alcohols, measurements made at 80% RH agree well with values previously reported by Davies et al.2
 | ||
| Fig. 2 (a) Evaporation coefficient for tetradecanol coated droplets, plotted as a function of trap RH. (b) Tetradecanol molecular footprint at the point of film formation, plotted as a function of trap RH. (c) Correlation between the measured evaporation coefficient and the calculated droplet surface temperature. | ||
Following the conclusions of Langridge et al., a change in the evaporation coefficient of a monolayer film coated droplet with RH would only be expected if there were some dependence of the droplet surface composition, in this case film morphology, on the environmental conditions. Davies et al. found that the value of γ for water evaporating through a fatty alcohol monolayer film depends on the molecular footprint of the surface active species (the packing density) and the length of their alkyl chain (the film thickness), with decreasing molecular footprint and increasing carbon chain length leading to smaller evaporation coefficients. In this work we observe a change in evaporation coefficient with RH for a single molecular species (i.e. only one alkyl chain length), therefore the film thickness could only be changing with RH through the formation of stacked monolayers at the droplet surface. To investigate this possibility, the footprints of the molecules in the tridecanol and tetradecanol films at the point of film formation were determined for each droplet evaporation event and are shown in Fig. 1(b) and 2(b), respectively. In the calculation, all fatty alcohol molecules were assumed to be present at the droplet surface at the onset of film formation due to their poor solubility in water. The calculated molecular footprints at all RHs are consistent with those previously reported by Davies et al. for fatty alcohol monolayer films, discounting any formation of stacked monolayers at the droplet surface.2 Thus, it is not a change in the surface film thickness which gives rise to the observed variation in γ with RH.
Fig. 1(b) and 2(b) show that, although small, there is an increase in the molecular footprint of the fatty alcohols with increasing measurement RH, suggesting instead that the observed increase in the evaporation coefficient with relative humidity may be caused by a change in the film packing density. This result is consistent with the conclusions of Davies et al., who found that the evaporation coefficient for hexadecanol monolayers increased as the fatty alcohol molecular footprint increased.2 In that study, the change in molecular footprint was driven by a coarse change in the droplet surface temperature through increasing the trap temperature from ∼284 to 328 K, leading to an increase in the molecular footprint by ∼0.16 nm2. In the work reported here all measurements were performed at a constant trap temperature of 293.15 K. However, it is important to consider the much finer temperature effect arising from the evaporative cooling of a droplet that depends on the magnitude of the evaporating mass flux. Identical droplets evaporating into different RH conditions exhibit different evaporative mass fluxes, due to changes in the magnitude of the gradient between the droplet water activity and the gas phase RH at infinite distance. This leads to different suppressions in the droplet surface temperature below that of the gas phase, with droplets injected into lower humidities experiencing a larger mass flux and, thus, a lower surface temperature.
The depression of a droplet surface temperature (Tdroplet) below ambient (Tgas) can be estimated using the equation:38
 | (3) |
III.ii. Mass transport through multi-component monolayer films
To study the impact of multi-component monolayer films on mass transport, measurements were made of the evaporation kinetics of water from aqueous droplets doped with two different fatty alcohols contained within the homologous series from tridecanol (C13H27OH) to hexadecanol (C16H33OH). Aqueous solutions (50/50 v/v ethanol/water) containing each of the individual fatty alcohols were prepared with concentrations on the order of 0.3 mM; these were then mixed together in different volume ratios, varying both the identities of the fatty alcohol compounds and their relative molar ratios. The evaporation of droplets generated from each mixture was then studied in the C-EDB, along with the relevant individual fatty alcohol solutions.The previous section demonstrated a dependence of evaporation coefficient on the gas phase RH due to differences in droplet surface temperature. In order to be able to directly compare the γ values for mixed component monolayer films with those of the relevant single component films, the RH must be constant within the trapping region for the duration of the measurements. Small drifts in RH at high humidities can lead to a marked change in the evaporation coefficient; a 3% increase in RH at 80% RH would increase the evaporation coefficient for tetradecanol by 26%. To identify any such drift, the trap humidity was continuously monitored during experiments through the use of probe droplets. The majority of experiments were performed at RHs in the range 50% to 70% where the value of γ is less sensitive to the effect of RH drift.
