
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-spin-state at a Li3PO4/LiCoO2 (104) interface†
Masato
Sumita
*a and
Takahisa
Ohno
*abc
aNational Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: SUMITA.Masato@nims.go.jp
bGlobal Research Center for Environment and Energy based on Nanomaterials Science (GREEN), NIMS, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
cInstitute of Industrial Science, University of Tokyo, Meguro, Tokyo 153-8505, Japan. E-mail: OHNO.Takahisa@nims.go.jp
First published on 19th January 2016
Abstract
We have found the disproportion between the intermediate spin (IS) and low spin (LS) configurations of Co atoms at a Li3PO4/LiCoO2 (104) interface through density functional molecular dynamics (DF-MD). The manifold of the spin state at the interface, however, does not affect the band alignment between the Li3PO4 and LiCoO2 regions.
All-solid state Li ion secondary batteries (ALBs) are regarded as next-generation rechargeable electronic cells.1 Although various materials have already been synthesized for electrolytes and electrodes, solid Li ion secondary batteries fabricated from these materials do not always show the expected performance because there are some problems caused by the interfaces between the electrolyte and electrode materials. Experimental results imply that the problems are attributed to harmful products at the atomistic level2 or to a space charge layer at electrode/electrolyte interfaces,3 which result in interfacial resistance. Investigation and the control of the interfaces at the atomistic level, therefore, are indispensable in ALB development.
In this communication, we report a theoretical study on the interface between amorphous Li3PO4 and LiCoO2, which is a typical electrode/electrolyte interface in ALBs, to give some insight into the interfacial properties. LiCoO2 is widely used as a 4 V-class electrode and Li3PO4 is also a fundamental electrolyte material. A battery that uses a Li/LiPON/LiCoO2 structure, where LiPON is nitrogen-doped Li3PO4,4 exhibits a high cyclic performance,5 indicating the existence of a stable electrolyte/electrode interface. Furthermore, Haruta et al.6 have recently succeeded in lowering the resistance at the LiPON/LiCoO2 interface via all-in-vacuum fabrication than that in a liquid electrolyte-based battery. In addition to the use as an electrolyte, Li3PO4 plays an important role as a coating material for enhancing the performance of LiCoO2.7 Li3PO4-coated LiCoO2 shows higher voltage than a pure LiCoO2 cathode with a polymer electrolyte.8
Here, we consider the LiCoO2 (104) surface, which is a stable active surface for Li+ intercalation/deintercalation. On the surface of nanosize stoichiometric LiCoO2, Co atoms have various spin configurations depending on the facets. The (104) surface has five-coordinated Co atoms (Co5C) whose spin configuration is reported to be the intermediate-spin (IS) state (S = 1, Fig. 1).9 The present calculations also indicate that the Co5C on the surface prefers the IS state to the low-spin (LS) configuration in vacuo (see the ESI†). However, the fate of radical electrons on the Co5C after forming the interface with the electrolyte remains unknown. We have investigated the spin configuration of Co5C on the (104) surface at the interface with the amorphous Li3PO4 phase using density functional molecular dynamics (DF-MD; molecular dynamics calculation with density functional theory) with an NPT ensemble. See the ESI† about the setup of an initial interface structure and the recipes for making a model structure for amorphous Li3PO4.
