
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation and stability of gas-phase o-benzoquinone from oxidation of ortho-hydroxyphenyl: a combined neutral and distonic radical study†
Matthew B.
Prendergast
a,
Benjamin B.
Kirk
b,
John D.
Savee
c,
David L.
Osborn
c,
Craig A.
Taatjes
c,
Kye-Simeon
Masters
d,
Stephen J.
Blanksby
e,
Gabriel
da Silva
f and
Adam J.
Trevitt
*a
aSchool of Chemistry, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: adamt@uow.edu.au
bLawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cCombustion Research Facility, Sandia National Laboratories, Livermore, CA 94551-0969, USA
dSchool of Chemistry, Physics and Mechanical Engineering, Faculty of Science and Engineering, Queensland University of Technology, Brisbane, QLD 4001, Australia
eCentral Analytical Research Facility, Queensland University of Technology, Brisbane, QLD 4001, Australia
fDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Melbourne, VIC 3010, Australia
First published on 19th October 2015
Abstract
Gas-phase product detection studies of o-hydroxyphenyl radical and O2 are reported at 373, 500, and 600 K, at 4 Torr (533.3 Pa), using VUV time-resolved synchrotron photoionisation mass spectrometry. The dominant products are assigned as o-benzoquinone (C6H4O2, m/z 108) and cyclopentadienone (C5H4O, m/z 80). It is concluded that cyclopentadienone forms as a secondary product from prompt decomposition of o-benzoquinone (and dissociative ionization of o-benzoquinone may contribute to the m/z 80 signal at photon energies ≳9.8 eV). Ion-trap reactions of the distonic o-hydroxyphenyl analogue, the 5-ammonium-2-hydroxyphenyl radical cation, with O2 are also reported and concur with the assignment of o-benzoquinone as the dominant product. The ion-trap study also provides support for a mechanism where cyclopentadienone is produced by decarbonylation of o-benzoquinone. Kinetic studies compare oxidation of the ammonium-tagged o-hydroxyphenyl and o-methylphenyl radical cations along with trimethylammonium-tagged analogues. Reaction efficiencies are found to be ca. 5% for both charge-tagged o-hydroxyphenyl and o-methylphenyl radicals irrespective of the charged substituent. G3X-K quantum chemical calculations are deployed to rationalise experimental results for o-hydroxyphenyl + O2 and its charge-tagged counterpart. The prevailing reaction mechanism, after O2 addition, involves a facile 1,5-H shift in the peroxyl radical and subsequent elimination of OH to yield o-benzoquinone that is reminiscent of the Waddington mechanism for β-hydroxyperoxyl radicals. These results suggest o-hydroxyphenyl + O2 and decarbonylation of o-benzoquinone serve as plausible OH and CO sources in combustion.
1. Introduction
Phenolic compounds, including alkylphenols, represent a substantial portion of lignin-derived biofuel stocks1 and the lighter fractions from lignite pyrolysis.2 They are also used as additives to enhance the oxidative stability of biodiesel and diesel.3–6 Phenol is a product of catechol thermal decomposition,7 benzene and hydroxyl radical reactions8 as well as phenyl9 and benzyl radical oxidation.10The pyrolysis of phenol proceeds with H-migration and CO elimination to produce cyclopentadiene or, at higher temperatures, H-loss to produce the phenoxyl radical.11 Investigations into the phenol + OH reaction report the H-abstraction product as the phenoxyl radical.12,13 However, at >390 K, H-abstraction from the phenyl ring and OH addition reactions are also expected with the former process resulting in hydroxyphenyl radicals.14 The o-hydroxyphenyl radical is an intermediate in the pyrolysis reaction reported for dimethoxybenzene (a model compound for the β-04 aryl ether unit within G-type lignin).15 The addition of O2 to the o-hydroxyphenyl radical site will produce the o-hydroxyphenylperoxyl radical, with its hydroxy H-atom within close proximity to the peroxyl radical substituent. As is the case for the o-methylphenylperoxyl radical,16,17o-hydroxyphenylperoxyl is expected to isomerise and eliminate OH via a phenoxyl QOOH intermediate to produce o-benzoquinone (o-BQ), a known precursor to cyclopentadienone (CPO) + CO.18–21 This mechanism was reported for the oxidation of protonated tyrosinyl radicals22 and has some similarities to the Waddington mechanism for β-hydroxyperoxyl radicals.23–25 Yet, to date, no direct experimental results have validated this mechanism for the o-hydroxyphenyl + O2 reaction system.
In this work, we report reactions of gas-phase o-hydroxyphenyl with O2 using two approaches: synchrotron-based time-resolved photoionisation mass spectrometry and distonic-ion mass spectrometry. The synchrotron-based method couples a slow-flow kinetic reactor to a time-of-flight mass spectrometer and VUV photoionisation that allows detection of reaction products with kinetic and isomeric details. The distonic ion approach exploits charge-tagged derivatives of neutral radical species to study radical kinetics by ion-trap mass spectrometry.26 These distonic ion oxidation experiments build on a framework provided by previous studies of distonic phenyl27 and o-methylphenyl radical oxidation.17 In combination, we show that OH elimination follows the reaction of o-hydroxyphenyl radicals with O2 to form o-BQ. The stability of this nascent o-BQ is also investigated.
2. Experimental
2.1 Synchrotron photoionisation mass spectrometry
The o-hydroxyphenyl + O2 reaction was investigated using time-resolved photoionisation mass spectrometry28 at the Chemical Dynamics Beamline29,30 at the Advanced Light Source (ALS at Lawrence Berkeley National Laboratories, USA). The apparatus comprises a slow-flow tube reactor, quasi-continuous vacuum-ultraviolet (VUV) synchrotron light source and an orthogonal acceleration time-of-flight mass spectrometer. o-Hydroxyphenyl radicals were generated within the flow tube by photolysis of o-bromophenol using a pulsed KrF excimer laser (248 nm) operating at 4 Hz with a fluence of ca. 50 mJ cm−2.The heatable quartz reactor flow tube is 62 cm long with a 1.05 cm inner diameter maintained at 4 Torr (533.3 Pa). Gas continuously escapes the reactor into a differentially pumped vacuum chamber through a 650 μm pinhole situated 37 cm along the flow tube. In the experiments reported here, o-bromophenol, O2 gas, and He gas are supplied to the reactor through separate mass-flow controllers at the overall rate of 202 sccm. The o-bromophenol was entrained in He gas using a fritted bubbler with the liquid sample maintained at 291 K (18 °C) and ∼573 Torr (76.4 kPa). The vapour pressure of o-bromophenol is roughly approximated at 291 K to be 0.17 Torr using Antoine parameters known for phenol.31 At 373 K and 4 Torr, number densities within the reactor are ca. 1.7 × 1012 molecule cm−3 for o-bromophenol, 7.7 × 1015 molecule cm−3 for O2 gas, and a total of 9.6 × 1016 molecule cm−3 for He gas. Reactions were conducted with the reactor temperature maintained at 373 K, 500 K and 600 K. The temperature profile of the reactor is such that the length ca. 20 cm above the pinhole is maintained at the set temperature. Gas flow velocities are as follows: 10.1 m s−1 at 373 K, 13.5 m s−1 at 500 K, and 16.2 m s−1 at 600 K. Total gas flow densities were: 1.0 × 1017 molecule cm−3 at 373 K, 7.7 × 1016 molecule cm−3 at 500 K, and 6.4 × 1016 molecule cm−3 at 600 K.
