
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The equilibrium molecular structures of 2-deoxyribose and fructose by the semiexperimental mixed estimation method and coupled-cluster computations†
Natalja
Vogt
*ab,
Jean
Demaison
a,
Emilio J.
Cocinero
c,
Patricia
Écija
c,
Alberto
Lesarri
d,
Heinz Dieter
Rudolph
a and
Jürgen
Vogt
a
aSection of Chemical Information Systems, Faculty of Sciences, University of Ulm, 89069 Ulm, Germany. E-mail: natalja.vogt@uni-ulm.de
bDepartment of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia
cDepartamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco (UPV-EHU), 48080 Bilbao, Spain
dDepartamento de Química Física y Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, 47011 Valladolid, Spain
First published on 23rd May 2016
Abstract
Fructose and deoxyribose (24 and 19 atoms, respectively) are too large for determining accurate equilibrium structures, either by high-level ab initio methods or by experiments alone. We show in this work that the semiexperimental (SE) mixed estimation (ME) method offers a valuable alternative for equilibrium structure determinations in moderate-sized molecules such as these monosaccharides or other biochemical building blocks. The SE/ME method proceeds by fitting experimental rotational data for a number of isotopologues, which have been corrected with theoretical vibration–rotation interaction parameters (αi), and predicate observations for the structure. The derived SE constants are later supplemented by carefully chosen structural parameters from medium level ab initio calculations, including those for hydrogen atoms. The combined data are then used in a weighted least-squares fit to determine an equilibrium structure (rSEe). We applied the ME method here to fructose and 2-deoxyribose and checked the accuracy of the calculations for 2-deoxyribose against the high level ab initio rBOe structure fully optimized at the CCSD(T) level. We show that the ME method allows determining a complete and reliable equilibrium structure for relatively large molecules, even when experimental rotational information includes a limited number of isotopologues. With a moderate computational cost the ME method could be applied to larger molecules, thereby improving the structural evidence for subtle orbital interactions such as the anomeric effect.
1. Introduction
The determination of accurate equilibrium structures for moderately large molecules remains a challenge, both from the experimental and theoretical points of view.1 The structure optimization by high-level ab initio methods allows us to obtain accurate structures, but it rapidly becomes too expensive when the size of the molecule increases. However, it is possible to obtain equilibrium structures more easily by using the semiexperimental (SE) method, which is generally considered the most accurate one for equilibrium structures (rSEe) of small molecules.2–4 This method derives the equilibrium rotational constants from experimentally determined (effective) ground-state rotational constants and theoretical corrections based on an ab initio cubic force field. The most complex molecule for which the rotational spectroscopy method has been tested is the amino acid proline (C5H9NO2: 17 atoms, 45 degrees of freedom).5 However, it was noticed in proline that the set of experimental rotational constants, although extensive, could not fix satisfactorily the molecular structure. This conclusion is quite general for molecules with many degrees of freedom because of the problem of statistical ill-conditioning. For this reason, ab initio constraints are required to analyze larger molecules. The compromise of these constraints is that they may induce systematic errors in the calculation, making it difficult to estimate the uncertainty of the resulting molecular structure.Recently, the predicate-regression mixed estimation (ME) method1,6,7 has proved successful in determining very accurate equilibrium structures for several medium-sized molecules.8,9 In the ME method the structure fitting uses simultaneously equilibrium moments of inertia together with bond lengths and bond angles from medium-level quantum chemical calculations. In this paper, we will demonstrate that it is possible to use this method for molecules larger than proline. We will first apply the ME method to the lowest-energy conformer of c-β-2-deoxy-D-ribopyranose-1C4-1,10 (Fig. 1, later abbreviated as deoxyribose), a 19-atom (C1) molecule with 51 degrees of freedom. The validity of the method will be checked for this molecule against high-level CCSD(T) ab initio calculations. Then, we will apply the ME method to the lowest-energy conformer of cc-β-D-fructopyranose-2C511,12 (Fig. 1, later abbreviated as fructose), a larger 24-atom (C1) molecule with 66 degrees of freedom. Both molecules represent the larger molecular systems for which equilibrium structures have been determined so far.
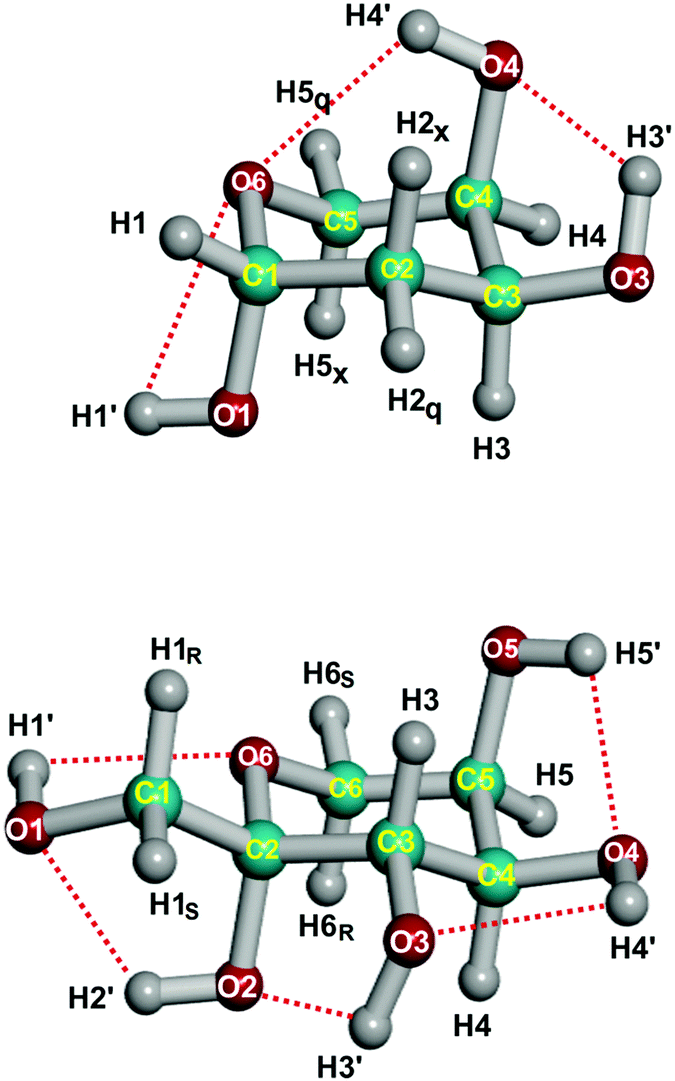 | ||
| Fig. 1 Lowest-energy conformations of deoxyribose (c-β-2-deoxy-D-ribopyranose-1C4-1, upper panel) and fructose (cc-β-D-fructopyranose-2C5), including atom numbering and intramolecular O–H⋯O hydrogen bond networks. | ||
Deoxyribose and fructose are representatives of 5/6-carbon-atom aldose/ketose monosaccharides, which make up carbohydrates. Carbohydrates constitute one of the most versatile biochemical constituents, playing important roles as energy resources, structural bio-scaffolds and signal transducers.13 In particular, deoxyribose is notably present in nucleotides forming DNA, while fructose is commonly attached to glucose to form sucrose. Both molecules exhibit dominant pyranose (six-membered) ring structures in the solid, liquid and gas phases, in contrast with the furanose (five-membered) ring observed for deoxyribose in DNA and other biologically active molecules or fructose in sucrose. The solid-state structure is known for both compounds,14,15 but there is no reliable gas-phase structure with which to assess the quality of the theoretical models used for other monosaccharides.