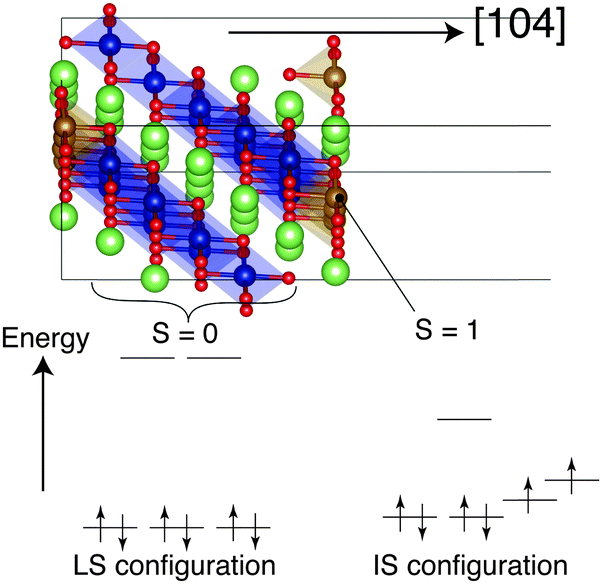 | ||
| Fig. 1 Low-spin (LS) configuration (S = 0) in the d orbitals of Co in bulk LiCoO2 (left) and intermediate-spin (IS) configuration (S = 1) of five-coordinated Co (Co5C) on the (104) surface (right).9 Orange balls indicate IS Co (Co5C) atoms and blue balls indicate LS Co atoms. | ||
We have performed three types of DF-MD calculation. One is the system where all Co5C atoms at the interface are set to the IS configuration. Hereafter, we refer to this system as I. Another is the system where all Co5C atoms at the interface are set to the LS state, which we refer to as II. After running a 100 ps DF-MD calculation, we have obtained the energetic equilibrium conditions during 40.0 ps for sampling. In I and II, some PO4 anions adsorbed onto the LiCoO2 (104) surface via two Co–O bonds (coverage; Θ = 0.75, including both sides of the LiCoO2 slab, see also Fig. 3), but one monolayer coverage (all Co5C atoms are covered by PO4 anions) is not reached during this DF-MD calculation. To explore the possibility of the monolayer-coverage situation (III), we have performed another DF-MD calculation whose initial structure has one monolayer, i.e., all Co5C atoms at the interface are covered by PO4 anions. For this one monolayer system, we have calculated only the LS state because PO4 adsorbed Co atoms prefer the LS configuration, as will be explained later.
The average total energies (EA), their standard deviations (SD), and average enthalpies [HA = EA + PVA; P (= 1.01325 bar) and VA are pressure and average volume] of all states are tabulated in Table 1 with the average cell parameters in the direction of the c-axis. Because the differences between the average cell parameters of the respective systems are within 3%, the comparison between the statistical energies of the systems is allowed. Indeed, there is no large discrepancy between EA and HA (including volume fluctuation). As shown in Table 1, the LS state (II) is more stable than the IS state (I) by 101.23 kJ mol−1. From this result, PO4 adsorbed Co atoms at the interface prefer the LS configuration. The PO4 adsorption is therefore expected to induce intersystem crossing from the IS state to the LS state. Although the one monolayer situation is maintained during the 40.0 ps DF-MD calculation, the one monolayer system III has no energetic advantage over the unsaturated LS state II because the one monolayer system III also shows a comparable energetic level with the system II. This indicates the possible existence of uncovered Co5C even at the interface with the amorphous Li3PO4 phase.
| I (IS) | II (LS) | III (monolayer) | |
|---|---|---|---|
| E A/kJ mol−1 (Eh) | 0.0 (−7330.67908) | −101.24 (−7330.71764) | −103.02 (−7330.71832) |
| SD/kJ mol−1 | 72.97 | 73.23 | 74.64 |
| c-axis/Å | 47.26 | 46.17 | 46.90 |
| H A/kJ mol−1 (Eh) | 0.0 (−7330.67901) | −101.24 (−7330.71757) | −103.02 (−7330.71825) |
As shown in Fig. 2, each distribution of energies of I–III (histograms) overlaps with each other. The IS and LS states have the possibility of taking an energetically and structurally common area by thermal fluctuation. This suggests that the Co5C atoms at the interface could thermally undergo a spin crossover between the LS and IS configurations similar to perovskite LaCoO3.10 Consequently, disproportionation involving LS and IS might appear at the interface.