The gas that escapes through the 650 μm pinhole is sampled by a skimmer to create a near-effusive molecular beam that is intersected by quasi-continuous vacuum-ultraviolet (VUV) synchrotron light. Ions produced by photoionisation are detected using a 50 kHz pulsed orthogonal-acceleration time-of-flight mass spectrometer. The photoionisation energy was typically scanned from 9 to 10 eV with 0.025 eV steps. Mass spectra are compiled into three-dimensional arrays of mass-to-change (m/z), reaction time, and photoionisation energy. All data are normalised for variations in the ALS photocurrent using a NIST-calibrated photodiode (SXUV-100, International Radiation Detectors Inc.). Background subtraction is achieved by subtracting the average signal during the 20 ms prior to the photolysis pulse from the dataset. The resulting photoionisation spectra and kinetic traces are normalised by the area under the curve and averaged together for each temperature. The error bars provided at a given photoionisation energy represent two standard deviations (2σ) for a mean of at least three measurements at 373 and 500 K, and two measurements at 600 K.
2.2 Ion-trap mass spectrometry
Distonic radical cation experiments were conducted on a modified Thermo Fisher Scientific LTQ ion-trap mass spectrometer (Thermo Fisher Scientific Inc., San Jose, USA) situated at the University of Wollongong. Radical precursor ions were generated by infusing methanolic solutions of 10 μM 2-bromo-4-aminophenol ([M + H]+ at m/z 188 and 190), 3-bromo-4-methylaniline ([M + H]+ at m/z 186 and 188), or 3-iodo-4-hydroxy-N,N,N-trimethylbenzenaminium iodide ([M − I]+ at m/z 278) into the electrospray ion source at 5 μL min−1 and were mass-selected using an isolation window of 5–6 Th (mass-to-charge) for brominated, and 1–2 Th for the iodinated cations with a q-parameter of 0.250. Mass spectra acquired with the ion-trap mass spectrometer and presented herein are an average of 50 scans unless otherwise stated. Typical instrumental settings: electrospray voltage (4–5 kV), capillary temperature (250 °C), and sheath gas flow at 10–15, auxiliary gas flow at 0–5 and sweep gas flow at 0 (arbitrary units). The normalised collision energy was typically 20–30% (ref. 32) for CID experiments with an activation time of 30 ms as defined within the control software.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ms (set by the control software). Background O2 resides in the trap due to air entrained by the atmospheric pressure ESI source. Reactions were also conducted with an increased O2 concentration by using a He bath gas doped with O2 (770 ± 45 ppm; BOC, Australia). Similarly, a He bath gas doped with oxygen-18 was used for isotopic labelling experiments.
000 ms (set by the control software). Background O2 resides in the trap due to air entrained by the atmospheric pressure ESI source. Reactions were also conducted with an increased O2 concentration by using a He bath gas doped with O2 (770 ± 45 ppm; BOC, Australia). Similarly, a He bath gas doped with oxygen-18 was used for isotopic labelling experiments.
The O2 concentration (molecule cm−3) within the ion-trap region was determined using the measured pseudo-first order rate coefficient for 3-carboxylatoadamantyl + O2 and its known second-order rate coefficient of 8.5 ± 0.4 × 10−11 cm3 molecule−1 s−1 with the O2 concentration determined for each experiment.37 The background O2 concentration within the ion trap is typically 6.4 × 109 molecule cm−3 and the increased O2 concentrations ranged from 1.6–2.2 × 1011 molecule cm−3 with an O2-doped bath gas. The effective temperature of ions stored within a linear quadrupole ion trap has been estimated at 318 ± 23 K,38 consistent with an earlier estimate of 307 K.37
The kinetic plots that show ion signal decay with increasing reaction time were produced by integrating the ion signal intensity over a selected mass-to-charge range and normalising it to the integrated total ion signal intensity. The normalised integrated ion signal intensity is then averaged for at least 10 scans and plotted against reaction time (0.030–10000 ms) to track changes in ion signal intensity due to reactions with O2. Measured pseudo-first order rate coefficients (k1st) were obtained by fitting eqn (1) to the average normalised integrated peak intensity against reaction time, for a select mass-to-charge range, using the Levenberg–Marquardt algorithm. Satisfactory fits with eqn (1) are consistent with pseudo-first order kinetic behaviour. Thus, allowing the second-order rate coefficient (k2nd) to be calculated using eqn (2) with a measured [O2]. The residual plots accompanying kinetic curves in Fig. 5, Fig. S7, and S8 (ESI†) show the difference between the average normalised integrated ion signal intensity and the expected value from eqn (1), i.e. the residuals, plotted as a function of reaction time.
| y = A0exp(−k1stt) + constant | (1) |
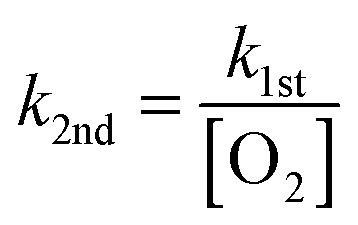 | (2) |
Statistical uncertainty from fitting pseudo-first order rate coefficients (k1st) to experimental decay curves was typically 2σ ≤ 10%. Systematic uncertainty in the ion-trap pressure and O2 concentration, including the generation of neutrals and charged species with mass-to-charge less than the low mass cut-off (50 Th) result in an upper limit of 50% uncertainty in the O2 concentration that is accumulated in reported second-order rate coefficients and reaction efficiencies.
