
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive picture of the ultrafast excited-state dynamics of retinal†
Oliver
Flender
,
Mirko
Scholz
,
Jonas
Hölzer
,
Kawon
Oum
* and
Thomas
Lenzer
*
Universität Siegen, Physikalische Chemie, Adolf-Reichwein-Str. 2, 57076 Siegen, Germany. E-mail: oum@chemie.uni-siegen.de; lenzer@chemie.uni-siegen.de
First published on 18th May 2016
Abstract
All-trans retinal is the chromophore of microbial rhodopsins initiating energy conversion and cellular signalling by subpicosecond photoinduced switching. Here, we provide detailed UV-Vis transient absorption experiments to disentangle the complex photochemistry of this polyene, which is governed by its terminal aldehyde group. After photoexcitation to the S2(1Bu+) state, the system exhibits polarity-dependent branching, populating separate S1(1Ag−) and intramolecular charge transfer (ICT) species. In all solvents, population of a singlet nπ* state from S1 is observed which represents the precursor of the T1 triplet state. While triplet formation dominates in nonpolar solvents (67% quantum yield), it is dramatically reduced in polar solvents (4%). The channel closes completely upon replacing the aldehyde by a carboxyl group, due to an energetic up-shift of 1nπ*. In that case, internal conversion via the ICT species becomes the main pathway, with preferential formation of the initially excited isomer.
1. Introduction
Derivatives of retinal play a key role as chromophores in rhodopsin photoreceptor proteins and fulfil diverse functions, such as energy conversion, cellular signalling, vision and photopigment regeneration.1–3 In microbial rhodopsins, all-trans retinal (= all-trans β-apo-15-carotenal, compound 1 in Fig. S1, ESI†) is connected as a protonated Schiff base (PSB) to an opsin apoprotein. Upon photoexcitation, the all-trans PSB undergoes excited-state photoisomerisation to the 13-cis isomer (compound 2 in Fig. S1, ESI†). Characterising light-triggered processes of retinyl chromophores and the influence of the environment on their dynamics is therefore of prime importance to understand their function in vivo.In the simplest model, the photoinduced dynamics of carotenoids involve three singlet electronic states: the S0(1Ag−) ground state, the one-photon-forbidden S1(1Ag−) first excited state, and the optically “bright” S2(1Bu+) second excited state absorbing in the blue-green spectral region. In apocarotenals, the conjugated terminal aldehyde group alters the electronic properties drastically, especially those of the S1 state. For instance, systems such as β-apo-12′-carotenal, show a strong decrease of the excited-state lifetime with increasing solvent polarity, in contrast to nonpolar carotenoids.4–7 This is ascribed to the formation of considerable intramolecular charge transfer (ICT) character in the S1 state.8–10
Earlier studies suggest that the photochemistry of retinal is more complex. For instance, Yamaguchi and Hamaguchi performed ultrafast transient absorption spectroscopy of all-trans retinal in n-hexane and evaluated their data based on a kinetic model involving branching in the S1 state:11 after S2 → S1 internal conversion (IC), S1 is depopulated via the “1nπ*/triplet channel” S1 → 1nπ* → T1 → S0, forming exclusively the all-trans isomer12 with 74% quantum yield, and via the “photoisomerisation/IC channel” S1 → p* → p → S0 involving perpendicular (90°) excited-state and ground state configurations p* and p, which lead to formation of trans and cis isomers. In a follow-up study, they reported transient absorption spectra in n-butanol,13 where the lifetime of the S1 state was 1.6 ps, and thus slower than in n-hexane (0.56 ps). In n-butanol, stimulated emission (SE) was observed above 620 nm, in agreement with SE kinetics previously detected in other polar solvents.14,15 Later on, Polívka et al.7 ascribed this red emission to an ICT state, based on an analogous assignment of SE features observed for β-apo-12′-carotenal, β-apo-8′-carotenal5–7 and β-apo-12′-carotenoic acid.16
Although these experiments have improved the understanding of retinal photochemistry, important pieces of information are still missing, such as (a) the complete spectral characterisation and ultrafast dynamics of the 1nπ* state in different solvents, (b) the determination of solvent-dependent lifetimes and quantum yields for the processes in the 1nπ*/triplet and photoisomerisation/IC channels, (c) the possible influence of ICT character in the excited electronic state manifold, and (d) an explanation for the fast excited-state decay of retinal which is a priori unexpected for such a short-chain polyene species. We will address all of these points and arrive at a comprehensive description of the excited-state dynamics of all-trans retinal, with the aid of complementary experiments for the closely related all-trans retinoic acid (compound 3 in Fig. S1, ESI†) and 13-cis retinal.
2. Experimental
2.1 Substances
All-trans retinal and all-trans retinoic acid were kindly provided in double-recrystallized form by BASF SE (Prof. Bernd Schäfer), 13-cis retinal was purchased from Toronto Research Chemicals and used without further purification. Purities were >97% for the all-trans retinoids, 95% for 13-cis retinal, and 99% or better for the organic solvents.2.2 Pump – supercontinuum probe (PSCP) spectroscopy
Ultrafast broadband transient absorption experiments were carried out using UV-Vis pump – supercontinuum probe spectroscopy at 920 Hz repetition frequency.17–19 All-trans retinal was excited by 400 nm pulses at every second laser shot (laser pulse fluence ca. 0.23 mJ cm−2). Extended spectral coverage into the deeper UV region was achieved by employing a 400 nm-seeded multifilament supercontinuum generated in a 2 mm thick translating CaF2 plate as probe pulse (260–660 nm, beam diameter ca. 130 μm). The relative pump–probe polarization was set at magic angle (54.7°). The pump–probe intensity cross-correlation time varied in the range 57–84 fs in the different experiments, with a time zero accuracy of ca. 10 fs. To avoid any influence of photoinduced sample degradation, 10–15 mL of a nitrogen-saturated solution of all-trans retinal, 13-cis retinal or all-trans retinoic acid in the organic solvent of interest was passed through a flow cell (path length 1 mm, window thickness 200 μm), so that the sample volume was exchanged after each pump–probe cycle. Solutions with an OD of 0.2 (at 400 nm, 1 mm path length) were employed. Under these conditions, the transient absorption signals were in the linear regime, as confirmed in separate experiments (Fig. S2–S7, ESI†). Steady-state absorption spectra of the sample solutions were recorded on a Varian Cary 5000 dual-beam spectrometer.2.3 Data processing and global kinetic analysis procedure
The PSCP spectra were chirp-corrected by using the coherent response of the respective pure solvents. The coherent solvent signals were in all cases subtracted from the transient absorption data of retinal to obtain the pure chromophore dynamics (Fig. S8, ESI†).Each data set, consisting of 512 kinetic traces, was subjected to a global modelling procedure employing the kinetic scheme discussed later on. Species-associated spectra were parametrised by a sufficiently large number of Gaussian functions. During the modelling, the known, separately measured absolute S0 absorption spectra for the trans/cis isomers of retinal and retinoic acid were kept fixed. In addition, the following boundary conditions based on existing experimental data were imposed: total isomerisation quantum yields of Φcis = 12% (n-hexane, n-hexadecane) and Φcis = 33% (ethanol), based on the measurements of Waddell et al. and Ganapathy and co-workers,20,21 were employed with an allowed variation of ±15%. Note that this only slightly influences the amplitude of the T1 triplet spectrum in the overlap region with the S0 ground state bleach (GSB), with the T1 peak being unaffected. Next we assumed that all-trans retinal does not isomerise in the triplet channel, as found in independent transient FTIR experiments of Yuzawa and Hamaguchi.12 Finally, absolute peak absorption coefficients (all in units of M−1 cm−1) for the T1 triplet band are available from pulsed laser photolysis experiments of all-trans retinal in a few solvents, albeit partially with either unknown or substantial uncertainties: 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (n-hexane),22–25 78600 (THF),26 68000 (methanol)26,27 and 63000 (acetonitrile).28 During the fitting procedure, the peak value of the fitted T1 spectrum was forced to stay within ±10% of these values.
