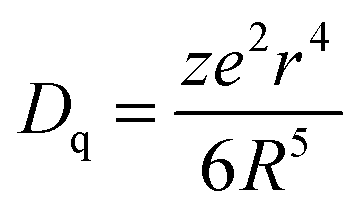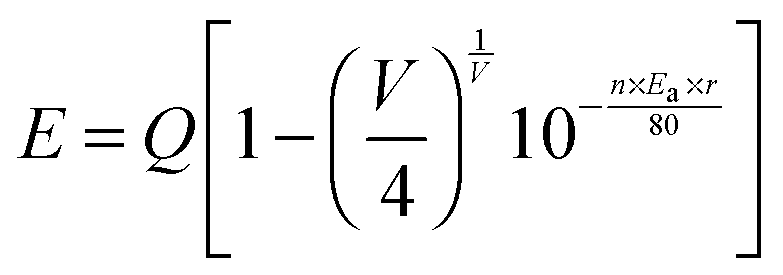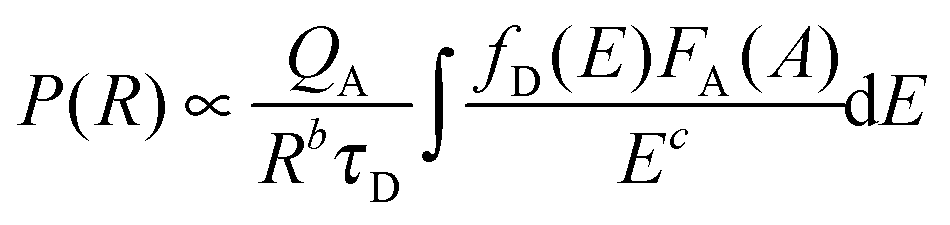A novel apatite, Lu5(SiO4)3N:(Ce,Tb), phosphor material: synthesis, structure and applications for NUV-LEDs†
Received
4th March 2016
, Accepted 24th April 2016
First published on 26th April 2016
Abstract
The lutetium containing nitride apatite Lu5(SiO4)3N was prepared by a solid state reaction at high temperature for the first time. Rietveld refinement indicated that the Lu5(SiO4)3N compound has a hexagonal space group of P63/m with cell parameters a = b = 9.700 Å and c = 7.238 Å. Additionally, the results revealed that there are two distinct lutetium sites in the Lu5(SiO4)3N host lattice, i.e. a Lu(1) site with nine coordination (Wyckoff site 4f) and a Lu(2) site with seven coordination (Wyckoff site 6h). Furthermore, the ratio of the number of Lu atoms in Lu(1) and Lu(2) sites is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The band gap for Lu5(SiO4)3N was determined to be 4.12 eV based on the density functional theory (DFT). In the Ce3+ doped Lu5(SiO4)3N:0.03Ce3+ compound, the emission peak centered at 462 nm was observed with the Commission International de I'Eclairage (CIE) coordinates of (0.148, 0.184), indicating blue-emission. Remarkably, in Ce3+ and Tb3+ co-doped Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ compounds, the color-tunability was observed with increasing Tb3+ co-doping rate on moving from blue at Tb3+ = 0.00 to green at Tb = 0.09, due to the energy transfer from Ce3+ to Tb3+ ions being matched well with the decay curve results. Under the excitation at 359 nm, the absolute quantum efficiency (QE) for Lu5(SiO4)3N:0.03Ce3+ was determined to be 42.13%. This phosphor material could be a platform for modeling a new phosphor and application in the solid-state lighting field.
:2. The band gap for Lu5(SiO4)3N was determined to be 4.12 eV based on the density functional theory (DFT). In the Ce3+ doped Lu5(SiO4)3N:0.03Ce3+ compound, the emission peak centered at 462 nm was observed with the Commission International de I'Eclairage (CIE) coordinates of (0.148, 0.184), indicating blue-emission. Remarkably, in Ce3+ and Tb3+ co-doped Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ compounds, the color-tunability was observed with increasing Tb3+ co-doping rate on moving from blue at Tb3+ = 0.00 to green at Tb = 0.09, due to the energy transfer from Ce3+ to Tb3+ ions being matched well with the decay curve results. Under the excitation at 359 nm, the absolute quantum efficiency (QE) for Lu5(SiO4)3N:0.03Ce3+ was determined to be 42.13%. This phosphor material could be a platform for modeling a new phosphor and application in the solid-state lighting field.
1. Introduction
Apatite compounds have been investigated as effective luminescent host materials in recent years because of their physical and chemical stability. In the past decades, they have been widely used in lighting and display areas.1–4 Apatite belongs to the hexagonal crystal system represented generally as M10(XO4)6Z2 (M = Ca2+, Sr2+, Ba2+, K+, Na+, Ce3+, La3+, Y3+, etc., X = P5+, Si4+, S6+, V5+, etc., and Z = O2−, F−, Cl−, Br−, OH−, S2−, etc.).5,6 In general, there are two kinds of non-equivalent crystallographic sites in apatite compounds, that is, a nine-coordinated 4f site M(I) with C3 point symmetry and a seven-coordinated 6h site M(II) with CS point symmetry, respectively.7,8 Thus, it is interesting and significant to investigate the relationship between the structure (substitution) and the luminescence properties of an apatite structure. Accordingly, many efforts have been made to obtain novel phosphors on the basis of different chemical compositions, such as Sr10(PO4)6O:Eu2+,9 Sr3GdNa(PO4)3F:Eu2+,Mn2+,10 and Ca9Mg(PO4)6F2:Eu2+,Mn2+.11
Meanwhile, the wavelength of excitation and emission bands for phosphors with an oxonitridosilicate structure is even longer than that of other phosphors because of their high covalency and strong crystal fields. (Si[O/N]4) units are stacked together by sharing their corners and edges forming condensed (Si[O/N]4) frameworks, making oxonitridosilicates excellent hosts for rare earth ions. Therefore, oxonitridosilicates have been used as hosts to obtain phosphors, such as: Y10(Si6O22N2)O2:Ce3+,Mn2+,12 Ca3Si2O4N2:Eu2+,13 and Y4Si2O7N2:Eu2+.14 Besides, the luminescence properties of Ce3+ ions can be strongly affected by their surrounding crystal field environment, due to their 5d electrons being unshielded from the crystal field by the 5s and 5p electrons in the excited state. Ce3+ doped oxonitridosilicates have been widely investigated, such as Ca3Si2O4N2:Ce3+,15 Y5(SiO4)3N:Ce3+,16 Sr3Si2O4N2:Ce3+,17 and Gd5Si3O12N:Ce3+.18 Among oxonitridosilicates, Lu–Si–O–N quaternary system compounds have been investigated in the previous literature, such as Lu4Si2O7N2.19,20 However, to the best of our knowledge, the Lu5(SiO4)3N compound (oxonitridosilicate) has not been identified and reported before.
In light of the above, a novel oxonitridosilicate Lu5(SiO4)3N with an apatite structure was obtained for the first time under 0.5 MPa atmospheric pressure. The crystal structure properties of Lu5(SiO4)3N were studied in detail. Rietveld refinement indicated that the Lu5(SiO4)3N compound has a hexagonal space group of P63/m with cell parameters a = b = 9.700 Å and c = 7.238 Å. Two distinct lutetium sites in the Lu5(SiO4)3N host lattice, i.e. a Lu(1) site with nine coordination (Wyckoff site 4f) and a Lu(2) site with seven coordination (Wyckoff site 6h), were revealed, and the ratio of the number of Lu atoms in Lu(1) and Lu(2) sites is 3:2. The band gap for Lu5(SiO4)3N was determined to be 4.12 eV based on the density functional theory (DFT). In addition, the luminescence properties and energy transfer behaviors between Ce3+ and Tb3+ were investigated systematically. The results indicated that the Lu5(SiO4)3N phosphor material could be a platform for modeling a new phosphor and application in the solid-state lighting field.
2. Experimental procedure
2.1 Materials and synthesis
Lu5(SiO4)3N hosts and Lu5(SiO4)3N:Ce3+,Tb3+ phosphors were prepared by a high temperature solid-state reaction. SiO2 (Aldrich, 99.9%), α-Si3N4 (Aldrich, 99.9%), Lu2O3 (Aldrich, 99.995%), CeO2 (Aldrich, 99.995%), and Tb4O7 (Aldrich, 99.995%) were weighed and used as starting materials according to the given composition of Lu5(SiO4)3N:Ce3+,Tb3+. The raw materials were fully mixed and ground in an agate mortar, and the mixture was placed into an alumina crucible and was heated at 1500 °C for 5 h in a reducing atmosphere with flowing gas (10% H2 + 90% N2) and 0.5 MPa atmospheric pressure. Eventually, the obtained samples were cooled to room temperature and ground again into powder for subsequent measurements.