A final argument for selecting these target molecules is that the rotational spectra have been observed for both compounds. Thus, experimental moments of inertia are available for the application of the ME method. The detection of the rotational spectra for the sugars used supersonic-jet microwave spectroscopy combined with picosecond UV laser desorption. For deoxyribose the experiment detected 6 different pyranoside forms in the gas phase.10 For the lowest-energy species the inertial data span the parent, all five monosubstituted 13C species and the endocyclic 18O species, which were observed in natural abundance. For fructose two pyranoside rotamers were detected and rotational data were available for the parent, all six monosubstituted 13C species and two single deuterated species of the lowest-energy conformation. However, data for the important endocyclic 18O species was missing.11,12
2. Methodology
2.1 Experimental
Previous experiments on fructose11,12 missed the detection of the endocyclic 18O6 isotopologue because it was too weak to be measured in natural abundance (ca. 0.2%). Since the coordinates of this ring atom are critical for the determination of the pyranose structure, we extended the rotational measurements to this species. For this purpose we used an enriched sample (>90%) of [18O6]-D-fructose (Omicron Biochemicals, USA) that was pressed into a cylindrical pellet. The solid target was vaporized by pulsed picosecond UV (355 nm) laser desorption, and the jet-cooled microwave spectrum was recorded in the region 6–18 GHz.11,12 Details of the Balle–Flygare-type Fourier transform microwave spectrometer (FT-MW) at the UPV-EHU have been reported before.16 The experimental rotational frequencies are given in Table S1 (ESI†).2.2 Computational
Different ab initio calculations were required for this work. The geometry optimizations were performed at the frozen-core (FC) and all-electron (AE) MP2 level17 with the cc-pVTZ, cc-pVQZ,18 cc-pwCVTZ19 and 6-311+G(3df,2pd)20 basis sets. The calculations were also performed at the levels of the density functional theory (B3LYP)21–23 with the 6-311+G(3df,2pd) basis set and the coupled-cluster method with single and double excitations (CCSD-FC)24 using the cc-pVTZ basis set. Moreover, the structure optimization for deoxyribose was possible at the level of the coupled-cluster method with a perturbative treatment of connected triples (CCSD(T)-FC)25 using the cc-pVTZ basis set. In order to determine the rovibrational contributions for both molecules, the anharmonic force field up to semidiagonal quartic terms was calculated at the MP2-FC/cc-pVTZ level of theory. This calculation was repeated for each isotopologue, as different isotopes require distinct vibrational corrections. The MP2, B3LYP and CCSD calculations were performed with the Gaussian 09 package,26 whereas the MolPro program27,28 was used for the CCSD(T) calculations.3. Results and discussion
It is well established that the quality of the structural fit is sensitive to the true accuracy of the ground-state rotational constants.1,29,30 For this reason, we first redetermined these parameters with the method of predicate observations, combining the experimental rotational frequencies with quartic centrifugal distortion constants derived from the ab initio force field.6,7 The uncertainty used for weighting the predicates was 10% of their value. The results are given in Tables S2 and S3 (ESI†) for deoxyribose and fructose, respectively. In order to obtain the semiexperimental equilibrium rotational constants, the experimental ground-state rotational constants were corrected using the vibration–rotation interaction constants (αi) derived from the ab initio MP2-FC/cc-pVTZ cubic force field. The derived rotational constants and the rovibrational corrections are given in Tables 1 and 2 for both molecules.| A 0 | B 0 | C 0 | A e − A0 | B e − B0 | C e − C0 | A e | B e | C e | |
|---|---|---|---|---|---|---|---|---|---|
| a The uncertainties used for weighting are (in MHz): 0.1, 0.05 and 0.05 for A, B and C, respectively. | |||||||||
| Parenta | 2437.825 | 1510.729 | 1144.980 | 28.839 | 16.273 | 14.042 | 2466.664 | 1527.002 | 1159.022 |
| 13C1 | 2432.691 | 1499.573 | 1139.154 | 28.630 | 16.140 | 13.946 | 2461.321 | 1515.713 | 1153.100 |
| 13C2 | 2417.585 | 1508.365 | 1141.810 | 28.490 | 16.150 | 13.905 | 2446.075 | 1524.515 | 1155.715 |
| 13C3 | 2428.912 | 1507.073 | 1141.768 | 28.721 | 16.069 | 13.898 | 2457.633 | 1523.142 | 1155.667 |
| 13C4 | 2428.003 | 1505.325 | 1141.295 | 28.441 | 16.274 | 14.023 | 2456.444 | 1521.600 | 1155.318 |
| 13C5 | 2410.436 | 1507.655 | 1139.739 | 28.575 | 16.104 | 13.923 | 2439.011 | 1523.759 | 1153.662 |
| 18O6 (ring) | 2408.851 | 1495.282 | 1131.440 | 28.368 | 16.005 | 13.794 | 2437.219 | 1511.287 | 1145.235 |
| A 0 | B 0 | C 0 | A e − A0 | B e − B0 | C e − C0 | A e | B e | C e | |
|---|---|---|---|---|---|---|---|---|---|
| a The uncertainties used for weighting are (in MHz): 0.1, 0.05 and 0.05 for A, B and C, respectively. b For definition of labeling R and S for hydrogen atoms, see Fig. 1. | |||||||||
| Parenta | 1465.278 | 770.570 | 609.969 | 16.983 | 7.741 | 5.789 | 1482.261 | 778.311 | 615.758 |
| 13C1 | 1461.740 | 764.218 | 606.475 | 16.975 | 7.624 | 5.723 | 1478.715 | 771.842 | 612.198 |
| 13C2 | 1465.322 | 769.506 | 609.303 | 16.937 | 7.694 | 5.740 | 1482.259 | 777.200 | 615.043 |