In Fig. 3 we report the change in droplet radius with time for aqueous droplets doped with only tridecanol, only pentadecanol, and with a mixture of both fatty alcohol species. The measurements were performed at an RH of 60% and at 293.15 K. As before, all droplets show an initial rapid loss of ethanol followed by swift water loss until a condensed monolayer film is formed around the droplet surface, inhibiting further mass loss (at ∼15 μm in radius, t = 3 s). As expected, the droplets doped only with tridecanol and pentadecanol show a linear change in droplet radius with time following film formation, corresponding to a constant value for the evaporation coefficient of the homogeneous monolayer film. By contrast, droplets doped with both fatty alcohol species, and thus exhibiting multi-component monolayer films, show a highly non-linear dependence of droplet radius on time following film formation, with the precise evaporation profile depending on the molar ratio of the alcohols present. This change in droplet evaporation rate with time following film formation indicates that the evaporation coefficient of the mixed monolayer film must be evolving as the droplet continues to evaporate, something which can only be caused by a change in the composition or morphology of the monolayer film with time.
 | ||
Fig. 3 Change in radius with time observed for aqueous droplets doped with tridecanol (green), pentadecanol (orange) and different molar ratios of tridecanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pentadecanol (4:1, blue; 3:2, red; 2:3, black; 1:4, purple) evaporating in to 60% RH at 293.15 K. Monolayer film formation is seen at a radius of ∼15 μm in all droplets. Data is averaged over multiple droplet evaporation events. :pentadecanol (4:1, blue; 3:2, red; 2:3, black; 1:4, purple) evaporating in to 60% RH at 293.15 K. Monolayer film formation is seen at a radius of ∼15 μm in all droplets. Data is averaged over multiple droplet evaporation events. | ||
As seen earlier, the droplet evaporation rate following film formation can be used to determine the evaporation coefficient for the monolayer film using the model of Kulmala et al. The following procedure was adopted in order to use the time-dependence of the evaporation rate of water through the multi-component monolayer film to determine a time-dependent value of the evaporation coefficient. First, a polynomial expression chosen to most closely represent the experimental radius versus time data following film formation was fitted to the data (Fig. 4(a)), and then differentiated with respect to time, giving the dependence of the droplet evaporation rate (dr/dt) on time at each point during the evaporation event (Fig. 4(b)). Using the semi analytical model of Kulmala et al. and the gas phase RH determined from the probe droplets, droplet evaporation profiles were then simulated for a range of γ values using an initial droplet radius representative of the measurements (Fig. 4(c)). The correlation between the gradient (dr/dt) of each modelled evaporation profile and the evaporation coefficient itself was used to create a calibration plot (Fig. 4(d)). This was used to transform the experimentally measured evaporation rate to a γ value, and thus obtain the variation in γ with time.
 | ||
| Fig. 4 Procedure for extracting γ for multi-component monolayer films. (a) 9th order polynomial fit (red) to raw droplet radius vs. time data (black) over the region where a monolayer film is observed. (b) Differential of 9th order polynomial fit (dr/dt) as a function of time. (c) Theoretical radius vs. time profiles calculated using the Kulmala et al. treatment for different values of γ at 58.2% RH (black, 2.5 × 10−5; red, 6 × 10−5; blue, 1 × 10−4; purple, 2.5 × 10−4). (d) Calibration plot showing the relationship between the evaporation coefficient and the rate of change of droplet radius with time (dr/dt). | ||
Fig. 5 shows how the evaporation coefficient for mixed component monolayer films containing different molar ratios of tetradecanol and hexadecanol, and tridecanol and pentadecanol varies following film formation as the droplet continues to evaporate. Each trace corresponds to an individual droplet evaporation event. The evaporation coefficients corresponding to homogeneous films of the parent fatty alcohols are also shown for comparison, including the effect of uncertainties in RH. Calculations of the average molecular footprint at the point of film formation indicate that both fatty alcohol species in the mixed component droplets must be present in their entirety at the droplet surface at the point of arrested evaporation in order for a condensed monolayer film to form. Fig. 5 clearly shows that the evaporation coefficient of the multi-component monolayer film at the point of film formation depends on the molar ratio of the fatty alcohol components it contains, with the γ value weighted towards the most abundant surface species. As the droplet continues to evaporate, a steady decrease in the value of the evaporation coefficient with time is observed until at long time the evaporation coefficient of the multi-component film is identical to that of a homogeneous film containing the longest chain fatty alcohol.