2.2 Characterization
Phase structures of the as-prepared samples were checked using an X-ray powder diffractometer (D/max-rA 12kw, Japan) with Cu Kα radiation (λ = 1.5418 Å) from 10° to 70° (2θ). The XRD data for the Rietveld analysis were collected in a step scanning mode with a step size of 0.02° and 3 s counting time per step from 10° to 75° (2θ). Rietveld refinement was performed using the computer software: general structure analysis system (GSAS).21 Computations were performed within the density functional theory (DFT) framework, using the Vienna ab initio software package (VASP), within the generalized gradient approximation by Perdew–Burke–Ernzher (GGA-PBE) and projector augmented wave (PAW) pseudopotentials. A cutoff energy of 520 eV and an appropriate k-point grid were chosen to ensure that total energies converged within 10−4 eV per formula unit. The supercell of Lu5(SiO4)3N used in this study includes 42 atoms. When the structures were optimized, all atoms were allowed to relax. Since the optimized lattices of the systems were determined using several structure parameters, it is time consuming to get the theoretical value of these parameters through the traditional DFT calculation. In this work, the structures were optimized until all residual forces were below 0.001 eV Å−1. Brillouin-zone integrations were performed using Monkhorst–Pack grids of special points with a mesh (2 × 2 × 3).22 The room-temperature excitation and emission spectra were measured on a Hitachi F-4500 spectrophotometer with a 150 W Xe lamp as the excitation lamp. The luminescence decay curves were obtained on a spectro-fluorometer (HORIBA JOBIN YVON FL3-21), and the 370 nm pulse laser radiation (370 nm Nano LED, model number 08254) was used as the excitation source. The particle morphology and microstructure of the Lu5(SiO4)3N and Lu4.97(SiO4)3N:0.03Ce3+ samples were examined by scanning electron microscopy (SEM) using a HitachiS-520 instrument with an accelerating voltage of 25 kV. The results of elemental analysis were gotten from an energy dispersive X-ray spectrometer (EDS) attached to the scanning electron microscope. The quantum efficiency (QE) was obtained using an absolute photoluminescence quantum yield measurement system (FluoroLog-3-2-iHR320, Horiba Jobin Yvon) with a F-3018 integral sphere at room temperature.
3. Results and discussion
3.1 Insight into Lu5(SiO4)3N
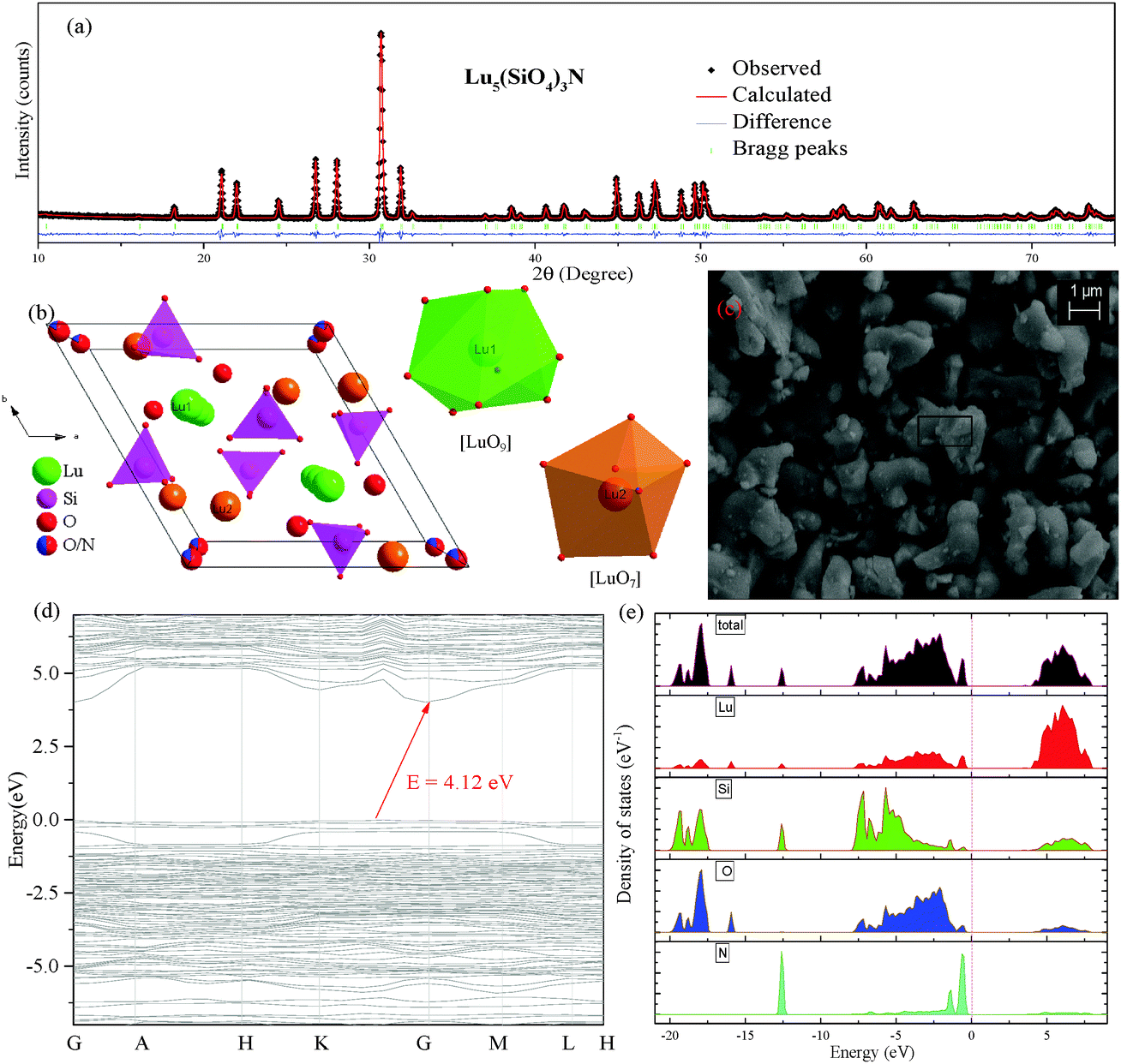
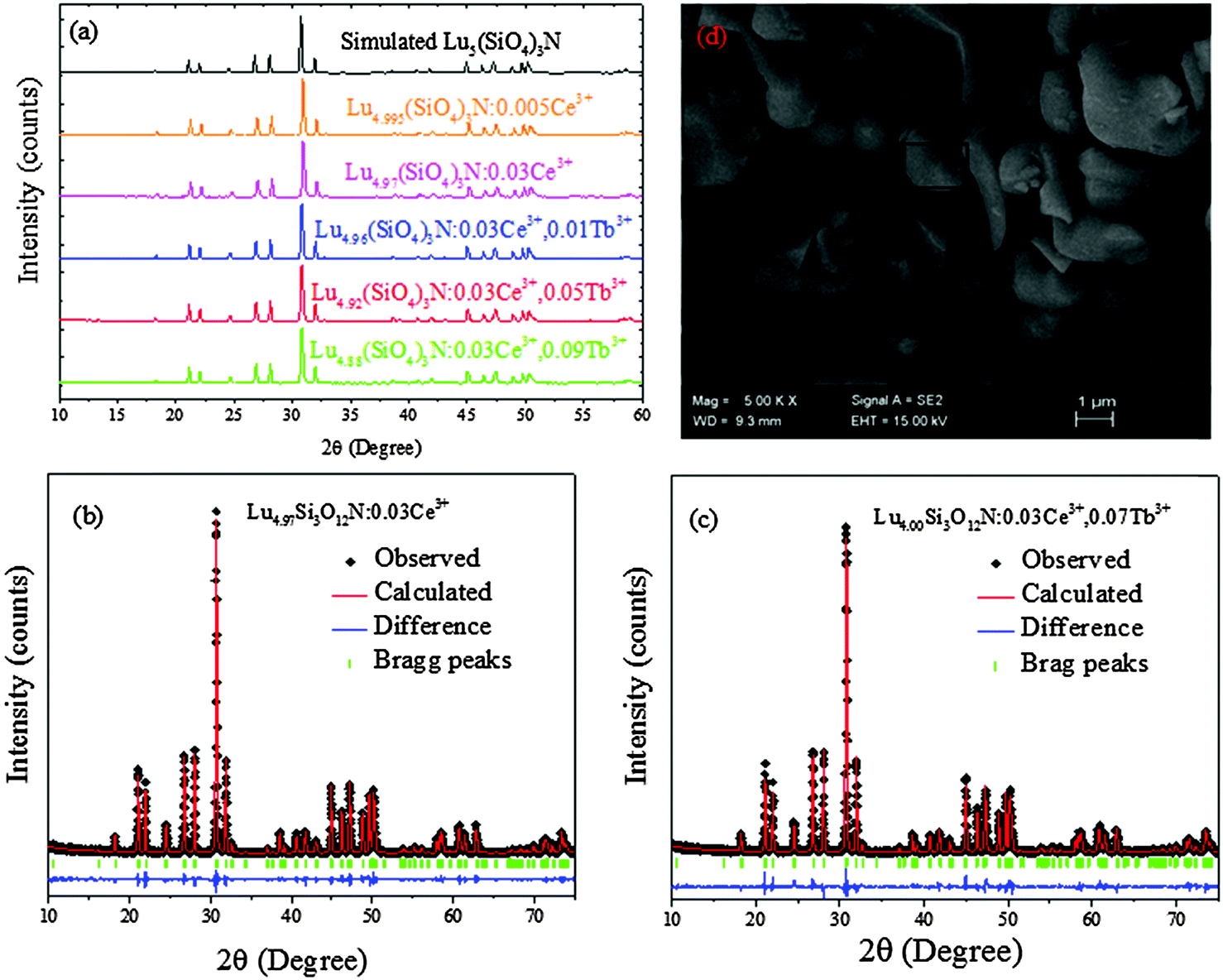
Fig. 1(a) shows the Rietveld structural refinement of the XRD pattern of the Lu5(SiO4)3N host using the GSAS program. As depicted in Fig. 1(a), the red solid line, black square points, green short vertical and blue lines represent the calculated pattern, the observed pattern from experiment, the Bragg positions of the calculated pattern, and the difference between the observed and calculated patterns, respectively. It is obvious that almost all the peaks of Lu5(SiO4)3N can be indexed by the hexagonal cell (P63/m). In addition, Lu5(SiO4)3N has similar XRD patterns to La5(SiO4)3N (apatite structure, ICSD no. 91850), which have been widely investigated.23–25 Therefore, La5(SiO4)3N was taken as a starting model structure for Rietveld refinement. The refinement was stable and gives low R-factors (Fig. 1(a) and Table 1), and the final refinement residual factors are Rwp = 8.88%, Rp = 5.99%, and χ2 = 1.63. Besides, the refinement results further confirm that the single-phase compound Lu5(SiO4)3N can be indexed in the hexagonal space group of P63/m with cell parameters a = b = 9.7005 Å, c = 7.2383 Å and V = 589.87 Å3. Furthermore, the fractional atomic coordinates, isotropic displacement parameters (Å2), as well as the main bond lengths (Å) for Lu5(SiO4)3N are illustrated in Tables 2 and 3. According to Table 2, O1 is located on site 2a with −6 site symmetry, while Si1, O2, and O3 are located on the mirror planes with site symmetry m (site 6h). O4 and N4 are on a general position (site 12i). Based on the results from Table 3, the average bond length of Lu(1)–O/N can be calculated to be 2.706 Å, and 2.543 Å for Lu(2) sites. Besides, the crystallographic information files (CIFs) of Lu5(SiO4)3N are also illustrated in the ESI.†Fig. 1(b) illustrates the crystal structure of Lu5(SiO4)3N viewed along the c-axis, and the coordination environment of cation sites for activators was also given. In the crystal lattice of this compound, Si atoms are tetrahedrally coordinated to form [SiO4] groups, which are isolated from each other. Lu5(SiO4)3N contains two non-equivalent lutetium sites, where Lu(1) is located on a 3-fold axis (Wyckoff symbol 4f), while Lu(2) is located on a mirror plane (site 6h). Lu(1) is nine coordinated by O/N atoms forming a tricapped trigonal-prismatic geometry, but Lu(2) is seven coordinated by O/N atoms forming an irregular polyhedron, respectively. Furthermore, the ratio of the number of Lu atoms in the two sites is 3:2.