2.3 Quantum chemical calculations
Reaction enthalpies were calculated from electronic energies computed with the G3X-K composite method40 in Gaussian 09.41 G3X-K is a modified G3SX composite method that uses M06-2X density functional theory in place of B3LYP and is parameterised for thermochemical kinetics. It is capable of reproducing barrier heights in the DBH24/08 test set42 to within 1 kcal mol−1, on average.40 The CBS-QB3 method was used for the calculation of adiabatic ionisation energies (AIE) and cation dissociation barriers, with an estimated error of 1 kcal mol−1 (0.05 eV) for AIEs43,44 and 2 kcal mol−1 for barriers from the DBH24/08 database.42 All stationary points were characterised as either minima (no imaginary frequencies) or transition states (one imaginary frequency whose normal mode projection approximates motion along a reaction coordinate). The assignment of a transition state between minima was verified by IRC calculations. The M06-2X geometries and frequencies and G3X-K energies were used to calculate preliminary product ratios within the MultiWell 2013 suite of programs.45 All reported energies include the zero-point energy correction for 0 K enthalpies and AIEs.2.4 Materials
3-Bromo-4-methylaniline, 4-amino-2-bromophenol, oxygen-18 (97%), and o-bromophenol (98%) were purchased from Sigma Aldrich. Gases and reagents obtained from commercial sources were used without further purification. The synthesis of 3-iodo-4-hydroxy-N,N,N-trimethylbenzenaminium iodide is described in Section 1 of the ESI.†3. Results and discussion
3.1 Synchrotron photoionisation mass spectrometry: o-hydroxyphenyl + O2
Fig. 1a is a product mass spectrum after photolysis of o-bromophenol with no O2 added to the reactor, serving as a background measurement. The spectrum is integrated from 0 to 20 ms after photolysis at a photoionisation energy of 10 eV with a reactor temperature of 373 K. Major photolysis product peaks are present at m/z 92 and 94. The photoionisation (PI) spectra for m/z 92 and 94 (not shown) are well matched to the integrated photoelectron spectrum for cyclopenta-2,4-dien-1-ylidenemethanone (C5H4CO, AIE = 8.09 eV)46–48 and the known photoionisation spectrum for phenol (C6H5OH, AIE = 8.49 eV),49–51 respectively. However, since the characteristic AIEs are below the photoionisation energy range scanned, these are tentative assignments for these background species. As an aside, cyclopenta-2,4-dien-1-ylidenemethanone (C5H4CO, m/z 92) may arise from o-bromophenol photolysis via HBr loss52 and phenol (C6H5OH, m/z 94) is probably formed via H-abstraction by the m/z 93 radical from the abundant o-bromophenol precursor.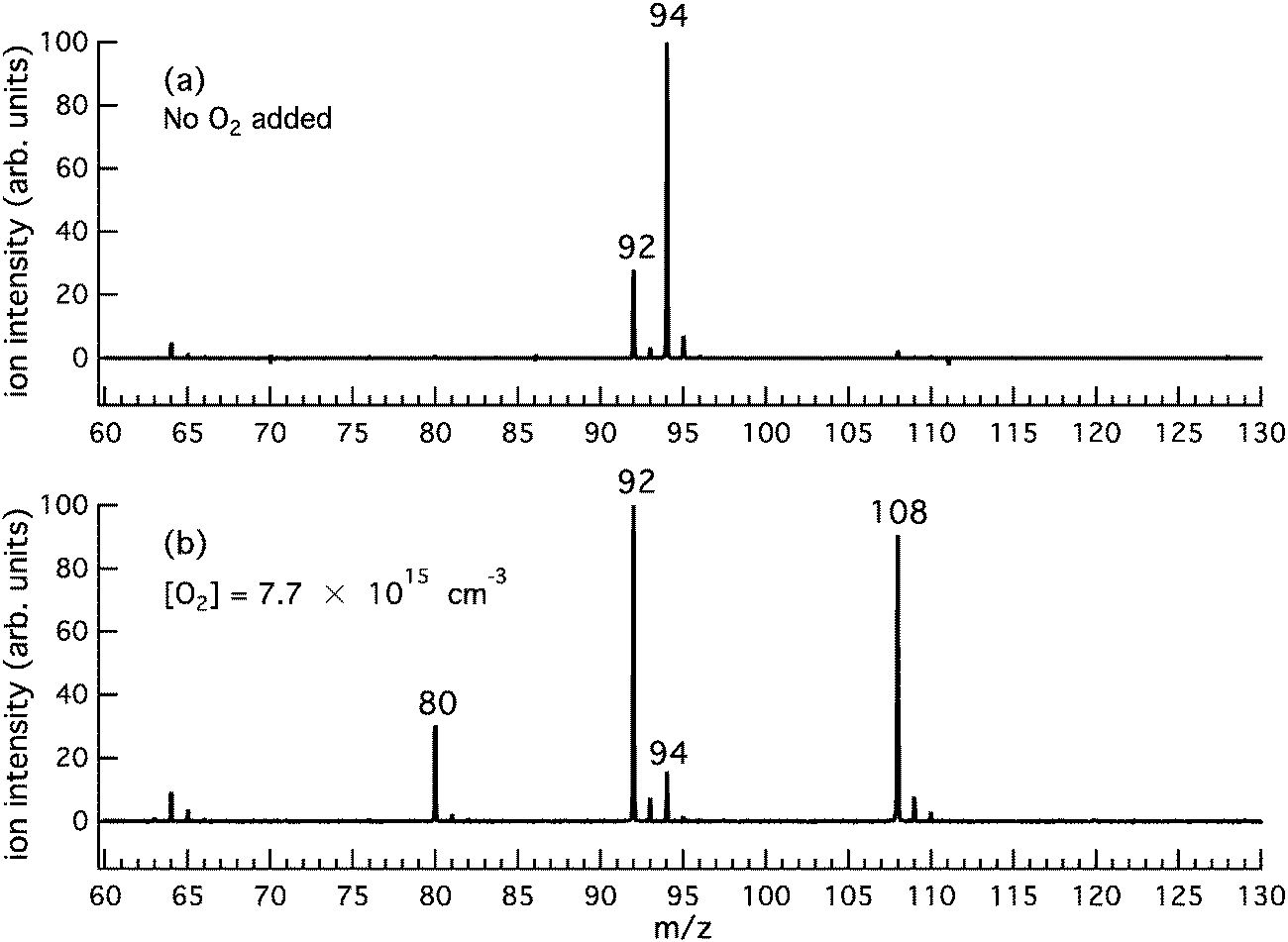 | ||
| Fig. 1 Product photoionisation mass spectra at 373 K and 10 eV integrated 0–20 ms after 248 nm photolysis of (a) o-bromophenol and (b) o-bromophenol in the presence of additional O2 (7.7 × 1015 molecule cm−3). In product spectrum (b) m/z 108 and 80 are reaction product peaks but, as discussed in the text, a portion of the m/z 80 signal may arise from dissociative ionisation of o-BQ (assigned to m/z 108). | ||
Fig. 1b is a product mass spectrum from photolysis of o-bromophenol in the presence of 7.7 × 1015 molecule cm−3 O2. The new product peaks at m/z 80, 108 and the minor peak at m/z 110 are consistent with C5H4O, C6H4O2, and C6H6O2 and are attributed to the o-hydroxyphenyl + O2 reaction. The PI spectra for m/z 80 and 108, integrated 0 to 20 ms after photolysis at 373 K, are provided in Fig. 2a and b. PI spectra at 500 K and 600 K are provided in the ESI† (Fig. S1 and S2, respectively). The PI onsets for m/z 80 at 9.4 eV and m/z 108 at 9.2 eV are in agreement with reference spectra for cyclopentadienone (CPO, m/z 80) and o-benzoquinone (o-BQ, m/z 108),53–55 and consistent with AIEs provided in Table 1. Ionization onsets for CPO and o-BQ were recently reported by Ormond et al.53 and compared within the inset of Fig. 2. The p-benzoquinone isomer can be excluded as a m/z 108 product contributor as its AIE is 9.96 eV with a sharp photoionisation onset,55,56 and there is no such feature in the PI spectrum up to 10 eV. The m/z 109 signal present in mass spectra obtained at 373, 500 and 600 K (ESI,† Fig. S3) could result, in part, from decomposition of o-hydroxyphenylperoxyl to o-hydroxyphenoxyl + O(3P). The hydroxyphenoxyl cation is expected at m/z 109, however unequivocal assignment of the m/z 109 species is confounded by the 13C isotope peak of the dominant m/z 108 product. In unpublished studies, we have observed phenoxyl radical decay that is kinetically matched to the growth of a +1 Da ion signal intensity. A m/z 110 product ion is present in Fig. S3 (ESI†) and kinetic traces in Fig. S5 (ESI†) show that the appearance of m/z 110 ions is delayed relative to m/z 108 ions (a primary product kinetic reference). Therefore, the delayed appearance of m/z 110 ions could be explained via H-abstraction by the o-hydroxyphenoxyl radical to produce o-catechol (C6H4OHOH, m/z 110).