| 13C3 | 1461.356 | 770.380 | 609.360 | 16.869 | 7.716 | 5.783 | 1478.225 | 778.096 | 615.143 |
| 13C4 | 1463.469 | 767.830 | 608.236 | 16.919 | 7.699 | 5.757 | 1480.388 | 775.529 | 613.993 |
| 13C5 | 1460.571 | 767.004 | 607.074 | 16.764 | 7.716 | 5.760 | 1477.335 | 774.720 | 612.834 |
| 13C6 | 1450.301 | 769.812 | 607.567 | 16.616 | 7.771 | 5.783 | 1466.917 | 777.583 | 613.350 |
| DR_C2b | 1450.487 | 762.130 | 607.208 | 16.696 | 7.582 | 5.726 | 1467.183 | 769.712 | 612.934 |
| DS_C2b | 1454.387 | 762.802 | 604.412 | 16.710 | 7.678 | 5.689 | 1471.097 | 770.480 | 610.101 |
| 18O6 (ring) | 1450.794 | 769.502 | 606.802 | 16.768 | 7.704 | 5.733 | 1467.562 | 777.206 | 612.535 |
The methodology used for determining the predicates was described before.31 Briefly, the CH bond lengths are computed at the MP2-FC/cc-pVTZ level of theory. Due to a compensation of errors, they are usually very close to the accurate equilibrium values. The CC bond lengths are also calculated at the same level. When the double bond character is negligible, these values are also a good choice for the predicates. The CO bond lengths are calculated at the B3LYP/6-311+G(3df,2pd) level and a small correction is applied to the calculated value.32 All these computed bond lengths are expected to have an accuracy of about 0.002 Å. The bond angles are first calculated at the MP2-FC level with the cc-pVTZ and 6-311+G(3df,2pd) basis sets with an expected accuracy of about 0.3–0.4°. From our previous work, it was found that the 6-311+G(3df,2pd) basis set gives slightly more accurate results.9 This outcome is confirmed here by comparing with the Born–Oppenheimer equilibrium structure, rBOe, (alternatively named in the literature as best estimated ab initio or CCSD(T)-based structure) determined below. The median absolute deviation (MAD) is 0.18° with the cc-pVTZ basis set and 0.09° with the 6-311+G(3df,2pd) basis set. For the dihedral angles, the CCSD-FC/cc-pVTZ level was used because the MP2 method has sometimes been found inaccurate.8,9,33,34 The estimated accuracy of the predicate dihedral angles is 0.7°. Comparison with the rBOe structure confirms this value, the MAD being 0.51°. For the bond angles, the accuracy of the MP2 and CCSD methods is similar. However, when the CCSD values are used for the predicates of the bond angles, the standard deviation of the fits is slightly smaller. For this reason, the CCSD-FC/cc-pVTZ values were also used for the predicates of all angles, but this choice has a negligible effect on the values of the fitted parameters. Actually, for deoxyribose, the CCSD-FC/cc-pVTZ and MP2-FC/6-311+G(3df,2pd) have the same MAD when compared to the rBOe structure. The structures calculated at these different levels of theory are given in Tables S4 and S5 (ESI†) for deoxyribose and fructose, respectively.
The ME method was applied in several steps. In the first step, the bond lengths and bond angles to all hydrogen atoms were held at their predicate values, while the parameters for the heavy atoms were fitted to the equilibrium rotational constants. This fit is the standard least-squares one. In the second step, a structure was fitted to both the equilibrium rotational constants and the full set of predicate values with their estimated uncertainties. This step leads to a considerable improvement in the accuracy of the structure. However, an inspection of the leverage values shows that they are close to unity for the predicates of many bond lengths, whereas they are distributed rather uniformly and are significantly below unity for the moments of inertia. It is obvious that the structural parameters of the hydrogen atoms (unsubstituted in most of the isotopologues) are almost exclusively determined by their predicate values. This outcome is not a problem because the predicates are expected to be accurate for these light atoms. To check that the predicates for the heavy atoms are compatible with the semiexperimental equilibrium moments of inertia, the errors of the predicates for the bond lengths of the heavy atoms of deoxyribose have been increased in a third step from 0.002 Å to 0.005 Å. This relaxation gives a fit compatible with the previous one, albeit with larger standard deviations (up to a factor of two) for some bond lengths. The results are given in Table 3 (Cartesian coordinates in Table S6, ESI†). The nice agreement of the derived (non-fitted) parameters with their predicate values indicates that the fit is likely to be of good quality. The exception is the C5–O6 bond length, worsened by an unfavorable propagation of errors. However, this problem is easy to point out because, in this case, the derived value is far from its predicate. This situation can be explained by underweighted predicates relative to the moments of inertia, so the fitted parameters remain sensitive to inaccuracies in the moments of inertia. In this particular case, a careful analysis indicates that the problem is mainly due to the small a coordinate of atom C5, aSE(C5) = −0.447(2) Å, to be compared to aBO = −0.430 Å. As a confirmation, an increase in the weight of the predicates increases the standard deviation of aSE(C5). Furthermore, there are different ways to circumvent this difficulty, the simplest one being to use another set of fitted parameters including C5–O6. In that case it results in 1.428(2) Å.