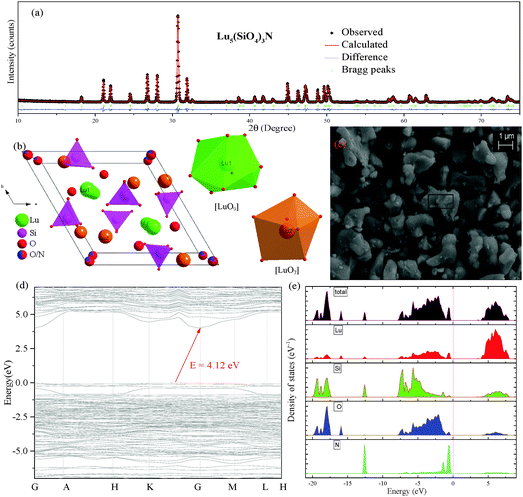 |
| | Fig. 1 Powder XRD patterns for the Rietveld refinement analysis of Lu5(SiO4)3N. Solid red lines are calculated intensities, the black square points are the observed intensities, and blue solid lines stand for the difference between the observed and the calculated intensities, and short green vertical lines below the profiles show the position of Bragg reflections of the calculated pattern (a); crystal structure diagrams of Lu5(SiO4)3N viewed in the c-direction and the coordination environment of cation sites for activators (b); SEM image of Lu5(SiO4)3N (c); calculated band structure of Lu5(SiO4)3N (d); electronic densities of states for Lu5(SiO4)3N, both total (TDOS) and partial (PDOS) onto the atomic orbital (e). | |
Table 1 Refinement parameters for Lu5(SiO4)3N obtained from the Rietveld refinement using X-ray powder diffraction data at room temperature
| Sample |
Lu5(SiO4)3N |
| Space group |
P63/m(176) |
|
a, Å |
9.7005(17) |
|
c, Å |
7.2383(3) |
|
V, Å3 |
589.87(4) |
| 2θ-interval, ° |
10–75 |
|
R
wp, % |
8.88 |
|
R
p, % |
5.99 |
|
χ
2
|
1.63 |
Table 2 Structural parameters for Lu5(SiO4)3N obtained from the Rietveld refinement using X-ray powder diffraction data at room temperature
| |
Wyck |
x
|
y
|
z
|
U (Å2) |
Occupation |
| Lu1 |
4f |
0.2310(8) |
−0.0153(9) |
1/4 |
0.0158 |
1 |
| Lu2 |
6h |
1/3 |
2/3 |
0.0007(4) |
0.0249 |
1 |
| Si1 |
6h |
0.3932(5) |
0.3788(1) |
1/4 |
0.0017 |
1 |
| O1 |
2a |
0 |
0 |
1/4 |
0.0568 |
1 |
| O2 |
6h |
0.3308(8) |
0.4682(7) |
1/4 |
0.0143 |
1 |
| O3 |
6h |
0.5871(9) |
0.4731(6) |
1/4 |
0.0014 |
1 |
| O4 |
12i |
0.3443(2) |
0.2570(5) |
0.0769(2) |
0.0027 |
5/6 |
| N4 |
12i |
0.3443(2) |
0.2570(5) |
0.0769(2) |
0.0027 |
1/6 |
Table 3 Main bond lengths (Å) of Lu5(SiO4)3N from Rietveld structure analysis, and the symmetry codes of the matching O/N element
| Bond |
Lengths (Å) |
Symmetry codes |
Bond |
Lengths (Å) |
Symmetry codes |
| Lu2–O1 |
2.3198(1) |
x, y, z |
Lu1–O3 |
2.6027(1) |
x − y, x, −0.5 + z |
| Lu2–O2 |
2.7223(1) |
−x + y, −x, z |
Lu1–O3 |
2.6027(1) |
1 − x, 1 − y, −0.5 + z |
| Lu2–O3 |
2.4911(1) |
1 − y, x − y, z |
Lu1–O3 |
2.6027(1) |
y, 1 − x + y, −0.5 + z |
| Lu2–O4/N4 |
2.6187(1) |
x, y, z |
Lu1–O4/N4 |
2.8857(1) |
1 − x, 1 − y, −z |
| Lu2–O4/N4 |
2.5149(1) |
y, −x + y, 0.5 + z |
Lu1–O4/N4 |
2.8857(1) |
y, 1 − x + y, −z |
| Lu2–O4/N4 |
2.5149(1) |
y, −x + y, −z |
Lu1–O4/N4 |
2.8857(1) |
x − y, x, −z |
| Lu2–O4/N4 |
2.6187(1) |
x, y, 0.5 − z |
Si1–O2 |
1.2823(1) |
x, y, z |
| Lu1–O2 |
2.6294(1) |
x, y, z |
Si1–O3 |
1.6295(1) |
x, y, z |
| Lu1–O2 |
2.6295(1) |
1 − y, 1 + x − y, z |
Si1–O4/N4 |
1.6215(0) |
x, y, z |
| Lu1–O2 |
2.6294(1) |
−x + y, 1 − x, z |
Si1–O4/N4 |
1.6215(0) |
x, y, 0.5 − z |
The morphology of the Lu5(SiO4)3N host prepared via solid state reaction is shown in Fig. 1(c). The SEM result shows that particles of the as-prepared sample are not uniform, which was caused by the agglomeration during sample sintering. Additionally, the SEM image indicated that the sample has a well-crystallized micropore with the diameter of about 3–6 μm. In order to confirm the chemical composition of the Lu5(SiO4)3N, the as-prepared sample was analysed by energy dispersive spectroscopy (EDS) analysis. Table 4 shows the results of EDS elemental analysis (wt%) within the rectangle area in Fig. 1(c). As illustrated in Table 4, Lu, Si, O, and N exist in the host Lu5(SiO4)3N. Besides, the weight ratio of each element in Lu5(SiO4)3N is Lu 70.80%, Si 7.06%, O 19.21% and N 2.93% according to the EDS results in Table 4.
Table 4 The results of energy dispersive spectroscopy (EDS) elemental analysis (wt%) of the Lu5(SiO4)3N sample within the rectangle area in Fig. 1(c) and Lu5(SiO4)3N:0.03Ce3+ within the rectangle area in Fig. 3(d)
| Lu5(SiO4)3N |
Lu4.97(SiO4)3N:0.03Ce3+ |
| Element |
Weight (%) |
Atomic (%) |
Element |
Weight (%) |
Atomic (%) |
| Lu |
70.80 |
23.13 |
Lu |
70.63 |
23.07 |
| Si |
7.06 |
12.20 |
Ce |
0.15 |
0.22 |
| O |
19.21 |
56.32 |
Si |
7.24 |
12.51 |
| N |
2.93 |
8.35 |
O |
18.96 |
55.59 |
|
|
|
|
N |
3.02 |
8.61 |
| Total |
100.00 |
100.00 |
Total |
100.00 |
100.00 |
The band structure of pure Lu5(SiO4)3N calculated by using VASP is presented in Fig. 1(d). The results reveal that the valence band maximum (VBM) and the conduction band minimum (CBM) are at different points, indicating that Lu5(SiO4)3N has an indirect broad band gap. In addition, the band gap was determined to be approximately 4.12 eV. As we know, electronic densities of states analysis were considered an effective way to assign the band structure and analyze the bonding interactions in the crystal. Therefore, the electronic densities of states of Lu5(SiO4)3N, both total (TDOS) and partial (PDOS), onto the atomic orbital were performed as illustrated in Fig. 1(e). It can be seen that the valence bands in the range of −19.89 to −17.35 eV originate from O, Si, and Lu. The peaks at about −15.93 eV correspond to Lu and O state bands. Furthermore, the valence bands close to the Fermi level (−0.61 to 0 eV) are dominated by the states of O and N. Finally, the conduction band (below 6.5 eV) is dominated by states of Lu. Therefore, the band gap of Lu5(SiO4)3N is determined by O/N and Lu atoms.