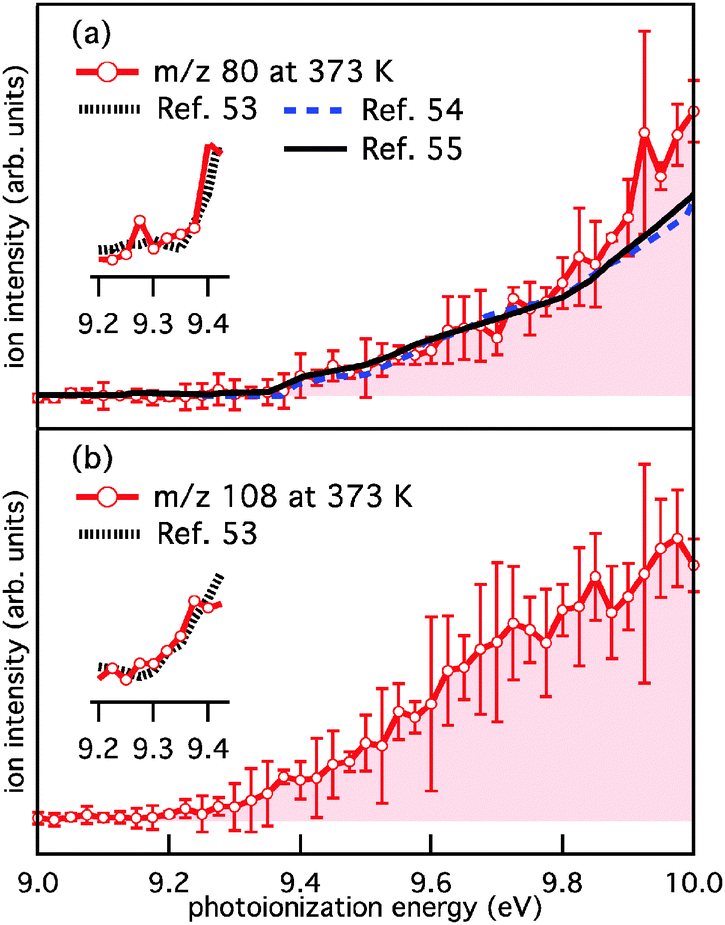 | ||
| Fig. 2 Photoionisation spectra integrated 0–20 ms after photolysis for (a) m/z 80 and (b) m/z 108 from o-hydroxyphenyl + O2 at 373 K. Each spectrum is an average of three PI spectra and the 2σ statistical uncertainty is represented by vertical error bars. Figures inset within (a) and (b) compare the experimental PI spectra near the onset to reference spectra for CPO and o-BQ from ref. 53 (1000 K). Reference PI spectra are also provided in (a) for CPO from ref. 54 and 55 (873 K). | ||
| Species | Measured (eV) | Calculated AIE (eV) | Literature values |
|---|---|---|---|
| Cyclopentadienone (CPO, m/z 80) | 9.4 | 9.41 | 9.41 ± 0.01 (AIE)53 |
| o-Benzoquinone (o-BQ, m/z 108) | 9.2 | 9.18 | 9.3 ± 0.1 (AIE)53 |
| o-Hydroxyphenoxyl radical (m/z 109) | 8.14 | ||
| o-Hydroxyphenol (catechol, m/z 110) | 8.14 | 8.56 (VIE)48,57 | |
| p-Benzoquinone (p-BQ) | 9.89 | 9.96 ± 0.01 (AIE)48,56 |
The detection of o-BQ (m/z 108) is rationalised by O2 addition to the o-hydroxyphenyl radical, followed by isomerisation of the hydroxyphenylperoxyl intermediate to hydroperoxyphenoxyl and subsequent OH loss to form o-BQ (Scheme 1). This pathway is analogous to the O2 addition and subsequent OH loss mechanism that operates in the o-methylphenyl + O2 reaction16,17 and OH loss in the Waddington mechanism for β-hydroxyperoxyl radicals.23,24Scheme 1 also includes pathways from o-BQ that lead to CPO and the CPO radical cation that will now be discussed.
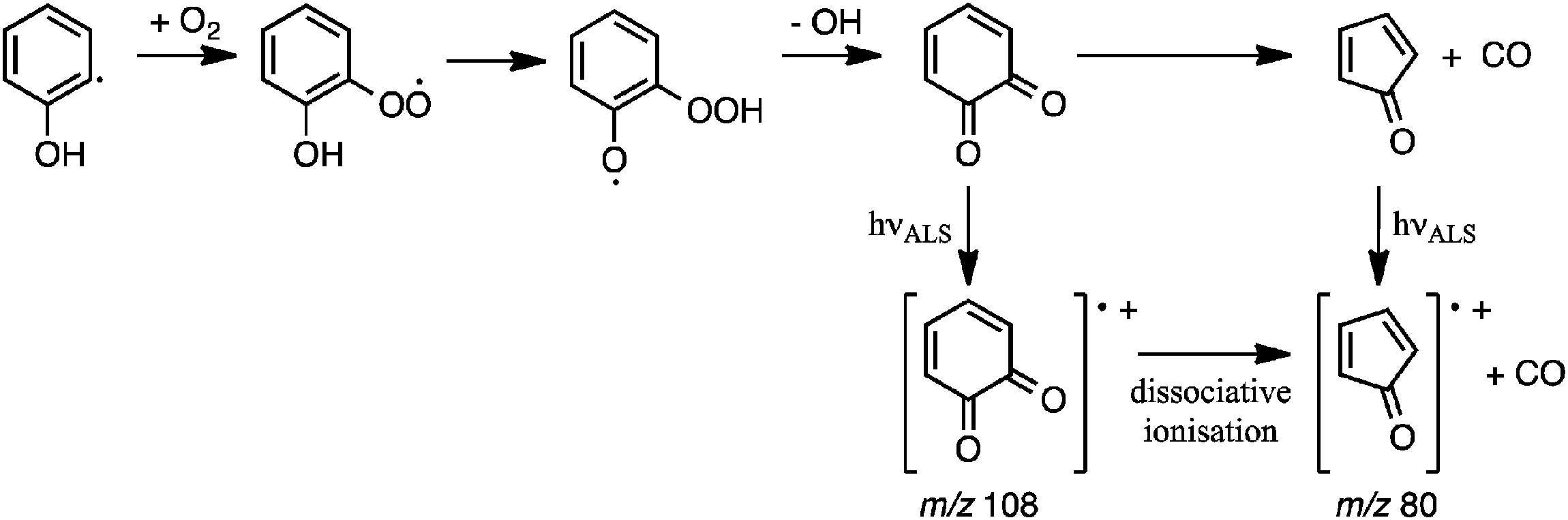 | ||
| Scheme 1 | ||
Included in Fig. 2a are reference PI spectra for CPO from Yang et al.54 and Parker et al.55 The close agreement between the m/z 80 and reference PI spectra shown in Fig. 2a from 9 to 9.8 eV support our assignments of m/z 80 as CPO. It is evident that at PI energies ≥9.8 eV all m/z 80 PI spectra diverge with the reference spectra under-predicting the current experimental data. Additional PI spectra acquired at 500 and 600 K (Fig. S1 and S2, ESI†) also diverge from the reference spectra at PI energies ≥9.8 eV.