 | ||
| Fig. 5 (a) Evaporation coefficient as a function of time since film formation for multi-component monolayer films containing different molar ratios of tetradecanol:hexadecanol; (from top to bottom) 3:1 (blue), 1:1 (red), 1:3 (black). Evaporation coefficients for homogeneous monolayer films of the parent compounds are also shown; tetradecanol (light grey shaded upper box) and hexadecanol (dark grey shaded lower box). (b) Evaporation coefficient as a function of time since film formation for multi-component monolayer films containing different molar ratios of tridecanol:pentadecanol; (from top to bottom) 4:1 (blue), 3:2 (red), 2:3 (black), 1:4 (purple). Evaporation coefficients for homogeneous monolayer films of the parent compounds are also shown; tridecanol (light grey shaded upper box) and pentadecanol (dark grey shaded lower box). | ||
These observations can be rationalised as follows. At the point of film formation, both fatty alcohol species in the droplet are present in their entirety at the droplet surface, giving a γ value for the multi-component film that is equal to the mole fraction weighted average of the γ values corresponding to homogeneous films of the two different fatty alcohols. As the droplet continues to evaporate and competition for space at the droplet surface increases, the fatty alcohol species with the shortest alkyl chain (associated with the largest evaporation coefficient) is preferentially expelled from the surface region into the bulk. This preferential expulsion of the shorter alkyl chain alcohol may be driven either by its higher solubility in the droplet bulk, or the fact that it will experience a lower adhesion within the monolayer film than the longer chain alcohol due to the reduced number of van der Waals interactions which its shorter alkyl chain can form. The expulsion of the shortest alkyl chain alcohol continues as the droplet evaporates further until only the longest alkyl chain fatty alcohol remains at the droplet surface. Thus, at long timescales a multi-component monolayer film becomes indistinguishable from a homogeneous film of the longest chain fatty alcohol present in the droplet (hexadecanol in Fig. 5(a), pentadecanol in Fig. 5(b)).
This hypothesis is supported by comparing the experimentally determined evaporation coefficient at the point of initial film formation with a predicted value (γtheo) based on the mole fraction of each fatty alcohol species known to be in the droplet (xA and xB) and the evaporation coefficients measured for homogeneous films containing those alcohols (γA and γB):
| γtheo = xAγA + xBγB | (4) |
:1 line, with a slight tendency to underestimate the experimental evaporation coefficient upon initial film formation. This is possibly due to the difficulty in accurately fitting the first few seconds of film formation (where the rate of change in droplet radius is most rapid) with a higher order polynomial which also encompasses the rest of the radial data.
 | ||
| Fig. 6 Correlation plot showing the agreement between the measured evaporation coefficient for multi-component monolayer films at the point of film formation and the theoretically calculated value, based on the molar ratio weighted average of the pure component film γ values. A 1:1 line (dashed red) is shown for comparison. | ||
III.iii. Combining surface and bulk phase influences on water evaporation: monolayer coated viscous aerosol
As discussed in the introduction, mass transport to and from a droplet may be limited either by the rate of gas phase diffusion, interfacial transfer or bulk phase diffusion. In the previous two sections we have considered the impact of slow interfacial transport; in this section we consider mass transport from a monolayer-coated viscous droplet, which can experience both reduced interfacial transport and bulk phase diffusion.Several studies have reported that secondary organic aerosol (SOA) can exist as ultra-viscous droplets, leading to bulk phase diffusion limitations in mass transport.46–49 As secondary organic aerosol contain a wide range of chemical species, it is highly likely that some of these will be surface active, leading to the possibility that SOA droplets could also contain a surface film which may inhibit mass transport. Here we study the water mass transport from a simplified surrogate system consisting of aqueous sucrose aerosol doped with different surface active fatty alcohols.
The evaporation rates of droplets formed from four aqueous solutions (50/50 v/v water/ethanol) were measured, one containing 60 g l−1 sucrose and three containing 60 g l−1 sucrose doped with ∼0.6 mM concentrations of either tridecanol, tetradecanol or pentadecanol. Droplets from each solution were injected in to the C-EDB trap at 293.15 K and ∼30% RH, conditions under which significant bulk phase diffusion limited mass transport would be expected for aqueous sucrose droplets.3 The change in droplet radius with time observed for droplets from each solution is shown in Fig. 7(a).