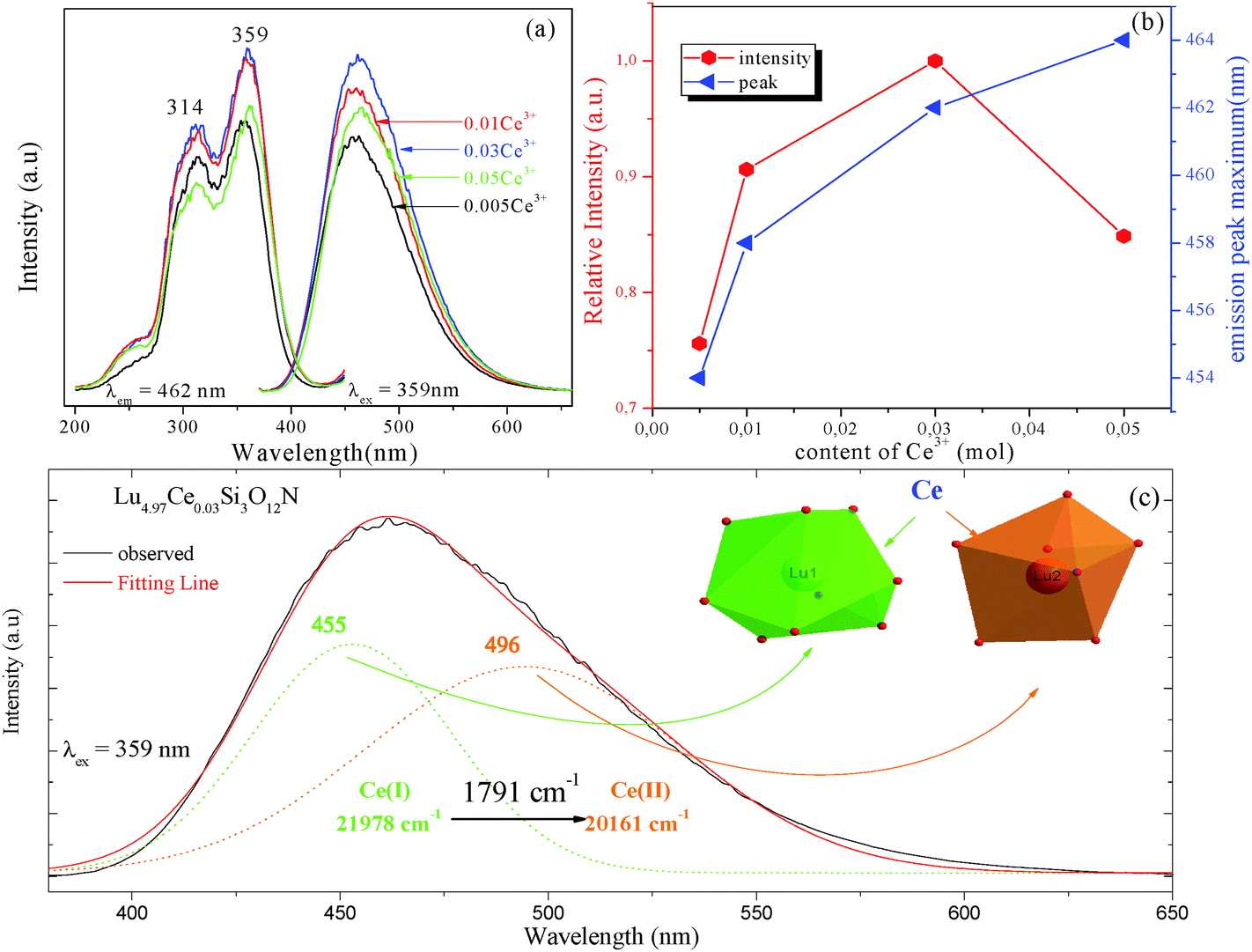
3.2 Luminescence properties of Lu5(SiO4)3N:Ce3+ phosphor
Fig. 2 depicts the photoluminescence excitation (PLE) and emission (PL) spectra of Lu5−x(SiO4)3N:xCe3+ (a), and the relative emission intensity and emission peak maximum (nm) as a function of Ce3+ concentration (b). Under the excitation at 359 nm, Lu5−x(SiO4)3N:xCe3+ exhibit a broad blue emission band peaking at about 460 nm. The PLE spectrum monitored at 462 nm shows a broad absorption from 245 to 400 nm, including two obvious broad bands centered at 314 and 359 nm attributed to the 4f → 5d transition of Ce3+.26,27 As depicted in Fig. 2(a) and (b), the emission intensity of Ce3+ increased firstly with the Ce3+ concentration x, and achieved the maximum value at x = 0.03 (mol), and then it decreased dramatically with further increase of concentration, which may be attributed to the concentration quenching effect. Therefore, the optimum doping concentration x of Ce3+ is 0.03. Moreover, the emission peak shows a slight red shift from 454 to 464 nm with increasing Ce3+ concentration, which may be due to the variation of crystal field strength. In addition, the correlation between the crystal field splitting (Dq) and the shape and the size of the polyhedron is given by the following equation:28| | 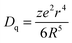 | (1) |
where Dq refers to the energy level separation, z is the charge or valence of the anion ligand, e stands for the charge of an electron, r represents the radius of the d wave function, and R represents the bond length. Assuming that z, e and r are the same for the d(Ce–O)-orbital and the d(Ce–N)-orbital, it can be considered that Dq is approximately proportional to 1/R5. Thus, if R is smaller, Dq has a larger value, and finally results in the red-shift of the emission peak. As the average bond length of the Lu1 site (2.706 Å, CN = 9) is somewhat longer than that of the Lu2 site (2.543 Å, CN = 7), it is presumed that the larger Ce3+ ion relative to the Lu3+ ion preferably enters the Lu1 site (more space with CN = 9) rather than the Lu2 site (less space with CN = 7). It is well-known that there are several factors that affect the extent of splitting of the d orbitals by ligands. Among several factors, the number and geometry of ligands have an important effect on the magnitude of the crystal field splitting (Dq). In comparison between an octahedral complex (Oh, CN = 6) and a tetrahedral complex (Td, CN = 4), it can be shown that for a point-charge model:29| | | 10Dq(Td) = 4/9[10Dq(Oh)] | (2) |
According to eqn (2), it is evident that the larger Ce3+ ion relative to the Lu3+ ion preferably enters the Lu1 site (more space, CN = 9) and the energy splitting of 5d orbitals of Ce3+ ions is larger than that in the Lu2 site (less space, CN = 7), implying that the emission band is gradually shifted to the red wavelength region with increasing Ce3+ concentration in Lu5−xCex(SiO4)3N phosphors.
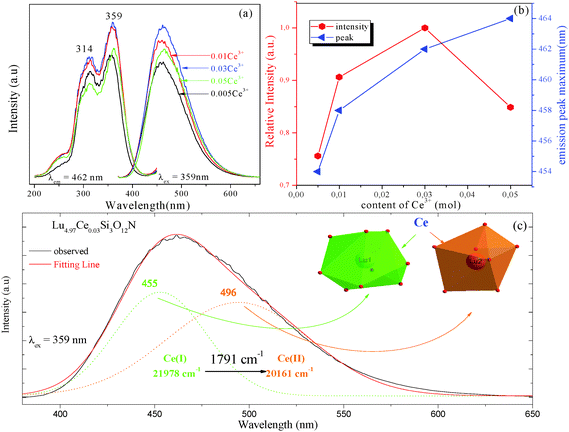 |
| | Fig. 2 The PLE and PL spectra of Lu5−x(SiO4)3N:xCe3+ (a); the relative emission intensity and emission peak maximum (nm) as a function of Ce3+ concentration (b); PL spectrum of Lu4.97Ce0.03(SiO4)3N fitted by Gaussian functions under the excitation at 359 nm (c). | |
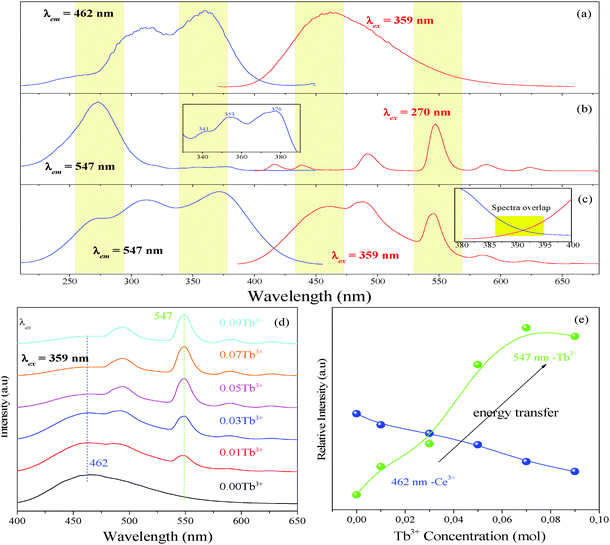
Furthermore, the PL spectrum of Lu4.97Ce0.03(SiO4)3N can be divided into two dotted bands centered at about 456 and 495 nm by using Gaussian fitting, as depicted in Fig. 2(c). In general, the emission of Ce3+ ions should contain two bands because of the spin–orbit splitting of the ground state (2F7/2 and 2F5/2 states) with an energy difference of about 2000 cm−1.30 However, the energy difference between 453 and 493 nm was calculated to be 1817 cm−1. Thus, these components can be ascribed to the contributions of Ce3+ ions at two different luminescent centers, which is inconsistent with the structure of Lu5(SiO4)3N. As far as we know, the emission position of Ce3+ ions is strongly affected by their local crystal environment, and the following equation can be used to determine the sites of Ce3+:31
| | 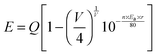 | (3) |
in which
E represents the position of the Ce
3+ ion emission peak (cm
−1),
Q is the energy position of the lower d-band edge for the free ions (50
000 cm
−1 for Ce
3+),
V stands for the valence of Ce
3+ (
V = 3 for Ce
3+),
n is the number of anions in the immediate shell around the Ce
3+ ion,
r is the effective radius of the host cations (La
3+) replaced by the Ce
3+ ion (Å), and
Ea is the electron affinity of the atoms that form anions in eV, which is constant in the same host. The effective ionic radius of Lu
3+ with nine-coordination is larger than that with seven-coordination. Therefore, the emission peaks at 453 nm and 555 nm are attributed to Ce
3+ ions with nine-coordination and seven-coordination as shown in the inset of
Fig. 2(c).