The possibility of other C5H4O isomers contributing to the m/z 80 ion signal was ruled out by calculating AIEs for closed-shell linear C5H4O isomers listed in Table S1 (ESI†). Isomers were excluded on the basis of having: an AIE <9.2 eV, or an AIE >10.0 eV and a relatively high formation enthalpy. As it stands, CPO is the only plausible isomer contributing to the m/z 80 PI spectra, however, the source of neutral CPO and the cause of the disparity around 9.8 eV in the m/z 80 PI spectra (Fig. 2a) require further examination.
The systematic differences between the m/z 80 signal and CPO reference spectra in Fig. 2a at photoionisation energies ≥9.8 eV could arise from dissociative ionisation of higher mass species, where o-BQ is a likely candidate. It is known that dissociative ionisation of 1,2-naphthoquinone and 9,10-phenanthrenequinone result in CO loss (both contain the o-BQ substructure).58 Fig. S3 (ESI†) shows product mass spectra at 373, 500 and 600 K integrated over two energy ranges; 9.40–9.75 eV (Fig. S3a–c, ESI†) and 9.85–10.00 eV (Fig. S3d–f, ESI†). These mass spectra reveal some variation in the product ratios but no additional product signals. Comparing the kinetic traces for m/z 80 and 108 at 500 K integrated over 9.40–9.75 eV (Fig. S4a, ESI†) shows that the kinetic traces are clearly different and consistent with the dominant fraction of each ion population arising from photoionisation of different neutrals. However, at higher energies (9.85–10.00 eV, Fig. S4b, ESI†), the m/z 80 and 108 kinetic traces appear more similar – this is consistent with a portion of C6H4O2 (108 Da) undergoing dissociative ionisation to yield product ions with m/z 80. Furthermore, the potential energy scheme for CO loss from the o-BQ radical cation (m/z 108) provided in Fig. 3 shows the cation dissociation barrier to be 9.7 eV relative to neutral o-BQ. These results support the proposition that at photoionisation energies ≥9.8 eV some of the m/z 80 signal arises from the dissociative ionisation of o-BQ. This contribution is in addition to the ionisation of CPO produced within the reactive flow.
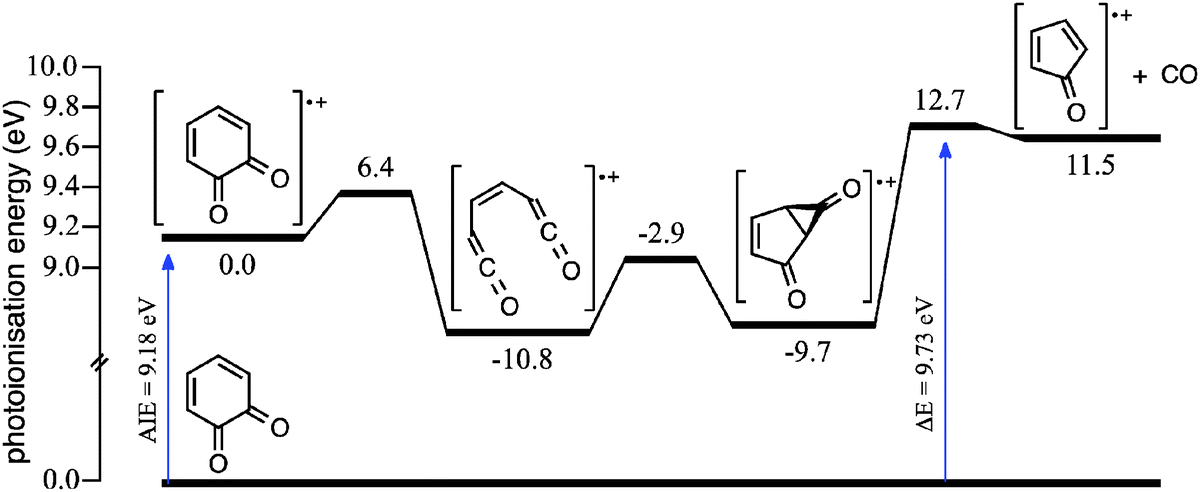 | ||
| Fig. 3 Potential energy schematic that depicts loss of CO from the o-BQ radical cation. CBS-QB3 0 K enthalpies are provided in kcal mol−1 relative to the o-BQ radical cation. CBS-QB3 AIE and dissociation barrier are provided in eV relative to o-BQ. | ||
Analogous to product pathways of the phenylperoxyl radical in phenyl + O2 reactions,59–62 CPO could be produced after decomposition of hydroxyl-substituted oxepinoxyl radicals. Unimolecular reaction pathways leading directly to CPO are discounted on the basis of experiments in Section 3.2 and prohibitively high energy pathways reported in Section 3.3.3. Ultimately, we propose that the m/z 80 and 108 products are generated according to processes summarised in Scheme 1: the o-hydroxyphenyl radical undergoes O2 addition to form the hydroxyphenylperoxyl radical and subsequent OH loss to produce o-BQ. And, a portion of the nascent vibrationally-excited o-BQ population then decomposes via decarbonylation to produce CPO.18–20 In addition, dissociative ionisation of o-BQ possibly contributes to the measured m/z 80 signal at energies ≥9.8 eV.
To further establish connections between the reaction products of o-hydroxyphenyl + O2 (cf.Scheme 1), charge-tagged derivatives of o-hydroxyphenyl radicals were prepared within an ion-trap mass spectrometer (at the University of Wollongong). The study of distonic radical ions can provide useful insight into the reactions of their neutral radical counterparts. The presence of a relatively unreactive charged substituent enables isolation and manipulation of reactive intermediates using ion-trap mass spectrometry, while products arise from reactions with the spatially separated radical moiety.17,63,64 Quantum chemical calculations were also conducted, and discussed later in Section 3.3, to rationalise experimental results for both the neutral and charge-tagged systems.
3.2 Ion-trap mass spectrometry: distonic o-hydroxyphenyl + O2
Photodissociation (PD, λ = 266 nm) of isolated m/z 188 and 190 ions ([M + H]+, assigned 3-bromo-4-hydroxybenzaminium cation) resulted in the m/z 109 signal in Fig. 4a. The m/z 109 ion, consistent with Br loss, was assigned to the 5-ammonium-2-hydroxyphenyl radical cation shown in Scheme 2. Isolation of this radical cation in the presence of background O2 (6.4 × 109 molecules cm−3) resulted in a major product ion at m/z 124 and a minor product ion at m/z 96 (<1%) that both grew in with increasing reaction times (0.030–10000 ms). A mass spectrum acquired with 2000 ms reaction time is shown in Fig. 4b. The m/z 124 product ion is rationalised by O2 addition to the charge-tagged 2-hydroxyphenyl radical followed by prompt OH elimination to yield 4-ammonium-2-benzoquinone.