000 (n-hexane),22–25 78600 (THF),26 68000 (methanol)26,27 and 63000 (acetonitrile).28 During the fitting procedure, the peak value of the fitted T1 spectrum was forced to stay within ±10% of these values.
The parameters, such as the position, width and height of the Gaussian functions describing the excited-state spectra and the rate constants, were optimised simultaneously to arrive at the best fit, which included convolution with the experimental time response, the latter one being determined from the pure solvent signal.
2.4 DFT/TDDFT calculations
Calculations for all-trans retinal and all-trans retinoic acid were performed based on the procedures described in our previous publications:29–31 the structure of the ground electronic state was optimized using DFT employing the B3LYP functional and a 6-31G(d) basis set. The energies of the three lowest excited states (vertical excitation) were determined by TDDFT using SVWN, BLYP, B3LYP, MPW1K and CAM-B3LYP functionals and a 6-31+G(d) basis set.3. Results and discussion
3.1 Ultrafast dynamics of all-trans retinal and all-trans retinoic acid in different solvents
UV-Vis steady-state absorption spectra of all-trans retinal and 13-cis retinal in various solvents are summarised in the ESI† (Fig. S9). The S0(1Ag−) → S2(1Bu+) band maximum is located slightly below 400 nm. The spectrum of the all-trans species has a larger absorption coefficient and red-shift than 13-cis. A discussion of the solvent-dependence of the absorption spectra is included in the ESI† (Fig. S9–S13), with the main finding that several factors, such as increasing solvent polarisability, polarity and hydrogen-bonding capability lead to an increased spectral red-shift.The 400 nm pump pulse used in the time-resolved experiments excites predominantly the S0 → S2 transition, with the weakly absorbing S1(1Ag−) state estimated to lie 2300 cm−1 below S2.32Fig. 1 shows transient UV-Vis PSCP broadband absorption spectra of all-trans retinal and all-trans retinoic acid in n-hexane. Fig. 2 compares the solvent dependence of the PSCP spectra of all-trans retinal in n-hexadecane, diisopropyl ether, THF and methanol as contour plots (corresponding transient spectra are included in the ESI,† Fig. S14–S19). The initially populated S2 state shows pronounced excited-state absorption (ESA) with a peak at 600 nm accompanied by an S0 → S2 GSB centred below 400 nm. Four central aspects are immediately clear upon inspection of the subsequent spectral development:
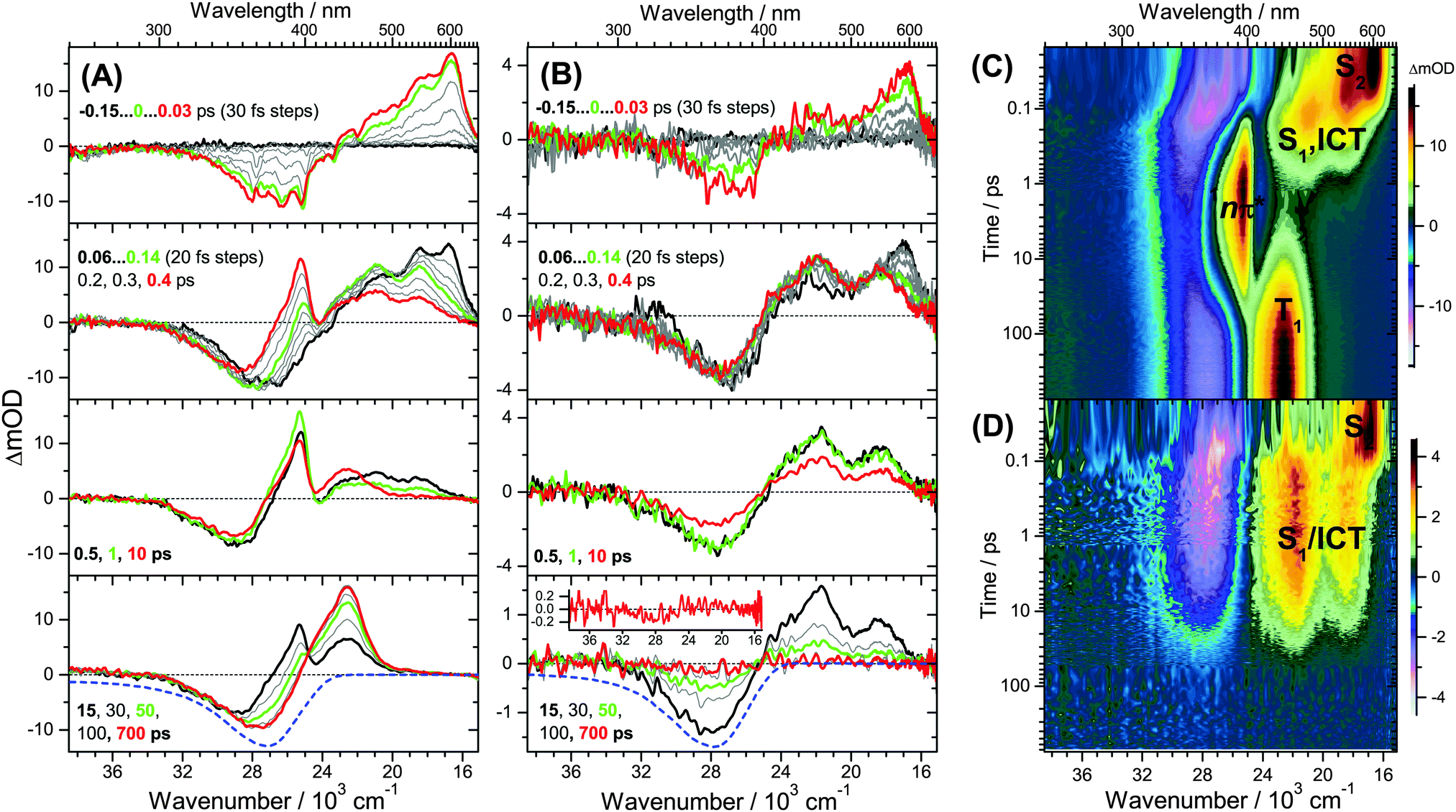 | ||