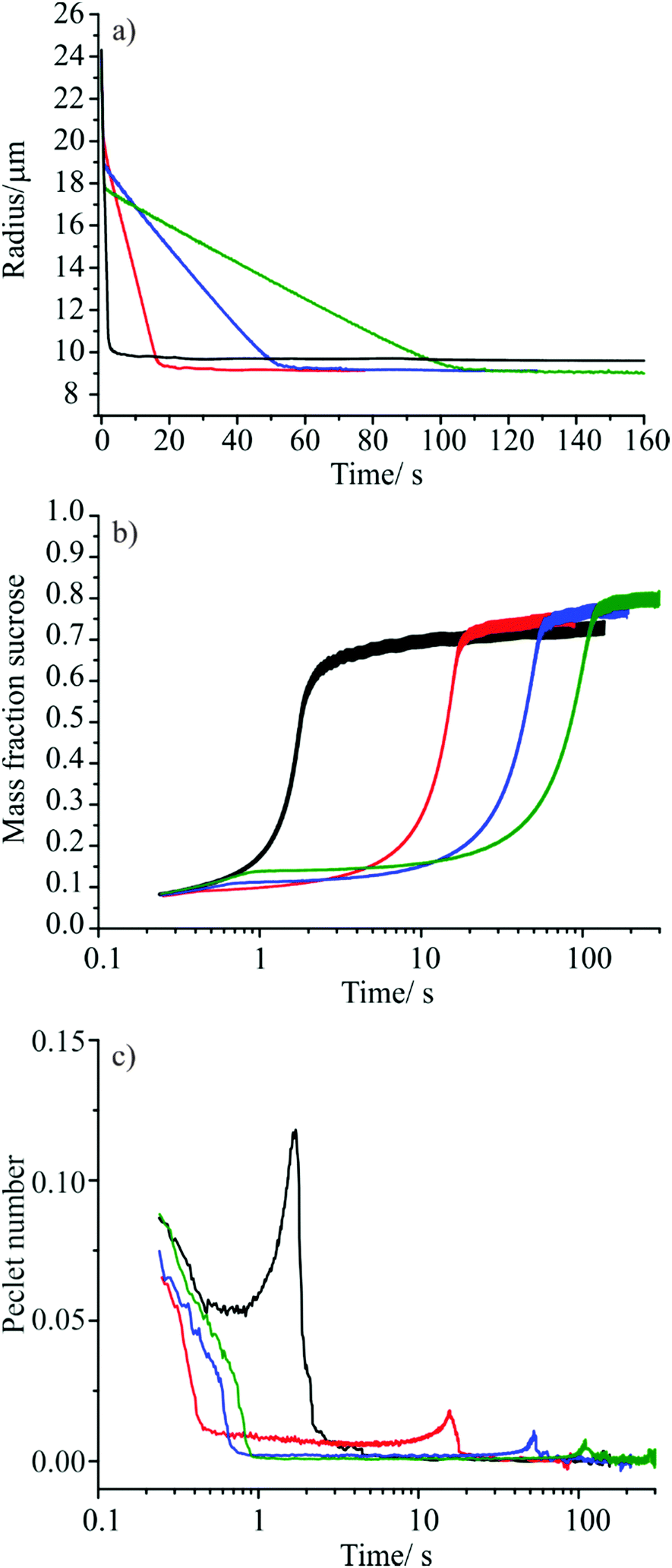 | ||
| Fig. 7 (a) Radius vs. time profiles for sucrose containing droplets evaporating in to 30% RH at 293.15 K; aqueous sucrose (black), aqueous sucrose doped with 0.6 mM tridecanol (red), aqueous sucrose doped with 0.6 mM tetradecanol (blue), aqueous sucrose doped with 0.6 mM pentadecanol (green). (b) Variation in bulk averaged mass fraction of sucrose in the droplet with time (colour key as in (a)). Line width indicates the level of agreement between 10 droplets of each composition. (c) Variation in droplet Péclet number with time for each of the aqueous sucrose systems under study (colour key as in (a)). | ||
In the uncoated aqueous sucrose droplets, rapid loss of water is observed until the droplet has decreased in radius to ∼10 μm, at which point an abrupt arrest in the evaporation rate is observed, corresponding to the beginning of a regime in which water mass transport is limited by slow diffusion within the bulk of the droplet. In this regime, the droplet continues to lose water very slowly over a period of several thousand seconds. The behaviour of the aqueous sucrose droplets doped with the fatty alcohols is different, exhibiting three distinct mass transport regimes. Droplets show an initial rapid loss of water controlled by the rate of gas phase diffusion, followed by slowing of the evaporation rate at t ∼ 0.7 s when the fatty alcohol forms a monolayer film around the droplet surface. At this point, water mass transport is controlled by interfacial transfer as reported previously in Sections III.i and III.ii. When the droplets reach a radius of around 9 μm, a further change in the evaporative flux is observed corresponding to the onset of bulk diffusion limitations on mass transport within the viscous sucrose bulk. As expected, the droplets containing tridecanol, tetradecanol and pentadecanol show different evaporation rates during the period where mass transport is controlled by interfacial transport due to their different γ values.