 | ||
| Fig. 2 Normalized histograms of total energies (in kJ mol−1 relative to the average energy of I) of system I (IS system), II (LS system), and III (monolayer LS system) during 40.0 ps DF-MD. | ||
 | ||
| Fig. 3 (a) Structure of PO4 anion adsorbed on the LiCoO2 (104) surface. (b) Schematic structure of PO4 adsorbed with two free Li ions shared by oxygen atoms of PO4 and LiCoO2. | ||
Three-dimensional and schematic structures of PO4 adsorbed on the LiCoO2 (104) surface are shown in Fig. 3. The average Co–O (PO4) bond lengths, which are relevant to PO4 adsorption to the LiCoO2 (104) surface, are tabulated in Table 2 with the Co–O bond lengths in the bulk region of the LiCoO2 (104) slab.
| I (IS) | II (LS) | III (monolayer) | |
|---|---|---|---|
| Co–O (PO4)/Å | 2.371 | 2.068 | 2.162 |
| Co–O (LCO)/Å | 1.941 | 1.940 | 1.933 |
The average Co–O (PO4) lengths in all the systems are estimated to be above 2.0 Å, which is larger than the average bond lengths of the bulk region of the LiCoO2 (104) slab [Co–O (LCO)]. The large bond length Co–O (PO4) is probably attributed to the Li ion neighbouring the oxygen of PO4. With the adsorption of the PO4 anion, one/two Li ions released from one PO4 anion exist because three Li ions electrostatically coordinate to four oxygen atoms of one PO4 anion, and two oxygen atoms of PO4 are used for the adsorption on the surface. Some Li ions can be located in a position that can be shared by two oxygen atoms: one is an oxygen atom of the adsorbed PO4 [broken lines in Fig. 3(b)] and the other is an oxygen atom on the LiCoO2 (104) surface [hashed lines in Fig. 3(b)].
The average Co–O (PO4) bond lengths of the LS systems II and III are estimated to be 2.07 and 2.16 Å, respectively. On the other hand, the average Co–O (PO4) bond length of the IS system I is estimated to be 2.37 Å, which is larger than that of the LS systems II and III. This indicates that the IS Co atom certainly prefers the PO4 desorption. Although PO4 adsorption on the (104) surface contributes to the destabilization of IS Co atoms on the surface, it stabilizes LS Co atoms. Consequently, the LS interface systems II and III lie at an energetically lower level than the IS interface system I.
The PO4 adsorption decreases the positive charge of Co atoms at the interface. As tabulated in Table 3, the average Mulliken charges of the interfacial Co atoms in the LS systems II and III decrease to the same extent as bulk Co (about 0.5), in contrast to the interfacial Co in the IS system I (0.65). Therefore, the PO4 adsorption induces the electron transfer from an oxygen atom of the PO4 anion to a Co atom.
Fig. 4 shows the contour maps of LDOS of alpha spin electrons along the c-axis at the average structures of all systems during the 40.0 ps DF-MD calculation. The IS system I has an interfacial level attributed to an unoccupied d orbital of IS Co at the interface, as shown in Fig. 4. On the other hand, the LS system II has an interfacial level due to the uncapped LS Co atoms at the interface (one side) that disappears in the one monolayer system III. For all these systems, band gaps in the bulk regions [LDOS in Fig. 4] of LiCoO2 (104) slabs and in the Li3PO4 phases are estimated to be approximately 2.3 eV [comparable to the experimental result (2.7 eV)11] and 5.0 eV, respectively. The band offset of the valence band maximum (VBM) between the bulk LiCoO2 and Li3PO4 phases is estimated to be 0.7 eV.