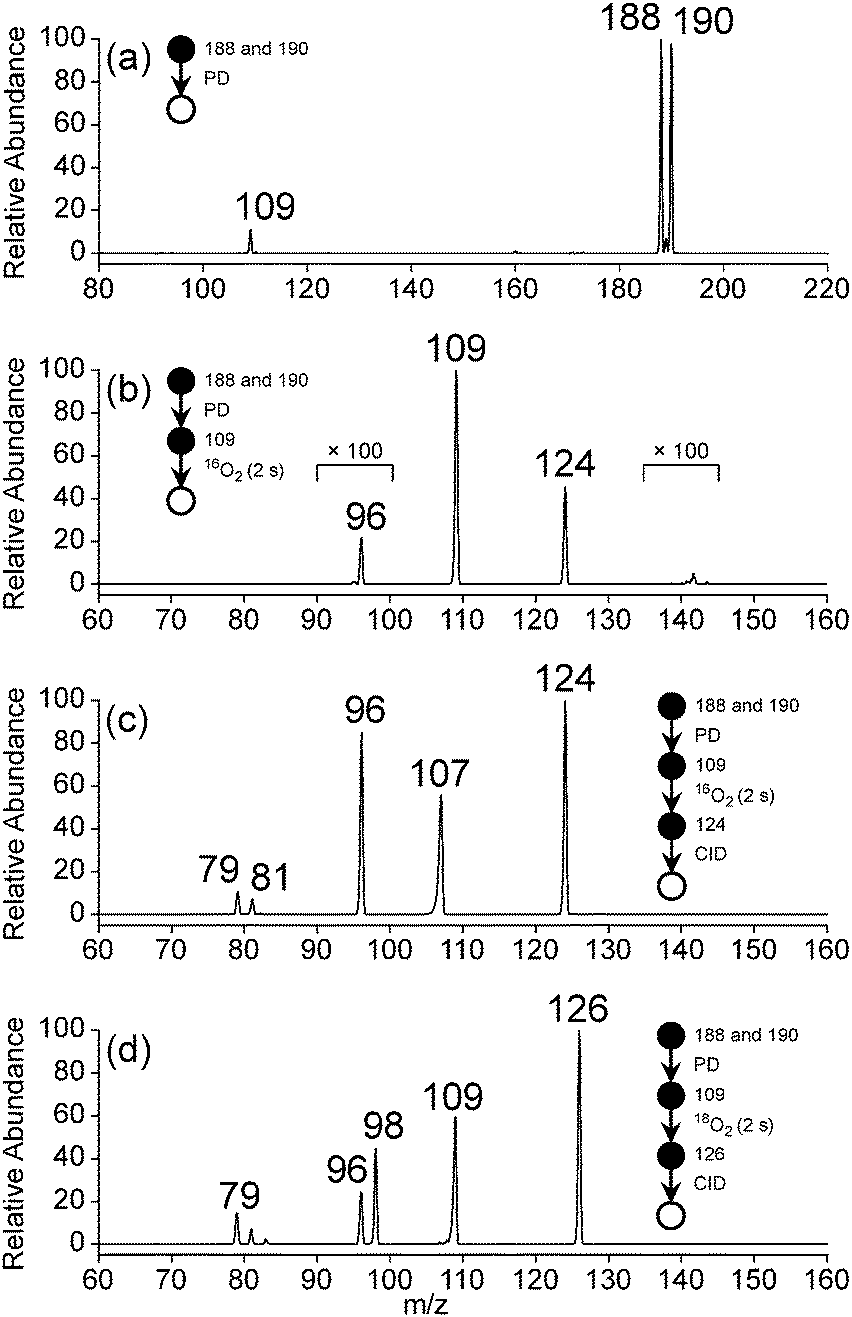 | ||
| Fig. 4 Mass spectra resulting from (a) PD of 3-bromo-4-hydroxybenzenaminium (m/z 188 and 190), (b) isolation of m/z 109 ions resulting from 266 nm PD of 3-bromo-4-hydroxybenzenaminium and storage for 2 seconds in the presence of background O2 (109 molecules cm−3), and (c) CID of the product ion at m/z 124. Experiments were repeated with 18O2 and (d) the product ion at m/z 126 was subjected to CID. | ||
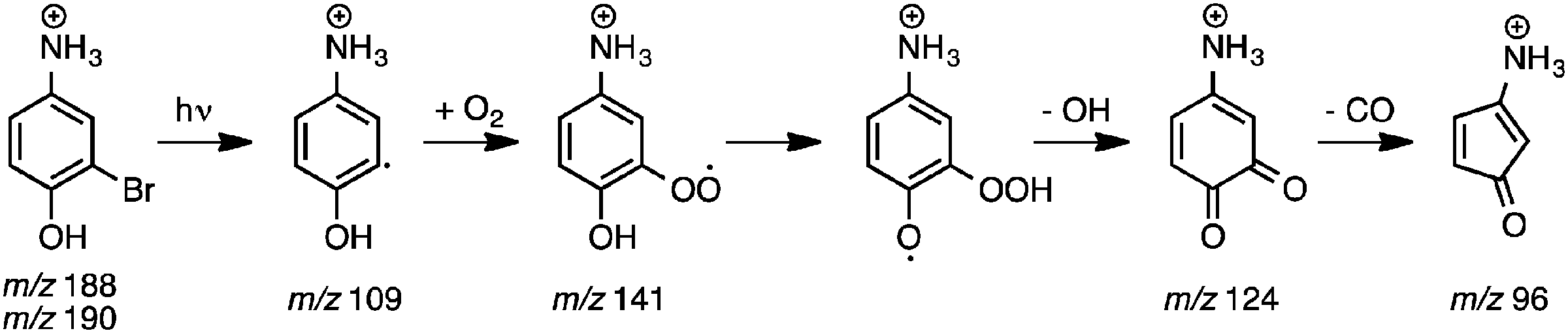 | ||
| Scheme 2 | ||
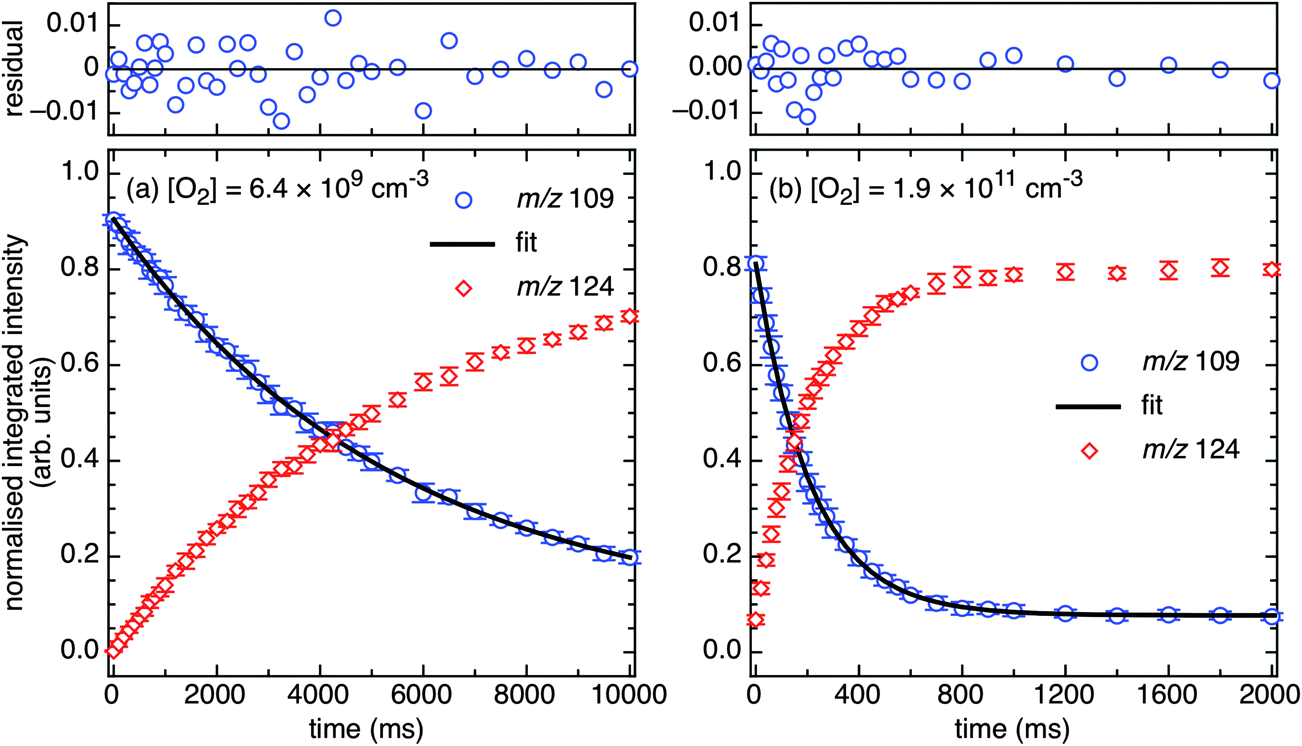 | ||
| Fig. 5 Kinetic curves for m/z 109 (solid blue circles), from PD of 3-bromo-4-hydroxybenzaminium cations, in reactions with (a) background O2 (6.4 × 109 molecules cm−3) and (b) increased O2 (1.9 × 1011 molecules cm−3). Residual plots from the fitting of eqn (1) are provided above. The m/z 124 product data are shown (red diamonds) and track with a rate coefficient in agreement with the m/z 109 decay (within 2σ). Error bars are 1σ. | ||
The mass spectrum in Fig. 4c, from isolation and subsequent collision-induced dissociation (CID) of m/z 124 product ions, shows major signals at m/z 96 and 107 and minor signals at m/z 79 and 81. The product ion at m/z 96 (−28 Da) is consistent with decarbonylation of ammonium-tagged o-BQ to yield ammonium-tagged CPO + CO. Fragment ions at m/z 107, 79, and 81 are assigned to loss of NH3 (−17 Da), NH3 + CO (−45 Da) and NC2H5 or C2H3O (−43 Da) from m/z 124, respectively. To verify these assignments, 18O2 was introduced into the ion trap and reacted with m/z 109 radical cations. The m/z 126 ions produced are consistent with 18O2 addition and 18OH loss (−19 Da, Fig. 4d) and exclude any contribution of NH3 loss (−17 Da). Isolation and subsequent CID of the m/z 126 ions resulted in fragments at m/z 96 and 98 consistent with loss of C18O and C16O from 18O-labelled o-BQ to yield CPO. Taken together, these data demonstrate a connection between the o-BQ intermediate (m/z 124) and the CPO structure (m/z 96) via processes summarised in Scheme 2. These data do not provide evidence for a “phenyl-like” oxidation mechanism for the direct formation of CPO via phenoxyl and oxepinoxyl radicals.59–62 Other fragment ions at m/z 79, 81, 83, and 109 are assigned to loss of NH3 + C18O (−47 Da), NH3 + C16O or C2H318O (−45 Da), NC2H5 or C2H316O (−43 Da), and NH3 (−17 Da) from m/z 126, respectively.
Potential energy schemes for formation of o-BQ and CPO are compared and discussed for both neutral and distonic cases in Section 3.3. Reactions of the PD generated 5-ammonium-2-hydroxyphenyl radical cation (m/z 109) with O2 were characterised further by kinetic measurements.