| Fig. 1 Transient UV-Vis PSCP absorption spectra of all-trans retinal (A) and all-trans retinoic acid (B) in n-hexane upon excitation at 400 nm. (Top panel) 0.15–0.03 ps with 30 fs steps; (2nd panel) 0.06–0.14 ps with 20 fs steps, and 0.2, 0.3 and 0.4 ps; (3rd panel) 0.5, 1 and 10 ps; (bottom panel) 15, 30, 50, 100 and 700 ps (inset in (B): magnified spectrum at 700 ps). Selected transient spectra are shown as thick coloured lines for guidance. The blue-dashed lines in the bottom panels are the scaled and inverted steady-state absorption spectra. The corresponding contour plots for all-trans retinal (C) and for all-trans retinoic acid (D) are presented with the symbols of the electronically excited species used in the global kinetic analysis. The concentration of retinoic acid was kept low (3 × 10−5 M) to avoid any influence of dimer formation on the spectra. This leads to a reduced signal-to-noise ratio. | ||
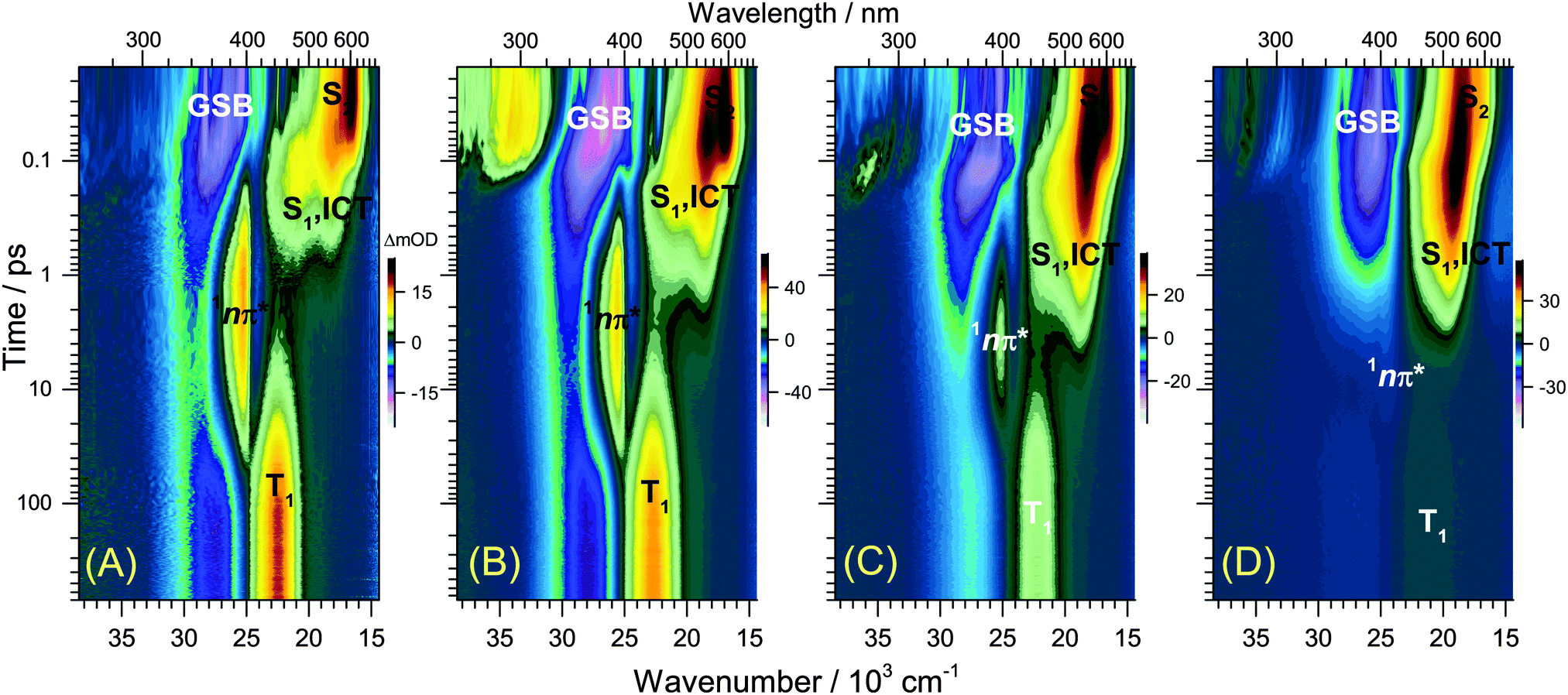 | ||
| Fig. 2 Contour plots of transient UV-Vis PSCP absorption spectra of all-trans retinal in n-hexadecane (A), diisopropyl ether (B), THF (C) and methanol (D) upon S0 → S2 excitation at 400 nm. Note the gradual disappearance of the 1nπ*/triplet channel with increasing solvent polarity. The absorption feature around 300 nm in (B) at short times (100 fs) is due to solvent contributions which cannot be removed perfectly by subtraction of the pure solvent spectrum. | ||
(a) For retinal in n-hexane, there is the emergence of a sharp absorption band centred at 396 nm (Fig. 1(A), second panel, and (C)). It is assigned to ESA of the 1nπ* singlet state formed after intermediate population of the S1 state (see below). The 1nπ* state represents the precursor of the T1 triplet state, and the latter one is formed at later times with a characteristic band peaking at 443 nm,26 see Fig. 1(A), bottom panel, and (C).
(b) The 1nπ*/T1 channel is closing down completely upon replacement of the terminal aldehyde group (retinal) by a carboxyl function (retinoic acid), see Fig. 1(B), second panel. There are no spectral fingerprints of the 1nπ* and T1 states.
(c) Spectral features of the 1nπ* state of all-trans retinal become weaker with increasing solvent polarity, see Fig. 2(A–D) and Fig. S14–S19 (ESI†). This suggests a strong reduction of the quantum yield for 1nπ* and T1 formation. Still, in methanol and acetonitrile, weak 1nπ* and well-visible T1 spectral features are present. A comparison of the solvent-dependent band shapes is shown in Fig. 3. In (A), at 0.17 ps the 1nπ* ESA band shows a sharp peak at 396 nm from nonpolar solvents up to diisopropyl ether. The band becomes much weaker in more polar solvents and also develops at later time (Fig. S16–S19, ESI†), already suggesting delayed formation of 1nπ* which is confirmed by the global analysis below. In (B), the long-lived T1 → Tn absorption develops from a narrow band in n-hexadecane/n-hexane into a broader band with a long-wavelength tail for polar solvents, such as methanol or acetonitrile.