3.3 Crystal structure of Lu5(SiO4)3N:Ce3+,Tb3+ phosphors
With 4f–4f forbidden transitions, Tb3+ can be effectively sensitized by Ce3+, which can make the emission color of phosphors tunable. Besides, phase purity is an important factor for phosphors to have stable luminescence properties. Thus, some Lu5−x−y(SiO4)3N:xCe3+,yTb3+ phosphors were selected for XRD measurements. Fig. 3(a) shows the XRD patterns of Lu4.995(SiO4)3N:0.005Ce3+, Lu4.97(SiO4)3N:0.03Ce3+, Lu4.96(SiO4)3N:0.03Ce3+,0.01Tb3+, Lu4.92(SiO4)3N:0.03Ce3+,0.05Tb3+, Lu4.88(SiO4)3N:0.03Ce3+,0.09Tb3+, as well as the simulated Lu5(SiO4)3N obtained from the cell refinement. It is shown that all XRD peaks of the samples are consistent with the simulated data for Lu5(SiO4)3N, indicating that all the samples are single-phase and the doped Ce3+ or Tb3+ ions were successfully introduced into the Lu5(SiO4)3N host lattice. In order to better understand the phase structure and the purity of the Ce3+/Tb3+ doped phosphors, Rietveld refinements were taken for Lu4.97Ce0.03(SiO4)3N and Lu4.90Ce0.03Tb0.07(SiO4)3N phosphors. The Ce3+ or Tb3+ ions were supposed to occupy both sites of Lu ions. The refinements were stable and gave low R-factors shown in Fig. 3(a) and (b), and Table 5. Besides, the lattice constants of Lu4.97Ce0.03(SiO4)3N and Lu4.90Ce0.03Tb0.07(SiO4)3N are close to that of Lu5(SiO4)3N, indicating that the doped Ce3+ or Tb3+ ions did not cause any significant changes in the crystal lattice of Lu5(SiO4)3N.
 |
| | Fig. 3 The XRD patterns of Lu4.995(SiO4)3N:0.005Ce3+, Lu4.97(SiO4)3N:0.03Ce3+, Lu4.96(SiO4)3N:0.03Ce3+,0.01Tb3+, Lu4.92(SiO4)3N:0.03Ce3+,0.05Tb3+, Lu4.88(SiO4)3N:0.03Ce3+,0.09Tb3+, as well as the simulated Lu5(SiO4)3N obtained from the structure refinement (a); powder XRD patterns for Rietveld structure analysis of the selected Lu4.97Ce0.03(SiO4)3N (b) and Lu4.90Ce0.03Tb0.07(SiO4)3N (c) phosphors based on the simulated Lu5(SiO4)3N phase model. Solid red lines are calculated intensities, the black circles are the observed intensities, and blue solid lines for the difference between the observed and the calculated intensities, and short green vertical lines below the profiles show the position of Bragg reflections of the calculated pattern; SEM image of Lu4.97(SiO4)3N:0.03Ce3+ (d). | |
Table 5 Refinement parameters for Lu4.97Ce0.03(SiO4)3N and Lu4.90Ce0.03Tb0.07(SiO4)3N from the Rietveld refinement using X-ray powder diffraction data at room temperature
| Samples |
Lu4.97Ce0.03Si3O12N |
Lu4.90Ce0.03Tb0.07Si3O12N |
| Space group |
P63/m(176) |
P63/m(176) |
|
a, Å |
9.6989(33) |
9.69885(25) |
|
c, Å |
7.2489(27) |
7.2592(03) |
|
V, Å3 |
590.35(5) |
591.99(24) |
| 2θ-interval, ° |
10–75 |
10–75 |
|
R
wp, % |
9.56 |
9.40 |
|
R
p, % |
6.73 |
7.09 |
|
χ
2
|
1.85 |
2.43 |
The SEM image of Lu4.97(SiO4)3N:0.03Ce3+ is demonstrated in Fig. 3(d), and the phosphor was also analyzed by EDS (Table 4). It is found that the particles have a smooth morphology with diameters of the particles being between 1 and 6 μm. In addition, the EDS results indicated the presence of Ce3+ in the sample.
3.4 Energy transfer investigations in Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+
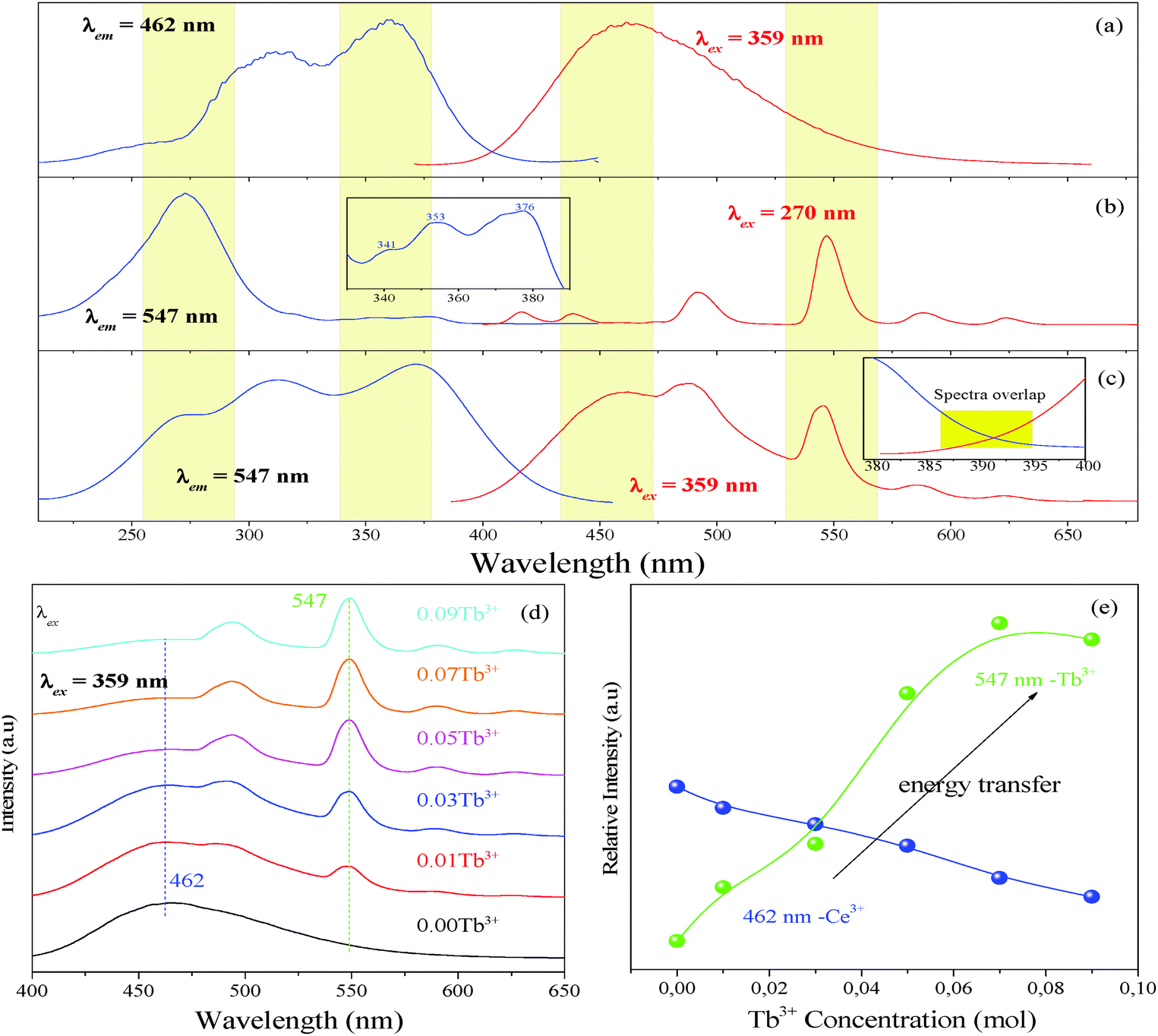
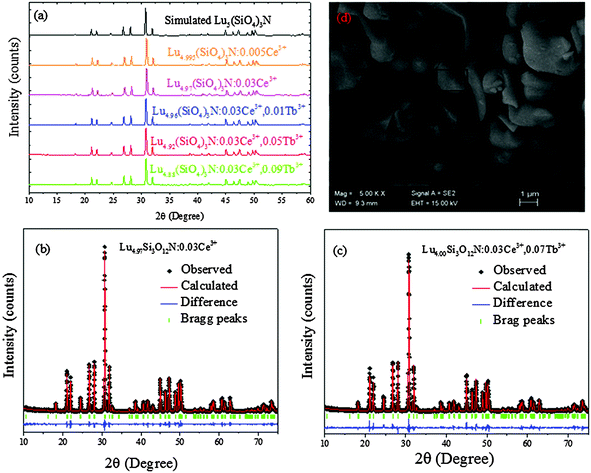
Fig. 4 presents the PLE and PL spectra of Lu4.97(SiO4)3N:0.03Ce3+ (a), Lu4.97(SiO4)3N:0.03Tb3+ (b), and Lu4.94(SiO4)3N:0.03Ce3+,0.03Tb3+ (c). The PLE spectrum of Lu4.97(SiO4)3N:0.03Tb3+ presents a broad band from 225 to 314 nm with a maximum at 270 nm corresponding to the 4f8–4f75d transition of Tb3+. As illustrated in the inset of Fig. 4(b), there are some weak lines at 341, 353, and 376 nm, corresponding to the transitions from the 7F6 ground state to the excited states (5D10, 5GJ, and 5L10 levels) of Tb3+ ions. Under 270 nm excitation, Lu4.97(SiO4)3N:0.03Tb3+ can emit strong green light, and the obtained emission spectrum gives typical characteristic transitions of Tb3+ ions, which are situated at about 417 nm (5D3–7F5), 440 nm (5D3–7F4), 491 nm (5D4–7F6), 547 nm (5D4–7F5), 589 nm (5D4–7F4), and 623 nm (5D4–7F3).32,33 As shown in Fig. 4(c), it includes three peaks located at 270, 314, and 359 nm in the excitation spectrum of Lu4.94(SiO4)3N:0.03Ce3+,0.03Tb3+ phosphor monitored with 462 nm. According to the above discussions, the peak at 265 nm belongs to the 4f8–4f75d1 transition of Tb3+, while the peaks at 306 and 365 nm correspond to the 4f–5d transition of Ce3+. Upon excitation at 359 nm, both the emission of Ce3+ and Tb3+ can be observed in the PL spectrum of the co-doped Lu4.94(SiO4)3N:0.03Ce3+,0.03Tb3+ sample as depicted in Fig. 4(c). Based on the Dexter theory, energy transfer requires a spectral overlap between the donor emission band and the acceptor excitation band.34 The inset of Fig. 4(c) shows the PLE spectrum of Lu4.97(SiO4)3N:0.03Tb3+ and the PL spectrum of Lu4.97(SiO4)3N:0.03Ce3+ ranging from 380 to 400 nm. It is clear that there is a spectral overlap in the range from 386 to 396 nm in combination with the emission band of Ce3+ (donor) and the excitation of Tb3+ (acceptor). Therefore, the energy transfer from Ce3+ to Tb3+ may occur in the Lu5(SiO4)3N host.