It is difficult to compare the behaviour of the four different droplet systems directly by examining the radius versus time data as small changes in the initial size of the droplets generated by the droplet-on-demand generator for each solution can mask fine changes in behaviour. In order to compare the droplets directly, the bulk averaged mass fraction of sucrose in each of the partially dry particles was estimated as a function of time (Fig. 7(b)) using the initial mass fraction of solute in the droplet and an aqueous sucrose density parameterisation based on a third order polynomial fit to bulk MFS data.50 This provides a way of comparing the total amount of water that has been removed from each of the particles by any given time point. It is important to note that the bulk-averaged mass fraction of sucrose reveals no information regarding concentration gradients that exist within the particle; previous studies have shown that slow bulk phase diffusion during evaporation leads to the formation of a solute rich shell and a water rich core.3,6 As such, the bulk-averaged mass fraction of sucrose at the point at which the particle begins to display bulk phase diffusion limited behaviour is not expected to match the sucrose mass fraction at 30% RH predicted by thermodynamic models.
Fig. 7(b) suggests that the presence of a monolayer surface film on an evaporating aqueous sucrose droplet increases the amount of water that can be removed from the droplet before bulk phase diffusion limitations begin to have an effect. As shown by the trend in the data for the tridecanol, tetradecanol and pentadecanol monolayers, the smaller the evaporation coefficient of the surface film, the greater the amount of water removed before mass transport begins to be inhibited by bulk phase effects. This observation can be rationalised by considering the variation in Péclet number, Pe, with time. The Péclet number is a dimensionless parameter that quantifies the ratio between the rate of mass transport by evaporation and the rate of diffusion within the bulk. More significant surface enrichments of solutes and larger concentration gradients occur when evaporation occurs at larger Péclet number. When considering the mass flux from an evaporating droplet, the Péclet number for mass transport is defined as:51
 | (5) |
The time-dependence of the Péclet numbers estimated for the evaporating droplets with and without a monolayer film are shown in Fig. 7(c). High Péclet numbers indicate that a surface boundary is receding rapidly, leading to significant enrichments in involatile solutes (in this case sucrose) at the droplet surface, a consequence of the inability of diffusional mass transport to maintain uniformity in composition. Fig. 7(c) shows that the much faster rate of the evaporative mass flux for the binary aqueous sucrose droplets (gas diffusion limited) compared with that of the monolayer coated droplets (surface exchange limited) leads to a higher peak in the Péclet number and occurs at earlier time. This can be interpreted as the uncoated aqueous sucrose particles displaying a greater non-uniformity in droplet composition with greater surface enrichment of sucrose than their monolayer coated counterparts, leading to a greater degree of water retention with the particle bulk once bulk diffusion through a viscous sucrose surface becomes the rate limiting step. While the difference in Péclet number between the uncoated and monolayer coated aqueous sucrose particles is most pronounced, a clear decrease in the Pe value and a shift in peak position to longer times is also observable as the chain length of the monolayer fatty alcohol is increased. This suggests that a more uniform droplet composition results from the slower mass transport prior to bulk phase arrest, allowing the removal of more water from the particle.
Fig. 8 shows a schematic that compares the limiting processes that determine the mass flux of water from an evaporating droplet based on our previous measurements of evaporative flux from droplets in the typical size range of these measurements (8–12 μm radius). In decreasing order of the resulting total mass flux (I) from the droplet these are: gas-diffusion limited evaporation of water, Ig (∼10−12 kg s−1);34 surface limited evaporation through formation of a monolayer film, Is (∼10−14 kg s−1);2 and bulk limited evaporation by slow diffusion of water from within the particle to the particle surface, Ib (∼10−16 kg s−1).32 For the droplet containing only aqueous sucrose, the rate of the initial loss of water is controlled by the mass flux in the gas phase as water transport within the droplet bulk and across the interface is rapid. Thus, the mass flux is determined by the value of Ig, i.e. gas phase diffusion is initially the limiting rate determining mass transport. As more water is removed from the particle and its mass fraction of sucrose increases, the diffusion coefficient of water in the droplet bulk decreases by many orders of magnitude. Then, the mass flux of water from the particle is determined by the value of Ib and the slow release of water from within the droplet core. Without the presence of a monolayer film, transport across the surface is facile and the evaporation rate is either determined by the gas diffusional transport or the slow release of water from within the droplet bulk, depending on the relative rates of the two processes.