| Parametera | Predicate | r SEe |
r
BOe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
r s | r 0 |
|---|---|---|---|---|---|
| a x = axial, q = equatorial. b See text and eqn (1). c Concerning discrepancy to the predicate value, see text. | |||||
| C1C2 | 1.5168(50) | 1.5182(14) | 1.5174 | 1.596(20) | 1.5220(32) |
| C2C3 | 1.5252(50) | 1.5258(14) | 1.5246 | 1.477(17) | 1.5299(32) |
| C3C4 | 1.5238(50) | 1.5226(14) | 1.5240 | 1.5205(49) | 1.5286(31) |
| C4C5 | 1.5130(50) | 1.5133(16) | 1.5148 | 1.5106(65) | 1.5147(36) |
| C1O6 | 1.4182(50) | 1.4183(14) | 1.4170 | 1.4187(33) | |
| C1O1 | 1.4079(50) | 1.4072(15) | 1.4049 | 1.4111(34) | |
| C3O3 | 1.4138(50) | 1.4145(15) | 1.4128 | 1.4180(35) | |
| C4O4 | 1.4273(50) | 1.4262(15) | 1.4249 | 1.4313(35) | |
| C1H1 | 1.0905(20) | 1.09050(70) | 1.0905 | 1.0906(15) | |
| C2H2x | 1.0906(20) | 1.09060(70) | 1.0917 | 1.0907(15) | |
| C2H2q | 1.0879(20) | 1.08789(70) | 1.0881 | 1.0879(15) | |
| C3H3 | 1.0897(20) | 1.08969(70) | 1.0904 | 1.0898(15) | |
| C4H4 | 1.0897(20) | 1.08970(70) | 1.0898 | 1.0898(15) | |
| C5H5q | 1.0872(20) | 1.08719(70) | 1.0873 | 1.0873(15) | |
| C5H5x | 1.0909(20) | 1.09090(70) | 1.0913 | 1.0910(15) | |
| O1H1 | 0.9604(20) | 0.96041(70) | 0.9601 | 0.9604(15) | |
| O3H3 | 0.9633(20) | 0.96330(70) | 0.9631 | 0.9633(15) | |
| O4H4 | 0.9624(20) | 0.96239(70) | 0.9622 | 0.9624(15) | |
| C1C2C3 | 111.72(50) | 111.747(81) | 112.20 | 110.13(74) | 111.99(18) |
| C2C3C4 | 109.69(50) | 109.559(85) | 110.12 | 110.59(55) | 109.99(19) |
| C3C4C5 | 109.89(50) | 110.107(93) | 109.70 | 109.82(34) | 109.89(21) |
| O6C1C2 | 111.70(50) | 111.755(83) | 111.56 | 111.75(18) | |
| O6C1O1 | 111.51(50) | 111.47(11) | 111.56 | 111.29(25) | |
| C2C3O3 | 111.36(50) | 111.33(13) | 111.16 | 111.50(30) | |
| C3C4O4 | 109.73(50) | 109.43(10) | 109.73 | 110.13(23) | |
| O6C1H1 | 104.09(50) | 104.09(17) | 104.02 | 104.08(39) | |
| C1C2H2x | 108.35(50) | 108.36(17) | 108.17 | 108.35(39) | |
| C1C2H2q | 109.78(50) | 109.79(17) | 109.84 | 109.80(39) | |
| C2C3H3 | 109.56(50) | 109.56(17) | 109.62 | 109.60(39) | |
| C3C4H4 | 109.46(50) | 109.46(17) | 109.55 | 109.48(39) | |
| C4C5H5q | 110.60(50) | 110.61(17) | 110.73 | 110.63(39) | |
| C4C5H5x | 110.30(50) | 110.30(17) | 110.26 | 110.32(39) | |
| C1O1H1 | 107.58(50) | 107.58(17) | 107.69 | 107.61(39) | |
| C3O3H3 | 105.66(50) | 105.67(17) | 105.72 | 105.71(39) | |
| C4O4H4 | 106.73(50) | 106.72(17) | 106.71 | 106.79(39) | |
| C1C2C3C4 | 49.87(70) | 49.67(12) | 49.05 | 52.7(12) | 49.76(28) |
| C2C3C4C5 | −53.25(70) | −53.26(14) | −52.77 | −56.4(12) | −53.38(32) |
| C5O6C1C2 | 58.65(70) | 58.93(14) | 58.45 | 59.32(32) | |
| O6C1C2C3 | −52.15(70) | −51.98(14) | −51.26 | −51.80(33) | |
| C1C2C3O3 | 172.72(70) | 172.65(15) | 171.97 | 173.43(34) | |
| C2C3C4O4 | 69.04(70) | 69.21(15) | 69.31 | 69.16(35) | |
| C5O6C1H1 | 178.53(70) | 178.54(24) | 178.46 | 178.50(55) | |
| O6C1C2H2q | −174.79(70) | −174.80(24) | −174.10 | −174.87(55) | |
| O6C1C2H2x | 67.79(70) | 67.79(24) | 68.62 | 67.88(55) | |
| C1C2C3H3 | −69.43(70) | −69.43(24) | −70.33 | −69.44(55) | |
| C2C3C4H4 | −174.54(70) | −174.55(24) | −174.18 | −174.49(55) | |
| C3C4C5H5q | 175.88(70) | 175.88(24) | 176.03 | 175.95(55) | |
| C3C4C5H5x | −63.19(70) | −63.17(24) | −62.75 | −63.31(55) | |
| O6C1O1H1 | −61.00(70) | −61.01(24) | −61.33 | −60.99(55) | |
| C2C3O3H3 | −78.67(70) | −78.67(24) | −79.40 | −78.65(55) | |
| C3C4O4H4 | −86.15(70) | −86.12(24) | −85.59 | −86.17(55) | |
| Derived parameters | |||||
| C5O6 | 1.4289 | 1.4186(22)c | 1.4268 | 1.4347(78) | 1.4473(51) |
| C4C5O6 | 110.07 | 110.06(10) | 110.00 | 110.33(47) | 110.02(23) |
| C5O6C1 | 112.80 | 112.609(93) | 112.71 | 113.02(46) | 112.72(21) |
| O1C1C2 | 107.79 | 107.64(12) | 107.68 | 108.05(29) | |
| O3C3C4 | 110.67 | 110.86(12) | 110.44 | 111.07(27) | |
| C3C4C5O6 | 59.13 | 59.48(16) | 59.54 | 58.69(72) | 59.00(37) |
| C4C5O6C1 | −62.38 | −62.44(16) | −62.97 | −61.39(69) | −62.06(38) |
| O1C1C2C3 | 70.69 | 70.73(14) | 71.11 | 70.96(33) | |
| O4C4C3O3 | −54.21 | −54.05(21) | −53.93 | −54.77(48) | |
To further check the accuracy of the equilibrium structure of deoxyribose, it was also calculated at the CCSD(T)-FC/cc-pVTZ level of theory. The small effect of further basis set enlargement (cc-pVTZ → cc-pVQZ) was then estimated at the MP2 level. The core-core and core-valence correlation correction was computed at the MP2 level using the cc-pwCVTZ basis set. The resulting rBOe estimate was:
| rBOe = re(CCSD(T)-FC/cc-pVTZ) + re(MP2-FC/cc-pVQZ) − re(MP2-FC/cc-pVTZ) + re(MP2-AE/cc-pwCVTZ) − re(MP2-FC/cc-pwCVTZ) | (1) |
The results of the different theoretical calculations are given in Table S4 (ESI†), and the derived rBOe structure is compared in Table 3 to the rSEe structure. For the bond lengths the largest difference is 0.002 Å for the C1–O1 bond. The largest differences in the bond and dihedral angles are 0.56° for C2–C3–C4 and 0.90° for C1–C2–C3–C4. The standard deviations (calculated from the MAD) are 0.0011 Å, 0.17°, and 0.75° for bond lengths, angles and dihedrals, respectively. This calculation confirms that the uncertainties chosen for the predicates are correct and that the rSEe structure is accurate. It has to be noted that for the angles C2–C3–C4 and C2–C3–C4–C5, the predicate values are closer to the rBOe structure than to the rSEe structure. This finding means that the small discrepancy is due to the semiexperimental rotational constants, not to the predicates.