 |
| | Fig. 4 The PL excitation and emission spectra of Lu4.97(SiO4)3N:0.03Ce3+ (a); the PL excitation and emission spectra of Lu4.97(SiO4)3N:0.03Tb3+ (b), and the inset of (b) shows PLE spectra of Lu4.97(SiO4)3N:0.03Tb3+ ranging from 330 nm to 390 nm; the PL excitation and emission spectra of Lu4.94(SiO4)3N:0.03Ce3+,0.03Tb3+ (c), and the inset of (c) illustrates the PLE spectrum of Lu4.97(SiO4)3N:0.03Tb3+ and the PL spectrum of Lu4.97(SiO4)3N:0.03Ce3+ in the range from 380 to 400 nm; the emission spectra of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, 0.05, 0.07, and 0.09) phosphors with an excitation wavelength of 359 nm (d); relative emission intensities of Ce3+ and Tb3+ ions in Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (e). | |
As discussed above, the optimum doping concentration of Ce3+ is 0.03 (mol). In order to further investigate the energy transfer process between the Ce3+ and Tb3+ ions in the Lu5(SiO4)3N host, a series of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, 0.05, 0.07, and 0.09) phosphors were prepared. Fig. 4(d) presents the emission spectra of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ phosphors with an excitation at 359 nm. It is clear that both the emission peaks of Ce3+ and Tb3+ can be observed in the as-prepared Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ phosphors except for Lu4.97(SiO4)3N:0.03Ce3+. As illustrated in Fig. 4(e), the relative emission intensities of the Ce3+ ions decreased remarkably, whereas the relative emission intensities of Tb3+ increased first with the Tb3+ concentration. Then, the emission intensity of Tb3+ in Lu4.97(SiO4)3N:0.03Ce3+,0.09Tb3+ decreased slightly with further increasing concentration of Tb3+, ascribed to the concentration quenching effect.
To further confirm the energy transfer between Ce3+ and Tb3+, the decay curves of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, and 0.05) were collected (λex = 370 nm and λem = 462 nm), which are shown in Fig. 5(d). Generally, energy transfer from Ce3+ to Tb3+ belongs to multipolar interaction between donor and acceptor ions. According to Dexter theory, the energy transfer probability through multipolar interaction can be calculated by eqn (4):35
| | 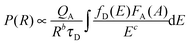 | (4) |
in which
b and
c are constant,
P represents the energy transfer probability,
τD is the decay time of the donor (activator Ce
3+) emission,
QA stands for the total absorption cross-section of the acceptor, and
R is the distance between the donor (Ce
3+) and the acceptor (Tb
3+). Thus, the energy-transfer probability
P is inversely proportional to the decay time
τD. Additionally, the decay curves for all the phosphors can be well fitted with a second-order exponential decay mode using
eqn (5):
36| | | I(t) = A1exp(−t/τ1) + A2exp(−t/τ2) | (5) |
where
I(
t) is the luminescence intensities at times
t,
A1 and
A2 are fitting constants, and
τ1 and
τ2 stand for the decay times for the exponential components. Based on
eqn (6), the average lifetimes (
τ*) can be calculated using the parameters in
eqn (5).
37| | | τ* = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (6) |
As illustrated in the inset of
Fig. 5(d), all the
R2 values are higher than 98%, which indicated that the fitting is relatively good. Therefore,
τ* values for Lu
4.97−y(SiO
4)
3N:0.03Ce
3+,
yTb
3+ (
y = 0, 0.01, 0.03, and 0.05) were determined to be 4.41, 3.74, 2.10, and 1.67 ns, respectively. Obviously, the decay time decreases gradually with the increase of Tb
3+ concentration, which strongly demonstrates the energy transfer from Ce
3+ to Tb
3+.
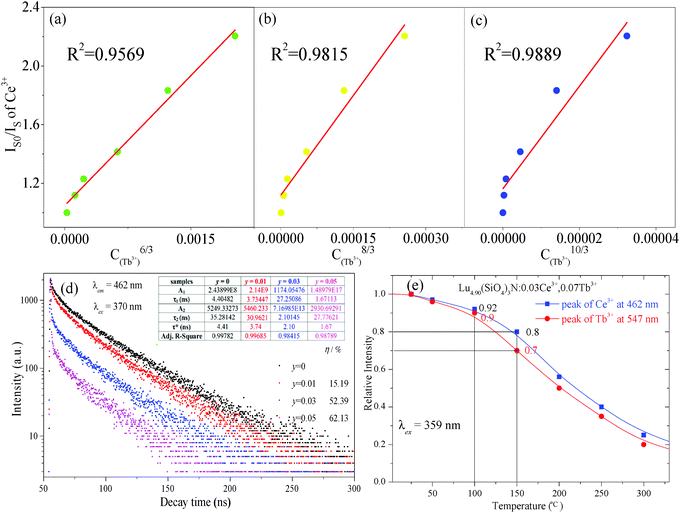 |
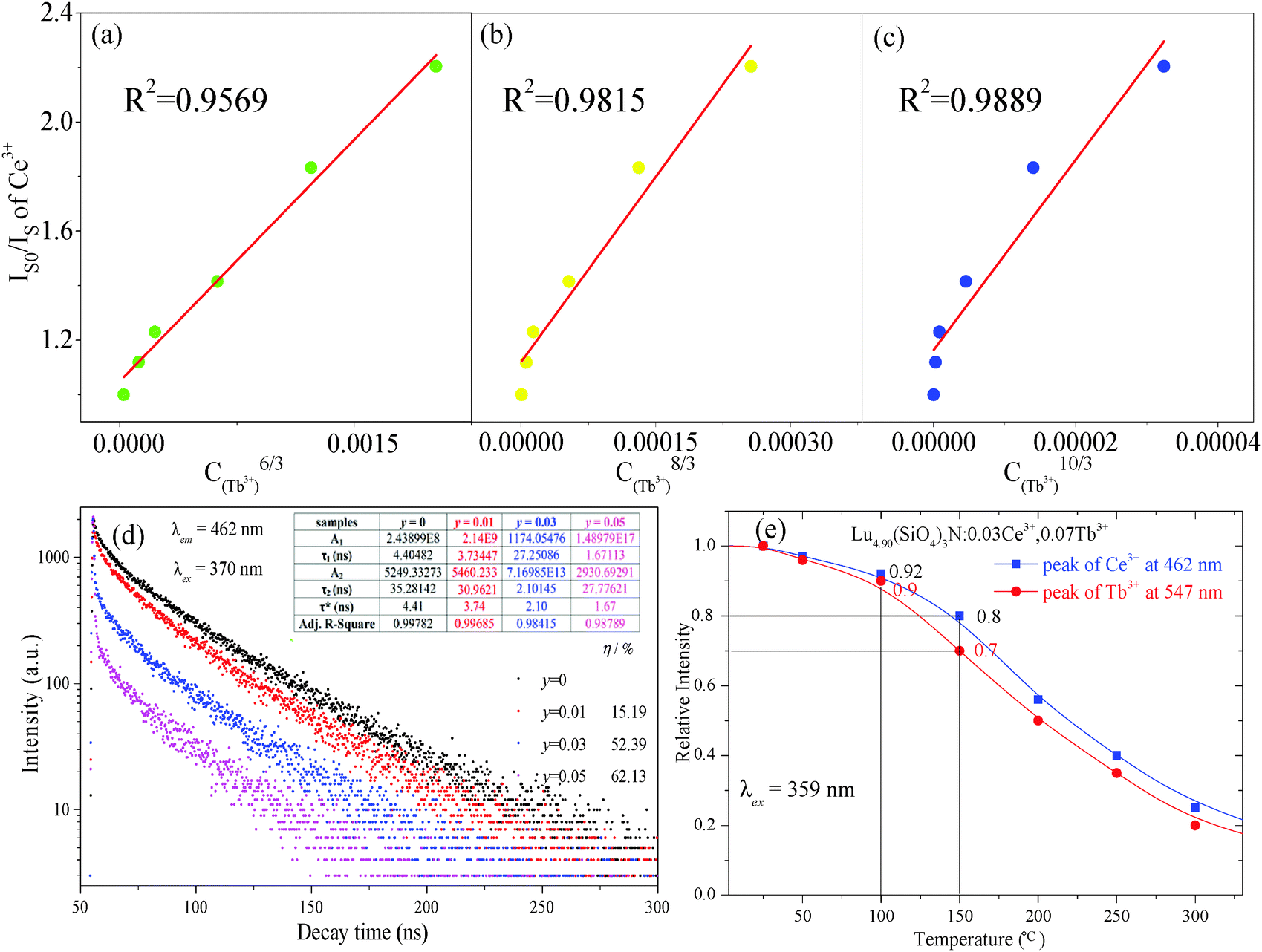
| | Fig. 5 Dependence of IS0/IS of Ce3+ on C6/3 (a); C8/3 (b); C10/3 (c); decay curves of Ce3+ emission in Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, and 0.05) phosphors under excitation at 370 nm monitored at 462 nm at room temperature, and the inset depicts the fitted lifetimes, fitting parameters, as well as the energy transfer efficiency calculated on the basis of lifetimes (d); the relative emission intensities centered at 462 nm (Ce3+) and 547 nm (Tb3+) of the selected sample as a function of temperature ranging from 25 °C to 300 °C (e). | |
In general, the energy transfer efficiency ηT from a sensitizer to an activator can also be expressed using the following equation by Paulose et al.:38,39
where
τS and
τ0 represent the luminescence lifetimes of the sensitizer Ce
3+ ion in the presence and the absence of the activator Tb
3+ ion, respectively. On the basis of the above calculated average values of the lifetimes (
τ*), the energy transfer efficiency
ηT can be calculated to be 15.19%, 52.39%, and 62.13% as given in
Fig. 5(d). With the increase of Tb
3+ concentration, the value of
ηT increased gradually and reached the maximum of 62.13% when
y equals to 0.05. Thus, an efficient energy transfer from Ce
3+ to Tb
3+ must have occurred in Lu
4.97−y(SiO
4)
3N:0.03Ce
3+,
yTb
3+.