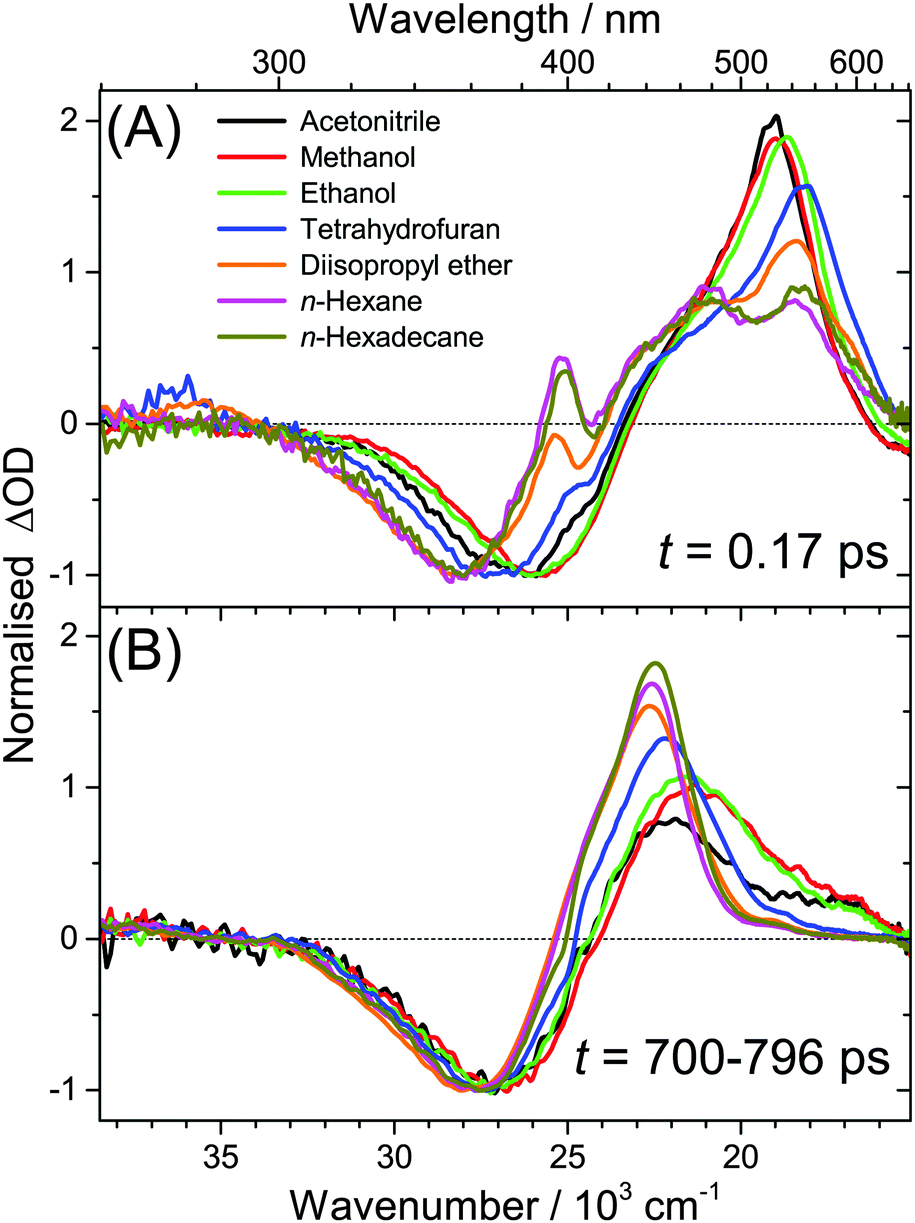 | ||
| Fig. 3 Comparison of broadband transient absorption spectra of all-trans retinal in the organic solvents n-hexadecane, n-hexane, diisopropyl ether, THF, ethanol, methanol and acetonitrile. Spectra are normalised at the maximum GSB amplitude. (A) Spectra at t = 0.17 ps: here, the spectral features of the S1 and ICT species (above 400 nm) and the rise of the 1nπ* absorption (around 396 nm) are observed. (B) Spectra averaged over the time range 700–796 ps: the long-lived T1 → Tn triplet absorption band above 400 nm is clearly visible. | ||
(d) The ESA band of all-trans retinoic acid (Fig. 1(B), second panel) featuring two peaks at 470 and 550 nm arises from an S1/ICT species, which is populated from S2, see below. A very similar ESA band is also observed for all-trans retinal, with peaks located at 480 and 540 nm (Fig. 1(A), second panel). The appearance of the S1 and ICT bands of all-trans retinal changes drastically with increasing polarity: from a double-peak structure in nonpolar solvents to a pronounced single peak band with a blue shoulder and an SE feature to the red (Fig. 3(A)). Most importantly, the kinetics in this ESA band exhibit a biexponential decay (e.g. at 530 nm), strongly suggesting that two distinguishable species contribute in this spectral region which decay independently via separate relaxation channels.
3.2 Kinetic model
Several kinetic schemes were explored to model the transient spectra of retinal. The best model reproducing all details of the dynamics is found in Fig. 4(A). As a central feature of the mechanism, separate S1 and ICT species are populated from S2 by IC. S1 and ICT decay independently to the ground electronic state, forming all-trans and cis isomers in S0. The S1 state is also the precursor of the 1nπ* state which forms the long-lived T1 state (all-trans isomer) by ISC. The resulting time constants and quantum yields are summarized in Table S2, ESI.