000 ms) to produce kinetic curves that describe decay of m/z 109 ion signal intensity due to reactions with O2.
A single exponential decay (eqn (1)) was satisfactorily fitted to the experimental data, in accord with pseudo-first order kinetic behaviour. Representative kinetic curves for m/z 109 and 124 ions are provided in Fig. 5 with fitted data and residuals from eqn (1) for m/z 109 signal decay. The k1st values for m/z 109 signal decay and m/z 124 signal growth are in agreement (e.g., in Fig. 5a, 1.9 ± 0.2 s−1 compared to 1.8 ± 0.1 s−1 within 2σ) and the m/z 124 intensity is well matched to the m/z 109 signal decay. This indicates that m/z 124 ions are the main reaction product from depletion of m/z 109 ions. As shown in Fig. 5b, at increased O2 concentrations the m/z 109 ion signal intensity ultimately approaches a constant value of ca. 10% at 1000 ms and remains constant up to a reaction time limit of 10000 ms. This indicates the presence of an unreactive isomer (or isomers) and is accounted for by the constant offset included in eqn (1).
Additional experiments that compare the oxidation kinetics of the ammonium-tagged o-hydroxyphenyl and o-methylphenyl radical cations along with trimethylammonium-tagged analogues are now described. Sample kinetic plots are provided in Fig. S7 (ESI†) for oxidation of 5-ammonium-2-methylphenyl radical cations (m/z 107) and in Fig. S8 (ESI†) for 5-(N,N,N-trimethylammonium)-2-hydroxyphenyl radical cations (m/z 151). For reactions of 5-ammonium-2-methylphenyl radical cations (m/z 107) the non-zero horizontal offset (shown in Fig. S7b, ESI†) is ca. 40% of the isolated m/z 107 ion population. Interestingly, k1st values for 5-ammonium-2-methylphenyl radical (m/z 107) and 5-ammonium-2-hydroxyphenyl radical (m/z 109) signal decay are separable with 2σ uncertainty, where the k1st for the 5-ammonium-2-hydroxyphenyl radical cations is reproducibly greater by ca. 15%. In the case of trimethylammonium-tagged o-hydroxyphenyl radical + O2 reactions, the m/z 151 ion population can be completely depleted by O2 reaction, suggesting that a pure population of trimethylammonium-tagged o-hydroxyphenyl radicals are formed from PD of the precursor. This observation is consistent with our previous investigation of trimethylammonium-tagged o-methylphenyl + O2 reaction kinetics17 and may be attributed to the greater number of internal degrees of freedom from the trimethylammonium substituent thus reducing the propensity for isomerisation.