 | ||
| Fig. 8 Schematic showing the three different mass transport processes which can be rate limiting in aerosol, along with typical values of the resulting total evaporative mass flux, I; (clockwise top right) gas phase diffusion (Ig), interfacial transport (Is), and bulk phase diffusion (Ib). | ||
For the monolayer coated sucrose droplets, the relative magnitudes of all three mass fluxes (gas-phase, surface and bulk) need to be considered in order to explain the observed behaviour. At the very beginning of the droplet evaporation, the evaporative flux is again controlled by the rate of gas phase diffusion. Once the monolayer is formed around the surface of the droplet, water loss is characteristic of that expected for evaporation limited by mass transport through the surface film, Is. It is this point at which the evaporative mass flux becomes dominated by the rate of surface exchange. For each of the fatty alcohol doped droplets, the transition from a surface inhibited mass flux to a bulk phase diffusion limited mass flux occurs when the near surface concentration of water in the droplet becomes significantly lower than that remaining within the particle core due to the reduced rate of bulk phase diffusion. The slower the rate of loss of water from a particle surface (for example due to a pentadecanol monolayer film as opposed to a tridecanol monolayer film), the slower the diffusional transport of water from the core of the particle to the surface can be whilst still maintaining the evaporative flux limited by the transport across the surface. This allows particles coated in longer chain alcohols to lose water for longer before the bulk content of water (and, thus, the bulk diffusion constant of water) has diminished to such an extent that the bulk can no longer supply water to the surface to even sustain the low flux demanded by the alcohol film.
IV. Conclusions
Quantifying the mass transport of water, and volatile components more generally, through organic monolayer films on droplet surfaces and from amorphous particles is of importance to fields as diverse as atmospheric chemistry, spray drying and drug delivery to the lungs. We have considered here the competition between mass transport limited by interfacial transport and bulk phase diffusion. For droplets coated in a monolayer condensed film of a single long chain alcohol, the evaporation coefficient has been shown to depend on the relative humidity of the gas phase. This is a consequence of the effect that the magnitude of the diffusional gradient in the gas phase has on the mass and heat flux from an evaporating droplet, with evaporation in to lower RHs leading to a larger suppression in the droplet surface temperature. This effects the coherence of the monolayer film, influencing the packing density and thereby impacting on the rate of interfacial transport. Lower surface temperatures lead to higher packing density and lower evaporation coefficients. The evaporation coefficient through monolayers of mixed composition is shown to depend on the mole fractions of the different organic components forming the monolayer, consistent with previous work.16,29–31 As an evaporating droplet diminishes in surface area, the more soluble, shorter alkyl chain component is found to preferentially partition into the bulk of the droplet, leading to a temporal dependence in evaporation coefficient with the value tending towards that observed through a monolayer containing only the least soluble, longer alkyl chain component.The drying kinetics of binary sucrose–water droplets show the onset of suppressed water loss within 2 s for the droplet sizes considered here. After this time, the evaporation rate of water is governed by slow diffusional transport of water from the particle core through a surface region strongly enriched in sucrose and characterised by considerably diminished diffusion constants.5,6 When a long chain alcohol is added to the droplet, the surface propensity of the alcohol leads to the formation of a surface film that slows the rate of water loss through a reduction in the evaporation coefficient. This leads to the droplet composition remaining more homogeneous through diffusional mixing for a longer period of time, even in excess of 100 s. By the time the sucrose concentration has reached a sufficiently high value, and the diffusion constant of water has decreased accordingly, a larger fraction of water has been removed from the monolayer coated droplet than in the binary aqueous sucrose droplet case. A greater degree of slowing in the evaporative flux achieved by increasing the chain length of the alcohol leads to a greater degree of dehydration.
Through detailed measurements of the evaporation rate of water from droplets of increasingly complex chemical composition, it is possible to resolve the factors that govern droplet drying rates and the potential morphology of the particles that are formed. In future studies, we will focus on expanding the operating range of droplet and gas phase temperature that can be accessed in such studies, the capability to probe evolving size and composition at much shorter evaporation times, and the additional complexity that will be apparent when studying drying droplets containing dispersions of insoluble inclusions.
Acknowledgements
This work was funded by the NERC through grant NE/I020075/1; Aerosol-Cloud Interactions – A Directed Program to Reduce Uncertainty in Forcing through a Targeted Laboratory and Modelling Programme (ACID-PRUF). Miss Hannah Macdonald is acknowledged for her early contributions towards this study. The experimental data presented in this paper are provided through the University of Bristol data repository at Reid, J. P. (2016): Water Evaporation Coefficient, University of Bristol, DOI: http://10.5523/bris.11ek298yyej9v1j4zxekv002gy.References
- R. Tuckermann, S. Bauerecker and H. K. Cammenga, Colloids Surf., A, 2007, 309, 198–201 CrossRef CAS.
- J. F. Davies, R. E. H. Miles, A. E. Haddrell and J. P. Reid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8807–8812 CrossRef CAS PubMed.