† Key parameters, such as the polarity dependence of quantum yields and selected time constants are highlighted in Fig. 4(B–E). Species-associated spectra (SAS) featuring absolute absorption coefficients are shown in Fig. 5. Additional SAS, species contributions to the spectra at selected times and kinetic traces including simulations are included in the ESI† (Fig. S20–S24).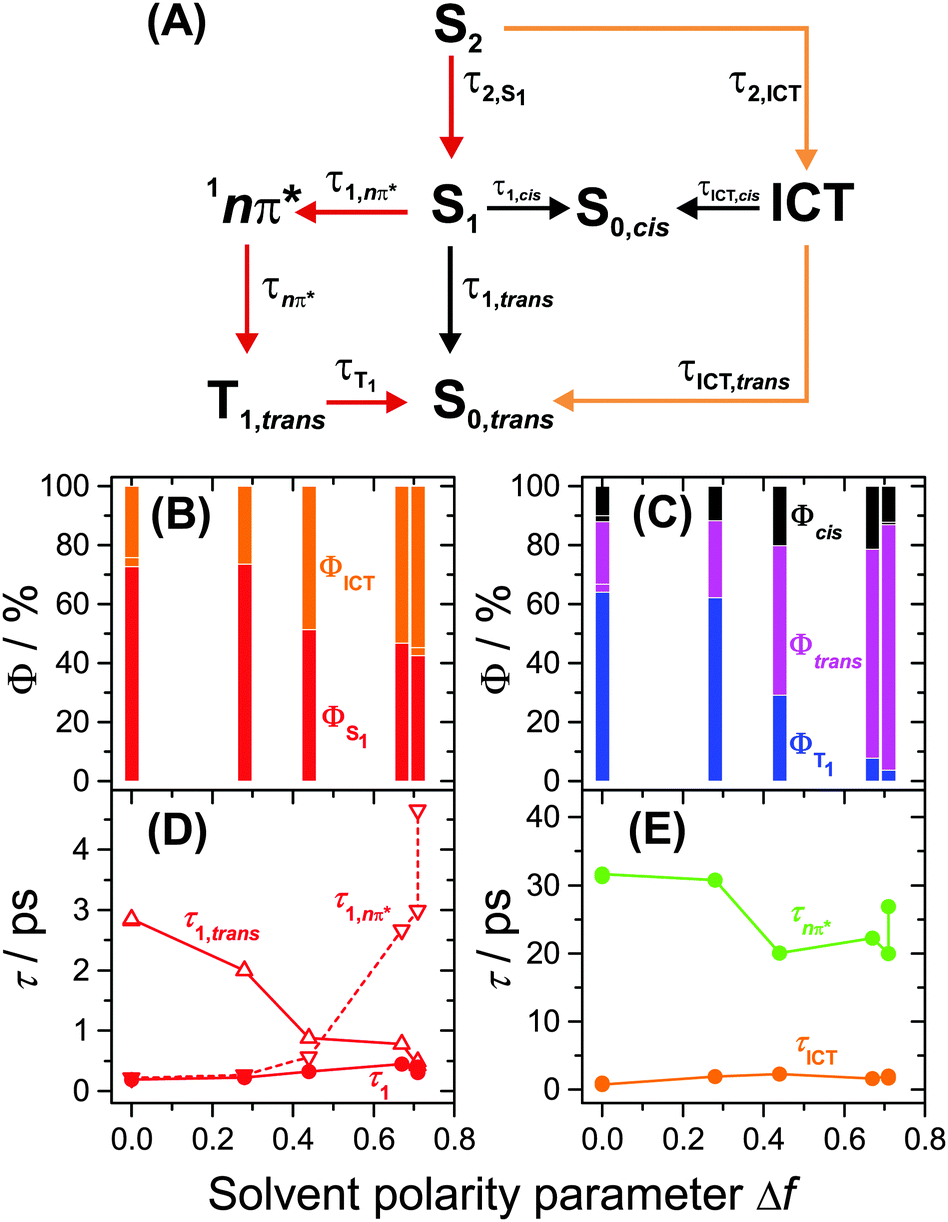 | ||
| Fig. 4 Kinetic model for all-trans retinal photochemistry and key parameters of the global kinetic analysis. (A) Kinetic model: S2 is initially populated from S0,trans by photoexcitation. The 13-cis isomer is the predominant cis isomerisation product, and according to the experiments of Yuzawa and Hamaguchi,12 the T1 species has an all-trans configuration. The red pathway dominates in nonpolar solvents, whereas the orange one prevails in highly polar solvents. (B) Branching ratios from S2 into S1 (red) and ICT (orange). (C) Branching ratios for formation of T1 (blue), S0,all-trans (magenta) and S0,cis (black). (D) Total lifetime τ1 of the S1 state and its contributions from the 1nπ*/T1 channel (τ1,nπ*) and internal conversion channel to S0,trans (τ1,trans). (E) Total lifetime of ICT (orange) and 1nπ* (green). | ||
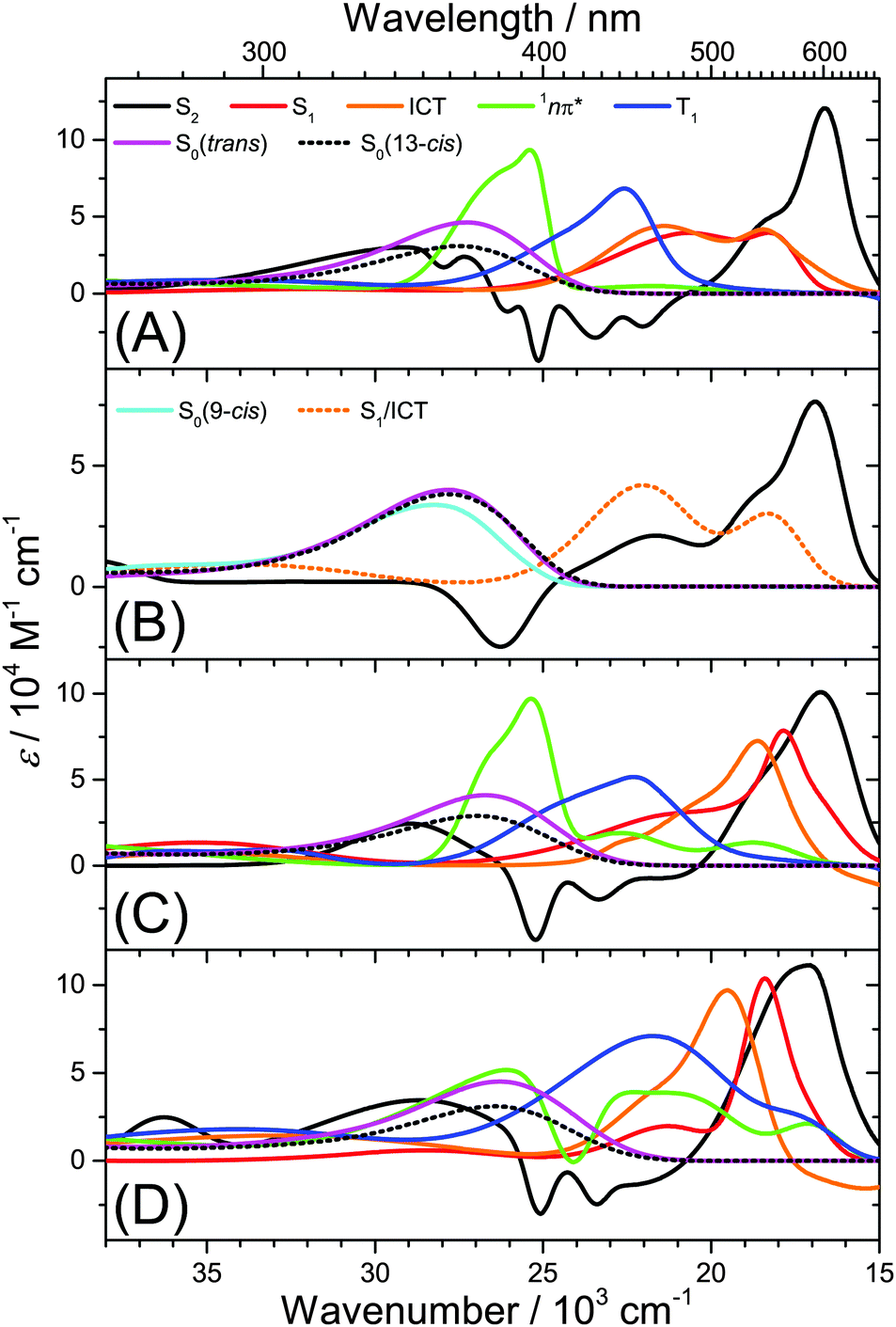 | ||
| Fig. 5 Species-associated spectra from global kinetic analysis. (A) All-trans retinal in n-hexane, (B) all-trans retinoic acid in n-hexane, (C) all-trans retinal in THF, and (D) all-trans retinal in methanol. | ||
We note that other kinetic schemes failed to reproduce central aspects of the spectral dynamics: for instance, a model employing branching from S2 forming the three species S1, ICT and 1nπ* was abandoned, because it did not reproduce the delayed rise of the 1nπ* band, suggesting that S1 must act as an intermediate for 1nπ* formation, see ESI† (Fig. S25).