The same procedure was used to calculate the semiexperimental structure of fructose. In the final fit the predicates for the bond distances connecting two substituted atoms in the set of experimental isotopologues were given a larger error of 0.005 Å instead of 0.002 Å. The predicates for the bond angles defined by three substituted atoms were given an error of 1.5° instead of 0.5°. Finally, the predicates for the torsional angles defined by four substituted atoms were given an error of 2.0° instead of 0.7°. This final fit is almost identical to the fit where the predicates have a larger weight. As a further check, the uncertainties of the predicates for the bond lengths of the heavy atoms have been increased by a factor 1.5. Introducing this change decreases the leverages but has no significant effect on the values of the fitted parameters. This observation gives us confidence in the accuracy of the derived results. The final structural parameters are given in Table 4 (Cartesian coordinates in Table S7, ESI†).
| Predicate | r SEe | r s | r 0 | |
|---|---|---|---|---|
| a For definition of HS and HR atoms, see Fig. 1. | ||||
| O1H1′ | 0.9617(20) | 0.96170(57) | 0.9617(13) | |
| C1O1 | 1.4193(20) | 1.41922(57) | 1.4195(13) | |
| C2C1 | 1.5182(50) | 1.5180(12) | 1.5244(29) | 1.5244(29) |
| H1SC1 | 1.0858(50) | 1.0853(13) | 1.0853(31) | 1.0853(31) |
| H1RC1 | 1.0904(50) | 1.0918(13) | 1.0920(30) | 1.0920(30) |
| O6C2 | 1.4109(50) | 1.4102(13) | 1.4125(13) | |
| H2′O2 | 0.9667(20) | 0.96670(57) | 0.9667(13) | |
| C3C2 | 1.5210(50) | 1.5206(13) | 1.5250(30) | 1.5250(30) |
| O3C3 | 1.4190(20) | 1.41896(57) | 1.4191(13) | |
| H3′O3 | 0.9639(20) | 0.96390(57) | 0.9639(13) | |
| H3C3 | 1.0888(20) | 1.08880(57) | 1.0888(13) | |
| C4C3 | 1.5183(50) | 1.5185(13) | 1.5212(31) | 1.5212(31) |
| O4C4 | 1.4190(20) | 1.41896(57) | 1.4194(13) | |
| H4′O4 | 0.9624(20) | 0.96240(57) | 0.9624(13) | |
| H4C4 | 1.0950(20) | 1.09496(57) | 1.0950(13) | |
| O2C2 | 1.4116(20) | 1.41169(57) | 1.4141(29) | 1.4141(29) |
| C6O6 | 1.4267(50) | 1.4263(13) | 1.4295(31) | 1.4295(31) |
| H6SC6 | 1.0866(20) | 1.08655(57) | 1.0866(13) | |
| H6RC6 | 1.0922(20) | 1.09215(57) | 1.0922(13) | |
| C5C6 | 1.5135(50) | 1.5136(13) | 1.5169(31) | 1.5169(31) |
| O5C5 | 1.4146(20) | 1.41460(57) | 1.4152(13) | |
| H5′O5 | 0.9629(20) | 0.96290(57) | 0.9629(13) | |
| H5C5 | 1.0961(20) | 1.09611(57) | 1.0961(13) | |
| C2C1O1H1′ | −65.36(70) | −65.45(14) | −65.43(34) | −65.43(34) |
| H1SC1O1H1′ | 175.71(70) | 175.78(17) | 175.48(40) | 175.48(40) |
| H1RC1O1H1′ | 55.80(70) | 55.83(17) | 56.07(40) | 56.07(40) |
| O2C2C1O1 | −52.79(70) | −52.82(17) | −52.52(41) | −52.52(41) |
| H2′O2C2C1 | 36.40(70) | 36.40(20) | 36.42(46) | 36.42(46) |
| C3C2C1O1 | −171.78(70) | −171.94(14) | −172.07(33) | |
| O3C3C2C1 | 64.33(70) | 64.38(19) | 64.20(44) | 64.20(44) |
| H3′O3C3C2 | 46.87(70) | 46.87(20) | 46.88(46) | 46.88(46) |
| H3C3C2C1 | −53.37(70) | −53.37(20) | −53.34(46) | −53.34(46) |
| C4C3C2C1 | −172.5(20) | −172.22(28) | −171.96(66) | −171.96(66) |
| O4C4C3C2 | 173.72(70) | 173.62(19) | 173.69(44) | 173.69(44) |
| H4′O4C4C3 | 43.89(70) | 43.88(20) | 43.89(46) | 43.89(46) |
| H4C4C3C2 | −64.83(70) | −64.82(20) | −64.83(46) | −64.83(46) |
| O6C2C1O1 | 67.63(70) | 67.75(16) | 67.70(39) | 67.70(39) |
| C6O6C2C1 | −179.64(70) | −179.76(15) | −180.57(34) | |
| H6SC6O6C2 | −177.46(70) | −177.46(20) | −177.48(46) | |
| H6RC6O6C2 | 64.54(70) | 64.53(20) | 64.54(46) | 64.54(46) |
| C5C6O6C2 | −57.7(20) | −57.83(22) | −59.31(52) | −59.31(52) |
| O5C5C6O6 | −67.10(70) | −67.01(16) | −67.01(39) | −67.01(39) |
| H5′O5C5C6 | 166.13(70) | 166.13(20) | 166.14(46) | 166.14(46) |
| H5C5C6O6 | 172.51(70) | 172.51(20) | 172.48(46) | 172.48(46) |
| C1O1H1′ | 106.37(50) | 106.37(14) | 106.39(33) | 106.39(33) |
| C2C1O1 | 109.38(50) | 109.36(11) | 109.74(26) | 109.74(26) |
| H1SC1O1 | 107.21(50) | 107.21(12) | 107.04(28) | |
| H1RC1O1 | 111.82(50) | 111.92(13) | 111.81(31) | |
| O2C2C1 | 109.75(50) | 109.77(13) | 109.89(31) | |
| H2′O2C2 | 105.79(50) | 105.79(14) | 105.84(33) | |
| C3C2C1 | 113.2(15) | 113.46(17) | 111.0(20) | 113.22(39) |
| O3C3C2 | 111.82(50) | 111.78(13) | 111.92(31) | |
| H3′O3C3 | 105.93(50) | 105.93(14) | 105.94(33) | |
| H3C3C2 | 108.50(50) | 108.50(14) | 108.51(33) | |
| C4C3C2 | 110.3(15) | 110.41(16) | 109.6(13) | 111.40(39) |
| O4C4C3 | 110.49(50) | 110.46(13) | 110.75(32) | |