As we know, there are two main aspects that are responsible for the resonance energy transfer mechanism: one is the multipolar interaction and the other is the exchange interaction. Meanwhile, the critical distance between the sensitizer and the activator should be shorter than 5 Å for the exchange interaction. To further determine the energy transfer mechanism in Lu5(SiO4)3N, the critical distance (RC) between Ce3+ and Tb3+ ions was calculated using the concentration quenching method proposed by Blasse.40,41
| | 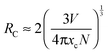 | (8) |
where
xc is the critical concentration,
V corresponds to the volume of the unit cell, and
N stands for the number of host cations in the unit cell. For the Lu
5(SiO
4)
3N host,
N = 10,
V is estimated to be 589.87 Å
3 as shown in
Table 1. Besides, the critical concentration is about 0.10 (mol) from the overall concentration of Ce
3+ (0.03) and Tb
3+ (0.07). Therefore, the critical distance
RC of energy transfer can be estimated to be about 10.405 Å (
xc = 0.10) on the basis of the above equation. The value of
RC is much larger than 5 Å, which indicated that the electric multipolar interaction can take place for the energy transfer between Ce
3+ and Tb
3+ in the Lu
5(SiO
4)
3N host lattice. In addition, the multipolar interaction can be determined by Dexter's energy-transfer expressions and Reisfeld's approximation given as follows:
42,43where
η0 and
η stand for the luminescence quantum efficiency of Ce
3+ in the absence and the presence of Tb
3+,
C corresponds to the total content of Ce
3+ and Tb
3+, and
n = 6, 8, and 10, respectively. The value
η0/
η is approximately calculated by the ratio of related luminescence intensities as (
I0/
IS). Besides,
n = 6, 8, and 10 correspond to the electric multipolar interaction of the electric dipole–dipole, dipole–quadrupole, and quadrupole–quadrupole, respectively. The
I0/
IS −
Cn/3 plots are further illustrated in
Fig. 5(a)–(c). By comparing the values of the fitting factors
R2, the optimal linear relationship is obtained when
n = 10 (
R2 = 0.9894). This indicates that the quadrupole–quadrupole interaction should be mainly responsible for the energy transfer from Ce
3+ to Tb
3+ ions in Lu
4.97−y(SiO
4)
3N:0.03Ce
3+,
yTb
3+.
As we know, the thermal stability of phosphor is one of the most important technological parameters for its application. Therefore, Lu4.90(SiO4)3N:0.03Ce3+,0.07Tb3+ was selected for PL measurements under 359 nm excitation at different temperatures ranging from 25 °C to 300 °C. The relative emission intensities centered at 462 nm (Ce3+) and 547 nm (Tb3+) of the selected sample as a function of temperature are shown in Fig. 5(e). The relative PL intensities centered at 462 nm (Ce3+) and 547 nm (Tb3+) decrease to 80% and 70% of the initial values (25 °C) with the temperature increasing to 150 °C, implying that the sample has high thermal stability.
3.5 CIE chromaticity diagram and quantum efficiency (QE) of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+
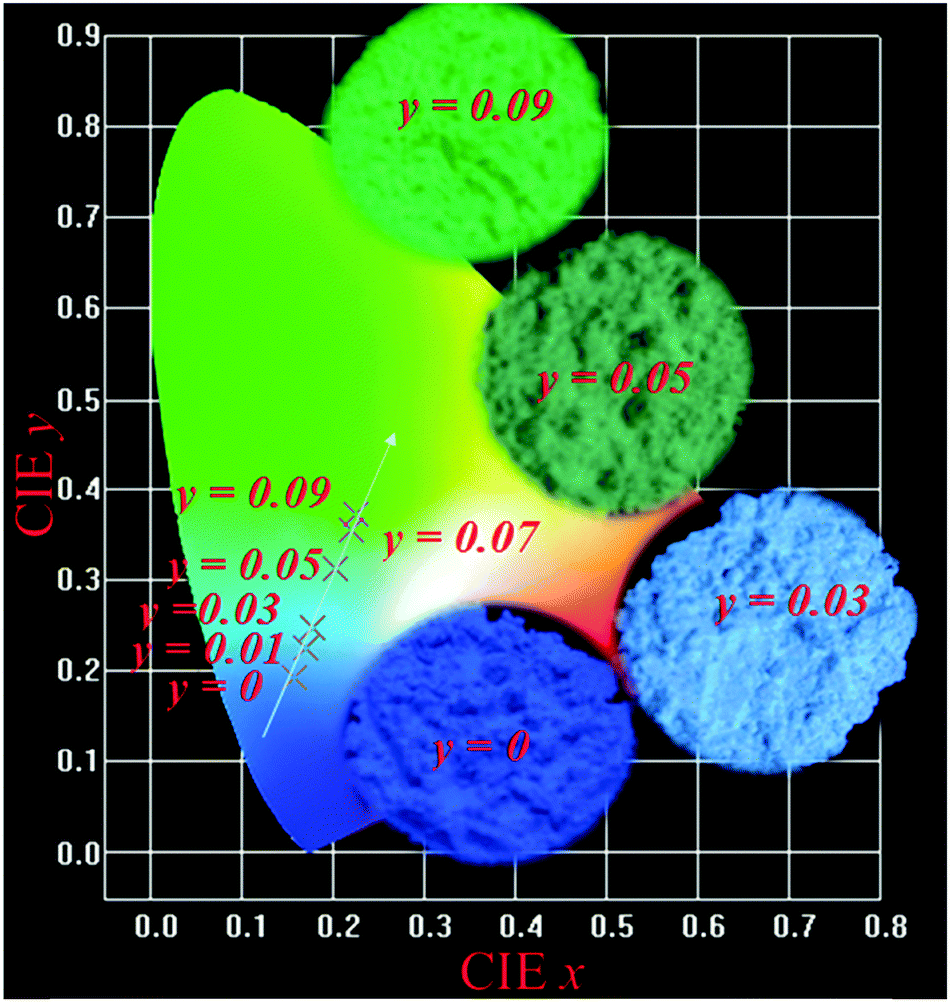
Fig. 6 depicts the CIE chromaticity diagram of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, 0.05, 0.07, and 0.09) phosphors with different Tb3+ concentrations under 359 nm excitation calculated by PL spectroscopy, and the insets of Fig. 6 show the digital photos of the selected samples (y = 0, 0.03, 0.05, and 0.09) under 365 nm UV lamp excitation. The CIE coordinates of the Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ phosphors shifted from the blue (0.1480, 0.1842) to green (0.2249, 3713) region with Tb3+ concentration, as indicated in Table 6. Therefore, the emission color of the phosphors can be tuned by adjusting the doping content of Tb3+ ions. In addition, the quantum efficiency of phosphors is another important factor for their application in LEDs. Therefore, the absolute quantum efficiency (QE) of the Lu4.97(SiO4)3N:0.03Ce3+ sample was investigated. Under the excitation at 359 nm, the absolute QE for the phosphor was determined to be 42.13%.