Furthermore, a model featuring a single S1/ICT state was tested for retinal. However, a single state should exhibit monoexponential decay behaviour. For instance, the build-up of population in the 1nπ* state in nonpolar alkanes and THF (τ1,nπ* ≤ 0.56 ps in Table S2, ESI†) then would quickly drain population from S1/ICT. It therefore immediately turned out that the second slower component observed in the spectral decay of the ESA band cannot be modelled, see Fig. S26, ESI.† Therefore separate S1 and ICT species are clearly identified. The obvious question then arises if the S1 and ICT species might be in equilibrium. Based on our modelling we conclude that a “fast” equilibrium with forward and backward reactions on the picosecond timescale is not compatible with the experimental data, because the fastest channel dictates the decay of the ESA band, and it becomes impossible to model the slower decay component. Yet, we cannot exclude the presence of a slow equilibrium (with time constants of several picoseconds for forward and backward reaction). This implies the presence of an appreciable barrier on the potential energy surface between S1 and ICT suppressing interconversion of the two species on the timescale of IC to the ground state. This further corroborates our final model in Fig. 4(A) assuming that the S1 and ICT species are effectively separated.
Lastly, the inclusion of trans–cis isomerisation in the mechanism of Fig. 4(A) is imperative for two reasons: the “persistent bleach” in the GSB region at 355 nm for all-trans retinoic acid in n-hexane in Fig. 1(B), bottom panel (in the absence of any triplet formation), directly proves the formation of cis-species exhibiting smaller absorption coefficients. We confirmed this by recording UV-Vis steady-state absorption spectra of different retinoic acid isomers (Fig. S27, ESI†). For all-trans retinal, the situation is more complicated, because there are long-lived spectral contributions of the triplet band and its corresponding GSB contribution. Yet, earlier experiments employing HPLC analysis of primary photoproducts of all-trans retinal provided an estimate for the quantum yield of cis-isomer formation and confirmed that trans–cis isomerisation in the excited state indeed takes place, with 13-cis representing the dominant product.20,21
3.3 Polarity-dependent excited-state dynamics of all-trans retinal
Having established the plausibility of the kinetic scheme in Fig. 4(A), we now turn toward the interpretation of the time constants and species-associated spectra obtained from the analysis. The lifetime of the S2 state is extremely short in all solvents, e.g. 55 and 60 fs in n-hexane and n-hexadecane, respectively. This supports our previously observed trend that the S2 lifetime of apocarotenals systematically decreases upon shortening of the polyene chain.33 In all cases, the S2 → Sn spectrum is characterised by a sharp peak at ca. 600 nm with a shoulder extending to shorter wavelengths.We observe a systematic decrease of the quantum yield for S1 formation with solvent polarity. For instance, it is 73% in n-hexane and only 43% in acetonitrile, thus population of ICT is favoured in polar solvents (Fig. 4(B)). The latter finding is in agreement with the characteristic change of the experimental ESA band shape and the appearance of pronounced SE above 600 nm from diisopropyl ether onwards (Fig. 5(C and D), orange lines, and Fig. S20, ESI†).
The species-associated spectra of S1 (red) and ICT (orange) exhibit systematic changes with polarity: in nonpolar solvents (n-hexane, n-hexadecane), the spectra contain two broad peaks and look similar. In polar solvents, the ICT spectra show a pronounced single peak with a shoulder toward shorter wavelengths and a distinct SE feature above 600 nm. The S1 spectra are more red-shifted and show no SE band. We therefore conclude that the S1 species is a higher-energy local minimum of the first electronically excited state (assuming a similar energetic position of the upper state in the ESA transitions of S1 and ICT). S1 has access to the 1nπ* state and is separated from the lower-energy ICT part of the potential by an appreciable barrier. The charge-transfer character of ICT is less pronounced in nonpolar solvents due to the missing dipolar solvent stabilization. Therefore the SE feature is likely hidden beneath the stronger ESA band due to its much smaller Stokes shift in nonpolar solvents.
The formation of 1nπ* from S1 significantly slows down with increasing solvent polarity, from 0.21 ps in n-hexane to 3.0 ps in acetonitrile and, at the same time, internal conversion to S0 accelerates (Fig. 4(D)). These two effects lead to a dramatic reduction of the triplet quantum yield from 64% in n-hexane to 4% in acetonitrile (Fig. 4(C)). It is reasonable to assume that a polarity-induced shift of the 1nπ*, S1 and ICT energy levels is responsible for this effect. Most importantly, an increase of polarity tends to increase the energy of 1nπ* species relative to S1 and ICT.34 This likely reduces the coupling between S1 and 1nπ* and slows down the IC rate for 1nπ* formation, favouring IC to S0 instead.
We completely characterise the 1nπ* state spectra in alkanes and diisopropyl ether (Fig. 5, green lines). The band is centred at 396 nm with a shoulder toward shorter wavelengths. In polar solvents, the shape of the 1nπ* spectrum should be taken with some caution, because its ESA band is weak (Fig. S17–S19, ESI†), due to the small quantum yield for 1nπ* formation and its subsequent decay to T1. Still, it is clearly visible that the 1nπ* band is located at about the same position as in nonpolar solvents and broadens.
The time constant for T1 formation from 1nπ* in alkanes and diisopropyl ether is ca. 31 ps. This ISC process accelerates somewhat with increasing solvent polarity (Fig. 4(E)). The T1 spectra in alkanes and diisopropyl ether are characterised by a main peak at ca. 443 nm (Fig. 5, blue lines, and Fig. 3), with a typical absorption coefficient of ca. 70000 M−1 cm−1. In polar solvents, the T1 spectra broaden significantly.