| H4′O4C4 | 106.63(50) | 106.62(14) | 106.65(33) | |
| H4C4C3 | 108.71(50) | 108.70(14) | 108.73(33) | |
| O6C2C1 | 104.9(15) | 104.75(17) | 105.51(42) | |
| C6O6C2 | 114.69(50) | 114.72(11) | 114.84(27) | |
| H6SC6O6 | 105.81(50) | 105.81(14) | 105.83(33) | |
| H6RC6O6 | 110.15(50) | 110.15(14) | 110.17(33) | |
| C5C6O6 | 112.4(15) | 112.09(15) | 112.02(35) | |
| O5C5C6 | 108.95(50) | 108.97(13) | 109.31(31) | |
| H5′O5C5 | 106.00(50) | 106.00(14) | 106.04(33) | |
| H5C5C6 | 108.35(50) | 108.35(14) | 108.35(33) | |
| Derived parameters | ||||
| C4C5 | 1.5135 | 1.5136(40) | 1.5100(60) | 1.5347(93) |
| C3C4C5 | 110.47 | 111.01(25) | 110.70(50) | 110.22(58) |
| C4C5C6 | 109.175 | 109.52(17) | 108.60(40) | 108.49(41) |
| C2C1HS | 109.74 | 109.61(15) | 112.5(13) | 109.79(35) |
| C2C1HR | 109.26 | 109.31(14) | 106.8(15) | 109.33(33) |
| C2C3C4C5 | 54.50 | 54.05(35) | 50.0(20) | 54.26(83) |
| C3C4C5C6 | −53.27 | −52.63(33) | −52.00(90) | −53.72(77) |
| C3C2C1HS | −54.43 | −54.67(20) | −49.7(10) | −54.68(47) |
| C3C2C1HR | 65.51 | 65.21(22) | 72.8(18) | 64.96(51) |
The determined structures for the two sugars are regarded as highly accurate. The standard deviation of the fitted parameters is a reliable indicator of their precision provided that the weights were correctly chosen and systematic errors were insignificant. From the present analysis and from our previous work,8,9 it is highly likely that the weights of the predicates have reasonably correct values. On the other hand, it is much more difficult to estimate the accuracy of the semiexperimental rotational constants. Furthermore, it is known that they are affected by a non-negligible systematic error.39,40 For these reasons, a conservative estimate of the accuracy of the fitted parameters can be stated as 0.002 Å for the bond lengths, 0.2–0.6° for the bond angles, and 0.5–0.9° for the dihedral angles.
The empirical substitution structures (rs) are also given in Tables 3 and 4 for comparison. As the range of the rovibrational corrections is quite small (0.37 MHz for A, 0.19 MHz for B, and 0.10 MHz for C, see Table 2 for fructose), the rs structure might be expected to be relatively accurate. Inspection of Tables 3 and 4 shows that such accuracy is not the case. This observation is confirmed by the examination of the Cartesian coordinates of fructose given in Table S8 (ESI†). This result is common for large molecules for which the isotopic shift of the rotational constants is generally small. We note that the results remain inaccurate even when the equilibrium rotational constants are used in the Kraitchman equations.8,9,31
It is also instructive to examine the quality of the effective structure (r0). In these molecules the number of ground-state rotational constants is not sufficient to determine a complete structure without multiple structural assumptions that render the results unreliable. On the other hand, using the same predicates as for the rSEe-fits, there is no difficulty in performing structural least-squares fits. The results are given in the last column of Tables 3 and 4. Obviously, the quality of the fits is only moderately good: the standard deviations of the fits and of the fitted parameters are about three times larger than in the rSEe-fits. Furthermore, an analysis of residuals shows that, contrary to the rSEe-fit, the predicates and the ground state rotational constants are not fully compatible and the distances between the heavy atoms are rather inaccurate. Nevertheless, the angles, although not very precise, are in fair agreement with the rSEe structures. In conclusion, the r0 structure permits the determination of approximate values for the bond and dihedral angles. However, interest in these structures is limited because it is not much more accurate than the predicates. Fig. 2 shows the deviations of semiexperimental and experimental parameters of the deoxyribose ring relative to the computed values, rBOe. It can be seen that there is an excellent agreement between the rSEe and rBOe structures, whereas the discrepancies between the rBOe and experimental structures, both rs and previously determined r0,10 denoted as r0(old), as well as between the rs and improved r0 (denoted as r0(new)) structures are very large. The ME method thus allows us to improve the fit of the experimental data and to considerably increase the accuracy of the experimental structure determination.