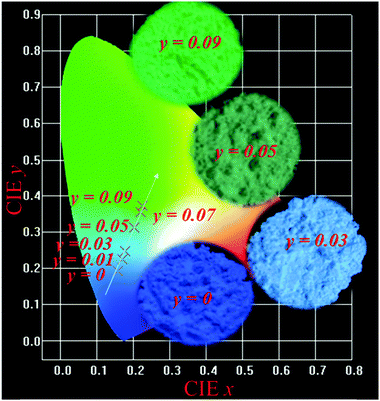 |
| | Fig. 6 The CIE chromaticity diagram of Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, 0.05, 0.07, and 0.09) phosphors under 359 nm excitation, and the insets of show the digital photos of the selected samples (y = 0, 0.03, 0.05, and 0.09) under 365 nm UV lamp excitation. | |
Table 6 CIE coordinates for Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ (y = 0, 0.01, 0.03, 0.05, 0.07, and 0.09) (λex = 359 nm)
| Samples |
CIE (x, y) |
|
y = 0 |
(0.1480, 0.1842) |
|
y = 0.01 |
(0.1586, 0.1937) |
|
y = 0.03 |
(0.1696, 0.2239) |
|
y = 0.05 |
(0.1783, 0.2483) |
|
y = 0.07 |
(0.2188, 0.3549) |
|
y = 0.09 |
(0.2249, 0.3713) |
4. Conclusions
In summary, novel single-phase Lu5(SiO4)3N compounds and Lu5(SiO4)3N:Ce3+,Tb3+ phosphors with an apatite structure were obtained for the first time via a high temperature solid state method with 0.5 MPa atmospheric pressure. The results indicated that Lu5(SiO4)3N crystallized in the hexagonal space group of P63/m with cell parameters a = b = 9.7005 Å and c = 7.2383 Å. Two non-equivalent lutetium sites exist in Lu5(SiO4)3N, that is, Lu(1) with nine coordination and Lu(2) with seven coordination. The ratio of the number of Lu atoms in Lu(1) and Lu(2) sites is 3:2. The band gap for Lu5(SiO4)3N was 4.12 eV. In the Ce3+ doped Lu5(SiO4)3N:0.03Ce3+ compound, the emission peak centered at 462 nm was observed with the Commission International de I'Eclairage (CIE) coordinates of (0.148, 0.184), indicating blue-emission. Remarkably, in Ce3+ and Tb3+ co-doped Lu4.97−y(SiO4)3N:0.03Ce3+,yTb3+ compounds, the color-tunability was observed with increasing Tb3+ co-doping rate on moving from blue at Tb3+ = 0.00 to green at Tb = 0.09, due to the energy transfer from Ce3+ to Tb3+ ions being matched well with the decay curve results. The energy transfer process from the Ce3+ to Tb3+ ions occurs in the host lattice by the quadrupole–quadrupole interaction mechanism. The excitation spectra of the Lu5(SiO4)3N:Ce3+,Tb3+ phosphors give a broad excitation band in the wavelength range from 235 nm to 435 nm. The absolute quantum efficiency (QE) for Lu5(SiO4)3N:0.03Ce3+ was determined to be 42.13%. All the above results demonstrate that Lu5(SiO4)3N could be a platform for modeling a new phosphor and application in the solid-state lighting field and that Lu5(SiO4)3N:Ce3+,Tb3+ samples could be used as blue-green phosphors.
Acknowledgements
This work is supported by the National Natural Science Foundations of China (Grant No. 41172053) and Chinese Scholarship Council.
Notes and references
- Y. Li, W. J. Liu, X. C. Wang, G. Zhu, C. Wang and Y. H. Wang, Dalton Trans., 2015, 44, 13196–13203 RSC.
- D. Kim, D. Park, N. Oh, J. Kim, E. D. Jeong, S. J. Jim, S. Kim and J. C. Park, Inorg. Chem., 2015, 54, 1325–1336 CrossRef CAS PubMed.
- F. Lu, X. Song and Q. Liu, J. Alloys Compd., 2011, 509, 2099–2104 CrossRef CAS.
- L. K. Bharat, J. Y. Park and J. S. Yu, Chem. Eng. J., 2014, 240, 179–186 CrossRef.
- M. F. Zhang, Y. J. Liang, S. Y. Xu, Y. L. Zhu, X. Y. Wu and S. Q. Liu, CrystEngComm, 2016, 18, 68–76 RSC.
- Z. L. Dong, T. J. White, K. Sun, L. M. Wang and R. C. Ewing, J. Am. Chem. Soc., 2015, 88, 184–190 Search PubMed.
- E. N. Bulanov, J. X. Wang, A. V. Knyazev, T. White, M. E. Manyakina, T. Baikie, A. N. Lapshin and Z. L. Dong, Inorg. Chem., 2015, 54, 11356–11361 CrossRef CAS PubMed.
- K. Fukuda, T. Asaka, S. Hara, M. Oyabu, A. Berghout, E. Bechade, O. Masson, I. Julien and P. Thomas, Chem. Mater., 2013, 25, 2154–2162 CrossRef CAS.
- Q. F. Guo, L. B. Liao, L. F. Mei and H. K. Liu, J. Solid State Chem., 2015, 226, 107–113 CrossRef CAS.
- M. M. Jiao, Y. C. Jia, W. Lu, W. Z. Lv, Q. Zhao, B. Q. Shao and H. P. You, J. Mater. Chem. C, 2014, 2, 90–97 RSC.
- K. Li, D. L. Geng, M. M. Shang, Y. Zhang, H. Z. Lian and J. Lin, J. Phys. Chem. C, 2014, 118, 11026–11034 CAS.
- D. L. Geng, H. Z. Lian, M. M. Shang, Y. Zhang and J. Lin, Inorg. Chem., 2014, 53, 2230–2239 CrossRef CAS PubMed.
- Y. C. Chiu, T. J. Lee, W. R. Liu, Y. T. Yeh, S. M. Jang and R. S. Liu, Opt. Express, 2011, 19, 331–339 CrossRef PubMed.
- G. Z. Chen, L. J. Yin, J. T. Dong, Y. Y. Feng, Y. Gao, W. D. He, Y. Jia and H. T. Hintzen, J. Am. Chem. Soc., 2016, 99, 183–190 CAS.
- C. H. Huang, Y. C. Chiu and W. R. Liu, Eur. J. Inorg. Chem., 2014, 3674–3680 CrossRef CAS.
- J. W. H. van Krevel, H. T. Hintzen, R. Metselaar and A. Meijerink, J. Alloys Compd., 1998, 268, 272–277 CrossRef CAS.
- X. M. Wang, C. H. Wang, X. J. Kuang, R. Q. Zou, Y. X. Wang and X. P. Jing, Inorg. Chem., 2012, 51, 3540–3547 CrossRef CAS PubMed.
- F. C. Lu, L. J. Bai, Z. P. Yang and X. N. Han, Mater. Lett., 2015, 151, 9–11 CrossRef CAS.
- X. Xu, C. Cai, L. Y. Hao, Y. C. Wang and Q. X. Li, Mater. Chem. Phys., 2009, 118, 270–272 CrossRef CAS.
- J. Takahashi, H. Yamane, M. Shimad, Y. Yamamoto, N. Hirosaki, M. Mitomo, K. Oikawa, S. Torii and T. Kamiyama, J. Am. Chem. Soc., 2002, 85, 2072–2077 CAS.
-
A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory, Report LAUR: Los Alamos, 1986, 86–748.
- G. Kresse and G. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- F. C. Lu and L. J. Bai, J. Am. Ceram. Soc., 2014, 97, 2397–2400 CrossRef CAS.
- B. Dierre, R. J. Xie, N. Hirosaki and T. Sekiguchi, J. Mater. Res., 2007, 22, 1933–1941 CrossRef CAS.
- S. Titeux, M. Gervais, P. Verdier and Y. Laurent, Mater. Sci. Forum, 2000, 325–326, 17–20 CrossRef CAS.
- M. Yang, H. P. You, K. Liu, Y. H. Zheng, N. Guo and H. J. Zhang, Inorg. Chem., 2010, 49, 4996–5002 CrossRef CAS PubMed.
- X. H. Gong, J. H. Huang, Y. J. Chen, Y. F. Lin, Z. D. Luo and Y. D. Huang, Inorg. Chem., 2014, 53, 6607–6614 CrossRef CAS PubMed.
- C. L. Zhao, Z. G. Xia and S. X. Yu, J. Mater. Chem. C, 2014, 2, 6032–6039 RSC.
-
J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, Harper Collins College Publishers, New York, 1993 Search PubMed.
- H. B. Liang, H. H. Lin, G. B. Zhang, P. Dorenbos and Q. Su, J. Lumin., 2011, 131, 194–198 CrossRef CAS.
- L. G. Van Uitert, J. Lumin., 1984, 29, 1–9 CrossRef CAS.
- T. Grzyb, K. Kubasiewicz, A. Szczeszak and S. Lis, Dalton Trans., 2015, 44, 4063–4069 RSC.
- L. Liu, Y. M. Liang, L. L. Li, L. C. Zou and S. C. Gan, CrystEngComm, 2015, 17, 7754–7761 RSC.
- Y. Chen, Y. Li, J. Wang, M. M. Wu and C. X. Wang, J. Phys. Chem. C, 2014, 118, 12494–12499 CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- D. Geng, M. Shang, Y. Zhang, H. Lian and J. Lin, Dalton Trans., 2013, 42, 15372–15380 RSC.
- C. H. Huang, T. M. Chen, W. R. Liu, Y. C. Chiu, Y. T. Yeh and S. M. Jang, ACS Appl. Mater. Interfaces, 2010, 2, 259–264 CAS.
- P. I. Paulose, G. Jose, V. Thomas, N. V. Unnikrishnan and M. K. R. Warrier, J. Phys. Chem. Solids, 2003, 64, 841–846 CrossRef CAS.
- W. Z. Lv, M. M. Jiao, Q. Zhao, B. Q. Shao, W. Lu and H. P. You, Inorg. Chem., 2014, 53, 11007–11014 CrossRef CAS PubMed.
- G. Blasse, J. Solid State Chem., 1986, 62, 207–211 CrossRef CAS.
- G. Blasse, Phys. Lett. A, 1968, 28, 444–445 CrossRef CAS.
- L. H. Liu, L. Wang, C. N. Zhang, Y. J. Cho, B. Dierre, N. Hirosaki, T. Sekiguchi and R. J. Xie, Inorg. Chem., 2015, 54, 5556–5565 CrossRef CAS PubMed.
- C. H. Huang, T. W. Kuo and T. M. Chen, ACS Appl. Mater. Interfaces, 2010, 2, 1395–1399 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information files (CIFs) of the Lu5(SiO4)3N compound. See DOI: 10.1039/c6cp01512c |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.