3.4 Influence of the type of terminal carbonyl group
Fig. 1(B and D) demonstrated that the 1nπ*/T1 channel is completely shut down upon replacing the terminal aldehyde by a carboxyl group. Consequently, global modelling for all-trans retinoic acid in n-hexane is possible using the SAS shown in Fig. 5(B). We note that a single spectrum for S1/ICT suffices, because the whole ESA band decays monoexponentially. It has a shape which is similar to the S1 and ICT species in all-trans retinal (Fig. 5(A)). The influence of the COOH group is twofold: it shifts the 1nπ* state to higher energies, so that access from S1 is no longer possible. Such an energetic upshift of 1nπ* states of carboxylic acids is known for shorter conjugated systems35 and was also found in our DFT/TDDFT calculations reported in Section 3.6. Secondly, the ICT character is weaker for a COOH group than for a CHO group, as suggested by our previous studies of longer-conjugated β-apocarotenals and β-apocarotenoic acids.5,6,16,36 Therefore the excited-state ICT character of retinoic acid should be reduced and the barrier between the S1 and ICT part should be small or even absent, leading to the monoexponential ESA decay. It also appears as if the larger energetic separation of the 1nπ* state from S1/ICT increases the IC lifetime for S1/ICT → S0 to 19 ps.3.5 Starting from the 13-cis isomer
For further testing of our kinetic model for retinal excited-state dynamics, we carried out selected transient absorption experiments starting from 13-cis retinal. We focus on the limiting cases in nonpolar and polar solvents. Transient absorption spectra in n-hexane and acetonitrile are included in Fig. S28, ESI.† Results of the global kinetic modelling are found in Table S2, ESI.† At first glance, the transient spectra look similar to those of the all-trans isomer. Inspection of the global modelling results revealed however significant differences: the 1nπ*/triplet quantum yield is reduced, and 13-cis formation dominates. The latter becomes immediately clear upon inspection of the transient absorption spectrum of 13-cis retinal in n-hexane: it is shifted toward shorter wavelengths (Fig. S29, ESI†). If a larger amount of all-trans isomer was formed, the triplet band should largely resemble the all-trans T1 band, which is obviously not the case. A similar argument holds for acetonitrile (Fig. S28(B), ESI†): if an appreciable amount of all-trans isomer was formed, there should be no GSB at long times but a weak absorption feature, because of the higher absorption coefficient of the all-trans isomer (Fig. S9, ESI†). This is also not the case. There is apparently a strong propensity for the recovery of the initially photoexcited isomer. We therefore conclude that the kinetic scheme in Fig. 4(A) is also applicable to the excited-state dynamics of 13-cis retinal, only the trans and cis denominations need to be exchanged.3.6 Comparison with DFT/TDDFT calculations
We also carried out selected DFT/TDDFT calculations for all-trans retinal and all-trans retinoic acid. Although there are well-known limitations and drawbacks of this method when applied to polyene systems with charge-transfer character,37,38 valuable semi-quantitative information was extracted regarding the influence of the terminal carbonyl group. Transitions and oscillator strengths are summarised in Table S3 and Fig. S30 (ESI†). Detachment and attachment electron densities39 for the three lowest excited singlet states are plotted in Fig. S31 (ESI†).We focus on the shifts of the electronic transitions upon replacement of the aldehyde by the carboxyl group. The optically dark S0 → 1nπ* transition is clearly identified in the detachment/attachment plots by the transfer of electron density from the oxygen lone pairs into the conjugated π-system (Fig. S31, ESI†). For all functionals, the S0 → 1nπ* transition is strongly shifted upward by 7000–10000 cm−1 (Table S3, Fig. S30, ESI†). In contrast, the S0 → 1Ag− and S0 → 1Bu+ ππ* transitions exhibit only a mild up-shift of 450–800 cm−1. The calculations therefore provide a rationale for the switching-off of the 1nπ*/triplet channel observed in the time-resolved experiments upon replacing the CHO by the COOH group, because the 1nπ* state is then no longer accessible from 1Ag−. The calculations also confirm the slight blue-shift observed in the steady-state absorption spectra upon replacement of the aldehyde by the carboxyl group (Fig. 1(A) and (B), bottom panels).
A closer look however reveals substantial discrepancies between experiment and TDDFT calculations. For instance, in the case of all-trans retinal, 1nπ* is no longer the lowest excited state for the functionals B3LYP, MPW1K and CAM-B3LYP. In addition, the spacing between the 1Bu+ and the 1Ag− states and their oscillator strengths are not consistent with experiment. For reaching quantitative agreement, more accurate quantum chemical calculations of these systems in different solvents will therefore be needed. This would require the accurate determination of the energetic positions of the S2, S1, ICT and 1nπ* species. However currently, such calculations are very difficult, because this would require the correct treatment of doubly excited configurations in these polyenes,37 a correct inclusion of the charge-transfer character introduced by the terminal carbonyl group,38 as well as an appropriate consideration of local solvent effects beyond a simple polarizable continuum approach, including solvent-dependent relaxation of the excited-state structures. Note also that, so far, even high-level gas-phase CASSCF and CASPT2 calculations for retinal strongly overestimate the energy of the bright S0 → S2 transition.40 Recent approaches, such as the one used by Frank, Birge and co-workers for the carbonyl carotenoid peridinin41 might be also valuable for a better understanding of the excited-state manifold of all-trans retinal and all-trans retinoic acid. In any case, we hope that the current experiments will serve as a useful benchmark for future high-level quantum chemical calculations of these systems.
4. Conclusions
The photochemistry of retinal is largely controlled by the 1nπ* state. According to El-Sayed's rule,42–44 only an 1nπ* → T1 ISC transition is sufficiently fast to efficiently compete with IC processes typically prevailing in carotenoids. We demonstrated that this channel is operative in all solvents and gradually closes with increasing solvent polarity. A complete suppression of the 1nπ* channel happens upon changing to a terminal substituent, such as carboxyl, which pushes up the energy of 1nπ*. Exactly this effect is also operative in the all-trans retinal Schiff base of microbial rhodopsins, as its 1nπ* state is known to be located at higher energy than for the corresponding aldehyde.45–50 Protonation of the Schiff base even completely removes its ability to form an 1nπ* state.51In all solvents, isomerisation of the initially photoexcited species is inefficient, e.g. <21% for all-trans retinal (Table S2, ESI†). Such an intrinsically low quantum yield is unwanted for retinal's function as an efficient photoswitch. In microbial rhodopsins, nature has optimized it to 64%,52,53 implying that the protein pocket promotes much more efficient trans–cis isomerisation.1
Acknowledgements
We acknowledge B. Schäfer from BASF SE for providing high purity all-trans retinal and all-trans retinoic acid samples for the current experiments. We are indebted to N. P. Ernsting (Humboldt University Berlin, Germany) and J. L. Pérez Lustres (University of Santiago de Compostela, Spain) as well as J. Troe, K. Luther, J. Schroeder, D. Schwarzer and A. M. Wodtke (Georg August University Göttingen, Germany) for their continuous support and advice.References
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126 CrossRef CAS PubMed.
- A. Wand, I. Gdor, J. Zhu, M. Sheves and S. Ruhman, Annu. Rev. Phys. Chem., 2014, 64, 437 CrossRef PubMed.
- P. D. Kiser, M. Golczak and K. Palczewski, Chem. Rev., 2014, 114, 194 CrossRef CAS PubMed.
- D. A. Wild, K. Winkler, S. Stalke, K. Oum and T. Lenzer, Phys. Chem. Chem. Phys., 2006, 8, 2499 RSC.
- M. Kopczynski, F. Ehlers, T. Lenzer and K. Oum, J. Phys. Chem. A, 2007, 111, 5370 CrossRef CAS PubMed.
- K. Oum, P. W. Lohse, F. Ehlers, M. Scholz, M. Kopczynski and T. Lenzer, Angew. Chem., Int. Ed., 2010, 49, 2230 CrossRef CAS PubMed.
- T. Polívka, S. Kaligotla, P. Chábera and H. A. Frank, Phys. Chem. Chem. Phys., 2011, 13, 10787 RSC.
- J. A. Bautista, R. E. Connors, B. B. Raju, R. G. Hiller, F. P. Sharples, D. Gosztola, M. R. Wasielewski and H. A. Frank, J. Phys. Chem. A, 1999, 103, 8751 CrossRef CAS.
- D. Zigmantas, T. Polívka, R. G. Hiller, A. Yartsev and V. Sundström, J. Phys. Chem. A, 2001, 105, 10296 CrossRef CAS.
- D. Zigmantas, R. G. Hiller, A. Yartsev, V. Sundström and T. Polívka, J. Phys. Chem. B, 2003, 107, 5339 CrossRef CAS.
- S. Yamaguchi and H. Hamaguchi, J. Chem. Phys., 1998, 109, 1397 CrossRef CAS.
- T. Yuzawa and H. Hamaguchi, J. Mol. Struct., 1995, 352/353, 489 CrossRef CAS.