 | ||
| Fig. 2 Histogram of absolute deviations of the rSEe, rs, r0(old data10) and r0(new data, present work) parameters relative to the rBOe values for deoxyribose. | ||
The accurate determination of the molecular structure allows us to obtain information on subtle electronic effects that are reflected in the molecular structure but are usually very difficult to notice, such as the anomeric effect. The anomeric effect is known to be present in both molecules: the hydroxy substituent on the anomeric carbon atom adjacent to the endocyclic oxygen atom prefers the axial orientation.41 Furthermore, the anomeric CO bond length is shorter than the standard single bond length, which is 1.417 Å in methanol.42 This parameter is 1.407 Å in deoxyribose and 1.410 Å in fructose. Finally, in the case of fructose, the C2–O6 bond adjacent to the anomeric C1–O1 bond is shorter (1.412 Å), whereas the O6–C6 bond is longer (1.426 Å). This result is in good agreement with the X-ray study of crystalline fructose15 and the ab initio calculations on methoxymethanol by Jeffrey et al.43
The structures of the title compounds are known to be further stabilized by intramolecular hydrogen bond networks. There are many different ways to point out the existence of a hydrogen bond.44,45 It may be defined on the basis of interaction geometries (short distances, fairly linear angles) or certain properties of the electron density distribution. Following the definition of Jeffrey44 and Steiner,45 the hydrogen bond D–H⋯A is possible if d(H⋯A) < 3.0 Å and if the angle θ = ∠(D–H⋯A) is larger than 90° or more conservatively 110°. However, if d > 2.2 Å and θ < 130°, the bond is considered as weak as is the case for pyranose and fructose. These results are in agreement with the conclusions about the low stability of the five-membered quasi-ring formed by hydrogen bond due to an unfavorable geometry of this ring (in comparison to the six-membered quasi-ring); see, for example; ref. 46.
Using this criterion, two weak H⋯O hydrogen bonds are present in deoxyribose, and in fructose there are five weak hydrogen bonds (see Fig. 1 and Table 5). Another consequence of the hydrogen bond is that the r(D–H) bond length is lengthened and that there is a correlation between r(D–H) and d(H⋯A). Indeed, there is a correlation between r(D–H) and d(H⋯A), the correlation coefficient being −0.86. This observation is consistent with r(O–H) bond lengths being longer than in methanol (0.957 Å).42 The d(H5⋯O4) in fructose is not determined accurately, and its value is likely to be too small, if this datum is eliminated, the correlation coefficient increases (in absolute value) to −0.93.
| d(H⋯O) | r(O–H) | ∠(O–H⋯O) | ||
|---|---|---|---|---|
| Deoxyribose | ||||
| H1⋯O6 | 2.514(3) | O1–H1 | 0.960(1) | 68.2 |
| H3⋯O4 | 2.257(4) | O3–H3 | 0.963(1) | 113.0 |
| H4⋯O6 | 2.366(6) | O4–H4 | 0.962(1) | 111.6 |
| Fructose | ||||
| H1⋯O6 | 2.437(4) | O1–H1 | 0.962(1) | 101.3 |
| H2⋯O1 | 2.175(6) | O2–H2 | 0.967(1) | 115.7 |
| H3⋯O2 | 2.270(7) | O3–H3 | 0.964(1) | 110.3 |
| H4⋯O3 | 2.381(7) | O4–H4 | 0.962(1) | 110.4 |
| H5⋯O4 | 2.208(8) | O5–H5 | 0.963(1) | 112.0 |
Bader's quantum theory of atoms in molecules (AIM) is frequently used to prove the existence of a hydrogen bond.47,48 According to this theory, the bond exists, if there is a point with minimal electron density along the bond path. This point is called a (3, −1) bond critical point (BCP). For detection of BCPs in deoxyribose, the required wave functions were generated for optimized geometries at the MP2 and B3LYP levels of theory with the cc-pVTZ basis set. The molecular graphs were computed with the AIM200049,50 program package, but no BCP nor associated ring critical point (RCP) could be found for the hydrogen bonds (see Fig. S1, ESI†). On the one hand, this might be explained by the fact that all the hydrogen bonds are weak. On the other hand, as it has been noted by Deshmukh et al.51,52 in the studies of alkanediols and sugars, the AIM method sometimes conflicts with experimental data. The explanation of this phenomenon requires further investigation that is not the purpose of the present study. We note that the stabilizing effects of the hydrogen bonds in fructose has been recently discussed from a theoretical point of view.53
Most of the C–C bond lengths are only slightly shorter than the value found for ethane, 1.522 Å.54 They are thus typical single bonds.55 However, the C4–C5 bond in deoxyribose at 1.513 Å and the C5–C6 bond in fructose at 1.514 Å are rather short, as seems to be the rule in aldohexoses for bonds that involve a C atom next to the ring O atom.15
4. Conclusions
We have demonstrated that the mixed regression method is more suitable for the accurate determination of the equilibrium structure of a moderately large molecule than either the pure high-level ab initio methods or the classical semiexperimental method. Another typical example of the superiority of this method is the structure of tropinone (34 degrees of freedom).31 The ME method combines two steps. First, high- or medium-level ab initio calculations furnish accurate values for the X–H bond lengths (X = C, N, O) and for bond angles, and more approximate values for the dihedral angles and for the distances between heavy atoms. Then, these data are supplemented by semiexperimental equilibrium rotational constants in a least-squares fit that allows us to check that the predicates are accurate and to improve their accuracy.Further work on the ME method will be directed to larger molecular systems, exploiting the synergy between experimental high-resolution rotational data and quantum chemical calculations.
Acknowledgements
JD, NV and JV thank the Dr Barbara Mez-Starck Foundation (Germany), AL and EJC thank the Spanish MINECO for funding (CTQ2014-54464-R, CTQ2015-68148-C2) and EJC thanks the MINECO for a Ramón y Cajal contract. The authors thank Prof. Norman C. Craig for very helpful suggestions.References
- J. Demaison, in Equilibrium Molecular Structures: From Spectroscopy to Quantum Chemistry, ed. J. Demaison, J. E. Boggs and A. G. Császár, CRC Press, Boca Raton, 2011, pp. 29–52 Search PubMed.
- J. Vázquez and J. F. Stanton, in Equilibrium Molecular Structures: From Spectroscopy to Quantum Chemistry, ed. J. Demaison, J. E. Boggs and A. G. Császár, CRC Press, Boca Raton, 2011, pp. 53–87 Search PubMed.
- K. L. Bak, J. Gauss, P. Jørgensen, J. Olsen, T. Helgaker and J. F. Stanton, J. Chem. Phys., 2001, 114, 6548–6556 CrossRef.
- F. Pawłowski, P. Jørgensen, J. Olsen, F. Hegelund, T. Helgaker, J. Gauss, K. L. Bak and J. F. Stanton, J. Chem. Phys., 2002, 116, 6482–6496 CrossRef.