- B. Zobrist, V. Soonsin, B. P. Luo, U. K. Krieger, C. Marcolli, T. Peter and T. Koop, Phys. Chem. Chem. Phys., 2011, 13, 3514–3526 RSC.
- F. H. Marshall, R. E. H. Miles, Y.-C. Song, P. B. Ohm, R. M. Power, J. P. Reid and C. S. Dutcher, Chem. Sci., 2016, 7, 1298–1308 RSC.
- H. J. Tong, J. P. Reid, D. L. Bones, B. P. Luo and U. K. Krieger, Atmos. Chem. Phys., 2011, 11, 4739–4754 CrossRef CAS.
- D. L. Bones, J. P. Reid, D. M. Lienhard and U. K. Krieger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11613–11618 CrossRef CAS PubMed.
- R. M. Power, S. H. Simpson, J. P. Reid and a. J. Hudson, Chem. Sci., 2013, 4, 2597–2604 RSC.
- C. E. Kolb, R. A. Cox, J. P. D. Abbatt, M. Ammann, E. J. Davis, D. J. Donaldson, B. C. Garrett, C. George, P. T. Griffiths, D. R. Hanson, M. Kulmala, G. McFiggans, U. Pöschl, I. Riipinen, M. J. Rossi, Y. Rudich, P. E. Wagner, P. M. Winkler, D. R. Worsnop and C. D. O'Dowd, Atmos. Chem. Phys., 2010, 10, 10561–10605 CrossRef CAS.
- J. F. Davies, R. E. H. Miles, A. E. Haddrell and J. P. Reid, J. Geophys. Res.: Atmos., 2014, 119, 10931–10940 Search PubMed.
- R. E. H. Miles, J. P. Reid and I. Riipinen, J. Phys. Chem. A, 2012, 116, 10810–10825 CrossRef CAS PubMed.
- J. Julin, M. Shiraiwa, R. E. H. Miles, J. P. Reid, U. Pöschl and I. Riipinen, J. Phys. Chem. A, 2013, 117, 410–420 CrossRef CAS PubMed.
- A. Morita, M. Sugiyama, H. Kameda, S. Koda and D. R. Hanson, J. Phys. Chem. B, 2004, 108, 9111–9120 CrossRef CAS.
- J. Vieceli, M. Roeselová, N. Potter, L. X. Dang, B. C. Garrett and D. J. Tobias, J. Phys. Chem. B, 2005, 109, 15876–15892 CrossRef CAS PubMed.
- S. Takahama and L. M. Russell, J. Geophys. Res., 2011, 116, D02203 CrossRef.
- S. Neshyba, E. Nugent, M. Roeselová and P. Jungwirth, J. Phys. Chem. C, 2009, 113, 4597–4604 CAS.
- L. M. Cosman and A. K. Bertram, J. Phys. Chem. A, 2008, 112, 4625–4635 CrossRef CAS PubMed.
- L. M. Cosman, D. A. Knopf and A. K. Bertram, J. Phys. Chem. A, 2008, 112, 2386–2396 CrossRef CAS PubMed.
- M. Hallquist, D. J. Stewart, J. Baker and R. A. Cox, J. Phys. Chem. A, 2000, 104, 3984–3990 CrossRef CAS.
- D. J. Henry, V. I. Dewan, E. L. Prime, G. G. Qiao, D. H. Solomon and I. Yarovsky, J. Phys. Chem. B, 2010, 114, 3869–3878 CrossRef CAS PubMed.
- X. Ma, P. Chakraborty, B. J. Henz and M. R. Zachariah, Phys. Chem. Chem. Phys., 2011, 13, 9374–9384 RSC.
- S. Sakaguchi and A. Morita, J. Chem. Phys., 2012, 137, 064701 CrossRef PubMed.
- W. Lin, A. J. Clark and F. Paesani, Langmuir, 2015, 31, 2147–2156 CrossRef CAS PubMed.
- A. Habartová, M. Roeselová and L. Cwiklik, Langmuir, 2015, 31, 11508–11515 CrossRef PubMed.
- H. Lee, S. K. Kandasamy and R. G. Larson, Biophys. J., 2005, 89, 3807–3821 CrossRef CAS PubMed.
- J. B. Gilman, T. L. Eliason, A. Fast and V. Vaida, J. Colloid Interface Sci., 2004, 280, 234–243 CrossRef CAS PubMed.
- E. C. Griffith, E. M. Adams, H. C. Allen and V. Vaida, J. Phys. Chem. B, 2012, 116, 7849–7857 CrossRef CAS PubMed.