- S. Yamaguchi and H. Hamaguchi, J. Phys. Chem. A, 2000, 104, 4272 CrossRef CAS.
- E. J. Larson, L. A. Friesen and C. K. Johnson, Chem. Phys. Lett., 1997, 265, 161 CrossRef CAS.
- E. J. Larson, S. J. Pyszczynski and C. K. Johnson, J. Phys. Chem. A, 2001, 105, 8136 CrossRef CAS.
- S. Stalke, D. A. Wild, T. Lenzer, M. Kopczynski, P. W. Lohse and K. Oum, Phys. Chem. Chem. Phys., 2008, 10, 2180 RSC.
- A. L. Dobryakov, S. A. Kovalenko, A. Weigel, J. L. Pérez Lustres, J. Lange, A. Müller and N. P. Ernsting, Rev. Sci. Instrum., 2010, 81, 113106 CrossRef CAS PubMed.
- K. Oum, T. Lenzer, M. Scholz, D. Y. Jung, O. Sul, B. J. Cho, J. Lange and A. Müller, J. Phys. Chem. C, 2014, 118, 6454 CAS.
- L. F. Tietze, B. Waldecker, D. Ganapathy, C. Eichhorst, T. Lenzer, K. Oum, S. O. Reichmann and D. Stalke, Angew. Chem., Int. Ed., 2015, 54, 10317 CrossRef CAS PubMed.
- W. H. Waddell and K. Chihara, J. Am. Chem. Soc., 1981, 103, 7389 CrossRef CAS.
- S. Ganapathy and R. S. H. Liu, J. Am. Chem. Soc., 1992, 114, 3459 CrossRef CAS.
- B. Veyret, S. G. Davis, M. Yoshida and K. Weiss, J. Am. Chem. Soc., 1978, 100, 3283 CrossRef CAS.
- R. Bensasson, E. J. Land and T. G. Truscott, Photochem. Photobiol., 1973, 17, 53 CrossRef CAS PubMed.
- T. Rosenfeld, A. Alchalel and M. Ottolenghi, J. Phys. Chem., 1974, 78, 336 CrossRef CAS.
- R. Azerad, R. Bensasson, M. B. Cooper, E. A. Dawe and E. J. Land, presented on the Int. Conf. at the Calouste Gulbenkian Foundation Centre, Lisbon, Portugal, 1974, in: Excited States of Biological Molecules, ed. J. B. Birks, Wiley (London), 1976, p. 531.
- W. Dawson and E. W. Abrahamson, J. Phys. Chem., 1962, 66, 2542 CrossRef CAS.
- M. M. Fisher and K. Weiss, Photochem. Photobiol., 1974, 20, 423 CrossRef CAS.
- P. K. Das and R. S. Becker, J. Am. Chem. Soc., 1979, 101, 6348 CrossRef CAS.
- K. Golibrzuch, F. Ehlers, M. Scholz, R. Oswald, T. Lenzer, K. Oum, H. Kim and S. Koo, Phys. Chem. Chem. Phys., 2011, 13, 6340 RSC.
- K. Oum, P. W. Lohse, J. R. Klein, O. Flender, M. Scholz, A. Hagfeldt, G. Boschloo and T. Lenzer, Phys. Chem. Chem. Phys., 2013, 15, 3906 RSC.
- K. Oum, O. Flender, P. W. Lohse, M. Scholz, A. Hagfeldt, G. Boschloo and T. Lenzer, Phys. Chem. Chem. Phys., 2014, 16, 8019 RSC.
- R. R. Birge, J. A. Bennett, L. M. Hubbard, H. L. Fang, B. M. Pierce, D. S. Kliger and G. E. Leroi, J. Am. Chem. Soc., 1982, 104, 2919 CrossRef.
- F. Ehlers, M. Scholz, J. Schimpfhauser, J. Bienert, K. Oum and T. Lenzer, Phys. Chem. Chem. Phys., 2015, 17, 10478 RSC.
- T.-i. Lai, B. T. Lim and E. C. Lim, J. Am. Chem. Soc., 1982, 104, 7631 CrossRef CAS.
- J. D. Coyle, Chem. Rev., 1978, 78, 97 CrossRef CAS.
- P. W. Lohse, R. Bürsing, T. Lenzer and K. Oum, J. Phys. Chem. B, 2008, 112, 3048 CrossRef CAS PubMed.
- J. H. Starcke, M. Wormit, J. Schirmer and A. Dreuw, Chem. Phys., 2006, 329, 39 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009 CrossRef CAS PubMed.
- M. Head-Gordon, A. M. Grana, D. Maurice and C. A. White, J. Phys. Chem., 1995, 99, 14261 CrossRef CAS.
- M. Merchán and R. González-Luque, J. Chem. Phys., 1997, 106, 1112 CrossRef.
- N. L. Wagner, J. A. Greco, M. M. Enriquez, H. A. Frank and R. R. Birge, Biophys. J., 2013, 104, 1314 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1962, 36, 573 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1964, 41, 2462 CrossRef CAS.
- K.-H. Grellmann, R. Memming and R. Livingston, J. Am. Chem. Soc., 1962, 84, 546 CrossRef CAS.
- W. H. Waddell, A. M. Schaffer and R. S. Becker, J. Am. Chem. Soc., 1973, 95, 8223 CrossRef CAS PubMed.
- R. S. Becker, G. Hug, P. K. Das, A. M. Schaffer, T. Takemura, N. Yamamoto and W. Waddell, J. Phys. Chem., 1976, 80, 2265 CrossRef CAS.
- R. S. Becker, P. K. Das and G. Kogan, Chem. Phys. Lett., 1979, 67, 463 CrossRef CAS.
- P. K. Das, G. Kogan and R. S. Becker, Photochem. Photobiol., 1979, 30, 689 CrossRef CAS.
- R. S. Becker, Photochem. Photobiol., 1988, 48, 369 CrossRef CAS PubMed.
- E. W. Abrahamson, R. G. Adams and V. J. Wulff, J. Phys. Chem., 1959, 63, 441 CrossRef CAS.
- R. Govindjee, S. P. Balashov and T. G. Ebrey, Biophys. J., 1990, 58, 597 CrossRef CAS PubMed.
- J. Tittor and D. Oesterhelt, FEBS Lett., 1990, 263, 269 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structures; pump fluence dependence for transient absorption signals of all-trans retinal in n-hexane and acetonitrile; subtraction of the solvent contribution in the broadband transient absorption spectra; steady-state absorption spectra of all-trans and 13-cis retinal, their characteristic parameters and solvatochromic analysis; UV-Vis transient broadband absorption spectra of all-trans retinal; overview of time constants and quantum yields from the global analysis; species-associated spectra for all-trans retinal in different solvents; spectral fits and kinetics for all-trans retinal in different solvents; comparison with fit results from other kinetic models; steady-state absorption spectra for isomers of retinoic acid; UV-Vis broadband transient absorption spectra of 13-cis retinal; results of DFT/TDDFT calculations for all-trans retinal and all-trans retinoic acid. See DOI: 10.1039/c6cp01335j |
| This journal is © the Owner Societies 2016 |