- W. D. Allen, E. Czinki and A. G. Császár, Chem. – Eur. J., 2004, 10, 4512–4517 CrossRef PubMed.
- D. A. Belsley, Conditioning Diagnostics, Wiley, New York, 1991, pp. 298–299 Search PubMed.
- L. S. Bartell, D. J. Romanesko and T. C. Wong, Chemical Society Specialist Periodical Report No. 20, ed. G. A. Sim and L. E. Sutton, The Chemical Society, London, 1975, vol. 3, pp. 72–79 Search PubMed.
- N. C. Craig, J. Demaison, P. Groner, H. D. Rudolph and N. Vogt, J. Phys. Chem. A, 2015, 119, 195–204 CrossRef PubMed.
- J. Demaison, N. C. Craig, P. Groner, P. Écija, E. J. Cocinero, A. Lesarri and H. D. Rudolph, J. Phys. Chem. A, 2015, 119, 1486–1493 CrossRef PubMed.
- I. Peña, E. J. Cocinero, C. Cabezas, A. Lesarri, S. Mata, P. Écija, A. M. Daly, Á. Cimas, C. Bermúdez, F. J. Basterretxea, S. Blanco, J. A. Fernández, J. C. López, F. Castaño and J. L. Alonso, Angew. Chem., Int. Ed., 2013, 52, 11840–11845 CrossRef PubMed.
- E. J. Cocinero, A. Lesarri, P. Écija, Á. Cimas, B. G. Davis, F. J. Basterretxea, J. A. Fernández and F. Castaño, J. Am. Chem. Soc., 2013, 135, 2845–2852 CrossRef PubMed.
- C. Bermúdez, I. Peña, C. Cabezas, A. M. Daly and J. L. Alonso, ChemPhysChem, 2013, 14, 893–895 CrossRef PubMed.
- P. C. Collins and R. J. Ferrier, Monosaccharides: Their Chemistry and Their Roles in Natural Products, Wiley, New York, 1995 Search PubMed.
- S. Furberg, Acta Chem. Scand., 1960, 14, 1357–1363 CrossRef.
- J. A. Kanters, G. Roelofsen, B. P. Alblas and I. Meinders, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 665–672 CrossRef.
- E. J. Cocinero, A. Lesarri, P. Écija, J.-U. Grabow, J. A. Fernández and F. Castaño, Phys. Chem. Chem. Phys., 2010, 12, 12486–12493 RSC.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- K. A. Peterson and T. H. Dunning, Jr., J. Chem. Phys., 2002, 117, 10548–10560 CrossRef.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, 1133–1138 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- G. D. Purvis, III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. C.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, A. Mitrushenkov, G. Rauhut, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, G. Hetzer, T. Hrenar, G. Knizia, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO program package, 2009 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef.
- H. M. Jaeger, H. F. Schaefer, III, J. Demaison, A. G. Császár and W. D. Allen, J. Chem. Theory Comput., 2010, 6, 3066–3078 CrossRef PubMed.
- H. D. Rudolph, J. Demaison and A. G. Császár, J. Phys. Chem. A, 2013, 117, 12969–12982 CrossRef PubMed.
- J. Demaison, N. C. Craig, E. J. Cocinero, J.-U. Grabow, A. Lesarri and H. D. Rudolph, J. Phys. Chem. A, 2012, 116, 8684–8692 CrossRef PubMed.
- J. Demaison and A. G. Császár, J. Mol. Struct., 2012, 1023, 7–14 CrossRef.
- N. Vogt, J. Demaison, W. Geiger and H. D. Rudolph, J. Mol. Spectrosc., 2013, 288, 38–45 CrossRef.
- N. Vogt, E. P. Altova, D. N. Ksenafontov and A. N. Rykov, Struct. Chem., 2015, 26, 1481–1488 CrossRef.
- N. Vogt, J. Demaison and H. D. Rudolph, Struct. Chem., 2011, 22, 337–343 CrossRef.
- N. Vogt, L. S. Khaikin, O. E. Grikina and A. N. Rykov, J. Mol. Struct., 2013, 1050, 114–121 CrossRef.
- J. Demaison, H. D. Rudolph and A. G. Császár, Mol. Phys., 2013, 111, 1539–1562 CrossRef.
- C. Puzzarini and V. Barone, Phys. Chem. Chem. Phys., 2011, 13, 7189–7197 RSC.
- N. Vogt, J. Vogt and J. Demaison, J. Mol. Struct., 2011, 988, 119–127 CrossRef CAS.
- N. Vogt, J. Demaison, J. Vogt and H. D. Rudolph, J. Comput. Chem., 2014, 35, 2333–2342 CrossRef CAS PubMed.
- E. Juaristi and G. Cuevas, Tetrahedron, 1992, 48, 5019–5087 CrossRef CAS.
- J. Demaison, M. Herman and J. Liévin, Int. Rev. Phys. Chem., 2007, 26, 391–420 CrossRef CAS.
- G. A. Jeffrey, J. A. Pople and L. Radom, Carbohydr. Res., 1974, 38, 81–95 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2001, 105, 10462–10467 CrossRef CAS.
- R. F. W. Bader, Atoms In Molecules, Oxford University Press, New York, 1990 Search PubMed.
- P. L. A. Popelier and R. F. W. Bader, Chem. Phys. Lett., 1992, 189, 542–548 CrossRef CAS.
- F. Biegler-König, J. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef PubMed.
- M. M. Deshmukh, N. V. Sastry and S. R. Gadre, J. Chem. Phys., 2004, 121, 12402–12410 CrossRef CAS PubMed.
- M. M. Deshmukh, L. J. Bartolotti and S. R. Gadre, J. Phys. Chem. A, 2008, 112, 312–321 CrossRef CAS PubMed.
- M. M. Deshmukh, S. R. Gadre and E. J. Cocinero, New J. Chem., 2015, 39, 9006–9018 RSC.
- C. Puzzarini and P. R. Taylor, J. Chem. Phys., 2005, 122, 054315 CrossRef PubMed.
- N. Vogt, L. S. Khaikin, O. E. Grikina, A. N. Rykov and J. Vogt, J. Phys. Chem. A, 2008, 112, 7662–7670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S8 and Fig. S1. See DOI: 10.1039/c6cp01842d |
| This journal is © the Owner Societies 2016 |