- E. C. Griffith, R. J. Perkins, D. M. Telesford, E. M. Adams, L. Cwiklik, H. C. Allen, M. Roeselová and V. Vaida, J. Phys. Chem. B, 2015, 119, 9038–9048 CrossRef CAS PubMed.
- E. C. Griffith, T. R. C. Guizado, A. S. Pimentel, G. S. Tyndall and V. Vaida, J. Phys. Chem. C, 2013, 117, 22341–22350 CAS.
- L. F. Voss, C. M. Hadad and H. C. Allen, J. Phys. Chem. B, 2006, 110, 19487–19490 CrossRef CAS PubMed.
- J. B. Gilman and V. Vaida, J. Phys. Chem. A, 2006, 110, 7581–7587 CrossRef CAS PubMed.
- D. K. Burden, A. M. Johnson and G. M. Nathanson, J. Phys. Chem. A, 2009, 113, 14131–14140 CrossRef CAS PubMed.
- J. F. Davies, A. E. Haddrell, R. E. H. Miles, C. R. Bull and J. P. Reid, J. Phys. Chem. A, 2012, 116, 10987–10998 CrossRef CAS PubMed.
- J. F. Davies, A. E. Haddrell and J. P. Reid, Aerosol Sci. Technol., 2012, 46, 666–677 CrossRef CAS.
- J. F. Davies, A. E. Haddrell, A. M. Rickards and J. P. Reid, Anal. Chem., 2013, 85, 5819–5826 CrossRef CAS PubMed.
- S. Corsetti, R. E. H. Miles, C. McDonald, Y. Belotti, J. P. Reid, J. Kiefer and D. McGloin, J. Phys. Chem. A, 2015, 119, 12797–12804 CrossRef CAS PubMed.
- W. J. Glantschnig and S. H. Chen, Appl. Opt., 1981, 20, 2499–2509 CrossRef CAS PubMed.
- P. Schiebener, J. Straub, J. M. H. Levelt Sengers and J. S. Gallagher, J. Phys. Chem. Ref. Data, 1990, 19, 677–717 CrossRef CAS.
- M. Kulmala, T. Vesala and P. E. Wagner, Proc. R. Soc. London, Ser. A, 1993, 441, 589–605 CrossRef CAS.
- N. A. Fuchs and G. A. Sutugin, Topics in Current Aerosol Research, Pergamon, 1971 Search PubMed.
- Y. Q. Li, P. Davidovits, C. E. Kolb and D. R. Worsnop, J. Phys. Chem. A, 2001, 105, 10627–10634 CrossRef CAS.
- P. Winkler, A. Vrtala, P. Wagner, M. Kulmala, K. Lehtinen and T. Vesala, Phys. Rev. Lett., 2004, 93, 075701 CrossRef PubMed.
- P. M. Winkler, A. Vrtala, R. Rudolf, P. E. Wagner, I. Riipinen, T. Vesala, K. E. J. Lehtinen, Y. Viisanen and M. Kulmala, J. Geophys. Res., 2006, 111, D19202 CrossRef.
- S. L. Clegg, P. Brimblecombe and A. S. Wexler, J. Phys. Chem. A, 1998, 102, 2155–2171 CrossRef CAS.
- Extended AIM Aerosol Thermodynamics Model (E-AIM), http://www.aim.env.uea.ac.uk/aim/aim.php.
- J. M. Langridge, M. S. Richardson, D. a. Lack, C. a. Brock and D. M. Murphy, Aerosol Sci. Technol., 2013, 47, 1163–1173 CrossRef CAS.
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirila, J. Leskinen, J. M. Makela, J. K. Holopainen, U. Poschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, Nature, 2010, 467, 824–827 CrossRef CAS PubMed.
- L. Renbaum-Wolff, J. W. Grayson, A. P. Bateman, M. Kuwata, M. Sellier, B. J. Murray, J. E. Shilling, S. T. Martin and A. K. Bertram, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8014–8019 CrossRef CAS PubMed.
- E. Saukko, A. T. Lambe, P. Massoli, T. Koop, J. P. Wright, D. R. Croasdale, D. A. Pedernera, T. B. Onasch, A. Laaksonen, P. Davidovits, D. R. Worsnop and A. Virtanen, Atmos. Chem. Phys., 2012, 12, 7517–7529 CrossRef CAS.
- C. Kidd, V. Perraud, L. M. Wingen and B. J. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7552–7557 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, 89th edn, 2008 Search PubMed.
- M. A. Boraey and R. Vehring, J. Aerosol Sci., 2014, 67, 131–143 CrossRef CAS.
- J. F. Davies and K. R. Wilson, Anal. Chem., 2016, 88, 2361–2366 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2016 |