 | ||
| Fig. 4 Contour maps of alpha spin LDOS of the systems I–III along the c-axis for the average structure obtained from all the structures during the 40.0 ps DF-MD calculation. Fermi levels are set to zero. Irrespective of the system, the gaps between the valence band maximum (BVM) and the conduction band minimum (CBM) of the LiCoO2 and Li3PO4 phases are estimated to be 2.3 and 5.0 eV respectively. | ||
| I (IS) | II (LS) | III (monolayer) | |
|---|---|---|---|
| Interface Co (Co5C) | 0.65 | 0.54 | 0.55 |
| Bulk Co | 0.51 | 0.50 | 0.49 |
Irrespective of the spin state, no reaction products grow during the 40.0 ps DF-MD calculations. Moreover, the IS Co atoms in the IS system I remain at the interface during the DF-MD calculation as shown in Fig. 4, that is, the radical electrons at the interface do not become one of the factors for producing some harmful products in this system at the current computational level. We speculate that a similar interface is obtained by fabrication under all-vacuum conditions6, which exclude impurities from the interface as much as possible.
Conclusions
Our simulation indicates that PO4 anions adsorb on the LiCoO2 (104) surface via two Co–O bonds without the growth of an impurity phase. With the PO4 adsorption, the IS Co atoms on the surface are destabilized, whereas the LS Co atoms on the surface are stabilized. Consequently, the LS system of the LiCoO2 (104)/Li3PO4 amorphous interface lies at an energetically lower level than the IS system; however, the one monolayer situation has no advantage in terms of energy against the unsaturated LS system. On the other hand, the difference in energy between the LS and IS interface systems is not large. Therefore, the multi-spin-state where the IS and LS state Co atoms coexist at the LiCoO2 (104)/Li3PO4 interface emerges similar to those in perovskite LaCoO3 that shows a thermally induced spin-crossover.10 The manifold of the spin state at the interface, however, does not significantly affect the band alignment between the Li3PO4 and LiCoO2 regions and the Li3PO4 electrolyte is found to have a wide electrochemical window against the LiCoO2 electrode. The manifold in the spin states of interfacial Co atoms does not affect this system significantly. We will investigate the effect on charging process and of sulfide electrolytes.Acknowledgements
We thank Dr. T. Suzuki for his advice on this communication. This work was supported by the JST ALCA project. The computations in this work were carried out on the supercomputer centers of NIMS.Notes and references
- K. Takada, Acta Mater., 2013, 61, 759 CrossRef CAS.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Chem. Mater., 2010, 22, 949 CrossRef CAS.
- K. Takada, N. Ohta, L. Zhang, K. Fukuda, I. Sakaguchi, R. Ma, M. Osada and T. Sasaki, Solid State Ionics, 2008, 179, 1333 CrossRef CAS.
- X. Yu, J. B. Bates, G. E. Jellison and F. X. Hart, J. Electrochem. Soc., 1997, 144, 524 CrossRef CAS.
- B. Wang, J. B. Bates, F. X. Hart, B. C. Sales, R. Z. Zuhr and J. D. Robertson, J. Electrochem. Soc., 1996, 143, 3203 CrossRef CAS.
- M. Haruta, S. Shiraki, T. Suzuki, A. Kumatani, T. Ohsawa, Y. Takagi, R. Shimizu and T. Hitosugi, Nano Lett., 2015, 15, 1498 CrossRef CAS PubMed.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Solid State Ionics, 1981, 3/4, 171 CrossRef.
- S. Seki, Y. Kobayashi, H. Miyashiro, Y. Mita and T. Iwahori, Chem. Mater., 2005, 17, 2041 CrossRef CAS.
- D. Qian, Y. Hinuma, H. Chen, L.-S. Du, K. J. Carroll, G. Ceder, C. P. Grey and Y. S. Meng, J. Am. Chem. Soc., 2012, 134, 6096 CrossRef CAS PubMed.
- A. Doi, T. Fukuda, S. Tsutsui, D. Okuyama, Y. Taguchi, T. Arima, Q. R. Baron and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 081109 CrossRef.
- J. V. Elp, J. L. Wieland, H. Eskes, P. Kuiper, G. A. Sawatzky, F. M. F. D. Groot and T. S. Turner, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 6090 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, cell parameters of the LiCoO2 (104) slab with thickness. See DOI: 10.1039/c5cp07735d |
| This journal is © the Owner Societies 2016 |