Second-order rate coefficients (k2nd, cm3 molecule−1 s−1) and reaction efficiencies (Φ%) derived from fitted pseudo-first order rate coefficients (k1st) are reported in Table 2. Collision frequencies were calculated using the Langevin collision model.39 Kinetic measurements were conducted at background O2 ([O2] = 6.4 ± 0.4 × 109 molecule cm−3) and increased O2 concentrations ([O2] = 1.6–2.2 × 1011 molecule cm−3). Repeated kinetic measurements provided consistent results and statistical uncertainties from fitting k1st were typically 2σ ≤ 10%. These results indicate stable absolute O2 concentrations within the ion trap as indicated by the linear relationship between k1st and [O2] (Fig. S9, ESI†). However, the uncertainty in the ion-trap pressure ultimately results in an upper limit of 50% uncertainty in the trap [O2], and consequently, a 50% uncertainty for the reported second-order rate coefficients and reaction efficiencies in Table 2.
| Distonic radical | [O2] (molecule cm−3) | k 2nd (cm3 molecule−1 s−1) | Collision frequency (cm3 molecule−1 s−1) | Φ (%) |
|---|---|---|---|---|
| a Rate coefficients reported in ref. 17. Ions of m/z 149 were generated by PD of the 3-bromo-N,N,N,4-trimethylbenzenaminium cation. | ||||
| 5-Ammonium-2-hydroxyphenyl | Low [109] | 2.9 × 10−11 | 5.9 × 10−10 | 4.9 |
| High [1011] | 3.2 × 10−11 | 5.5 | ||
| 5-Ammonium-2-methylphenyl | Low [109] | 2.6 × 10−11 | 5.9 × 10−10 | 4.4 |
| High [1011] | 2.6 × 10−11 | 4.4 | ||
| 5-(N,N,N-Trimethylammonium)-2-hydroxyphenyl | 6.6 × 109 | 2.5 × 10−11 | 5.7 × 10−10 | 4.4 |
| 5-(N,N,N-Trimethylammonium)-2-methylphenyla | 2.2 × 109 | 2.9 × 10−11 | 5.7 × 10−10 | 5.1 |
| 8.5 × 109 | 2.6 × 10−11 | 4.5 | ||
Reaction efficiencies for all species reported in Table 2 are all approximately equal to 5%, similar to reported reaction efficiencies for neutral phenyl radicals and a range of positively charged distonic phenyl radical ions, including trimethylammonium and pyridinium-tagged phenyl radicals,27 and distonic o-methylphenyl radicals.17 For the 5-ammonium-2-hydroxyphenyl + O2 reaction mechanism discussed further below, proximity of the ortho-OH-substituent to the peroxyl-radical site in the o-hydroxyphenylperoxyl radical provides a notably low-energy reaction pathway (refer to Fig. 6) that competes with dissociation of the peroxyl radical intermediate toward separated reactants, however, it does not appear to significantly affect measured reaction efficiencies compared to the other values reported in Table 2 and for the phenyl-type radical + O2 reactions cited above. This moderate reaction efficiency of ∼5%, consistent for a range of phenyl-type radicals,17,27 indicates that the rate of reaction is not controlled by the microcanonical rate for forward dissociation pathways. Instead, it may result from an entropic bottleneck after formation of the non-covalent complex between the phenyl radical and O2 (ref. 65–67) that reflects reaction flux back to the free reactants. More experiments and insights are required to address this question.
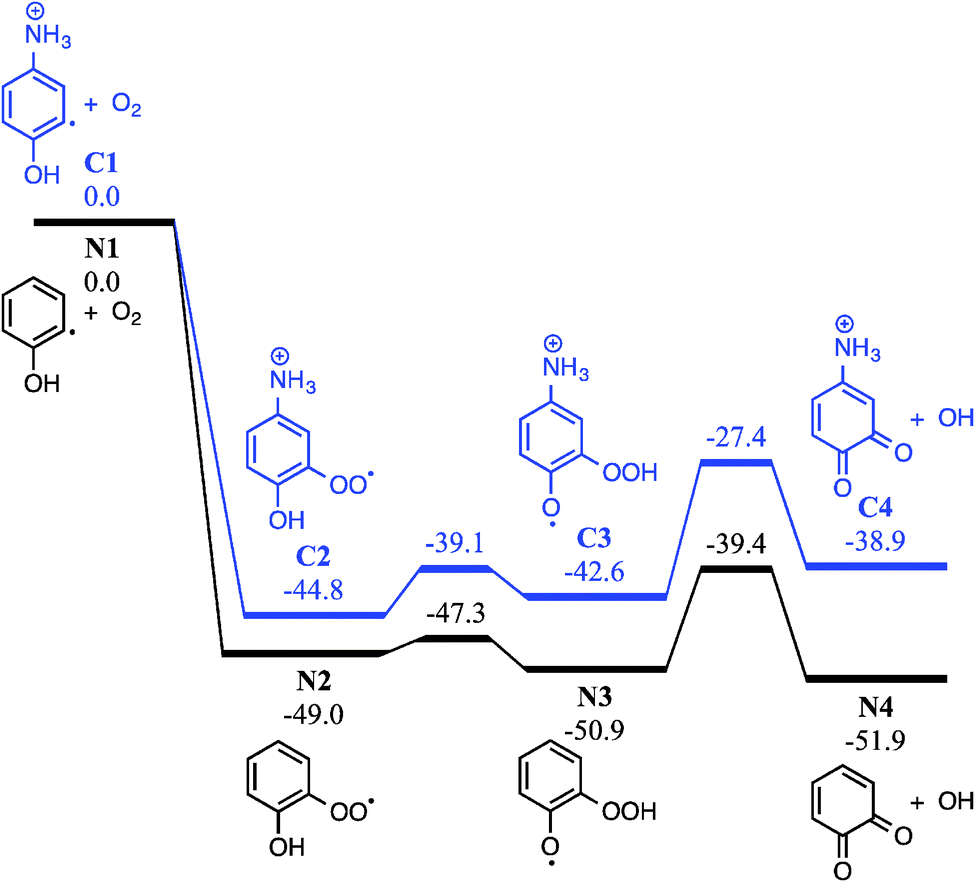 | ||
| Fig. 6 Potential energy schematic depicting the peroxyl → hydroperoxy radical isomerisation and subsequent OH elimination for both neutral (black) and ammonium-tagged (blue) o-hydroxyphenyl + O2. G3X-K 0 K enthalpies are provided in kcal mol−1 relative to the respective o-hydroxyphenyl + O2 reactants. | ||
3.3 Reaction mechanism
Addition of O2 to the neutral o-hydroxyphenyl radical produces the o-hydroxyphenylperoxyl radical species (N2) that is 49.0 kcal mol−1 below the energy of separated reactants (N1), as shown in Fig. 6. Close proximity of the OH substituent to the peroxyl radical site in the o-hydroxyphenylperoxyl radical (N2) allows for a 1,5-H shift viaTSN2 → N3 with an incredibly small 1.7 kcal mol−1 barrier to the o-hydroperoxyphenoxyl radical (N3). Elimination of OH from the hydroperoxyl group in N3viaTSN3 → N4 (11.5 kcal mol−1 barrier) results in o-BQ + OH with a reaction exothermicity of 51.9 kcal mol−1. Comparing this to the charge-tagged case, the o-hydroxyphenylperoxyl radical analogue (C2) is 44.8 kcal mol−1 below the energy of the separated reactants (C1). The barrier to the 1,5-H shift in C2 and subsequent OH loss from C3 is 27.4 kcal mol−1 below reactants and the resulting 4-ammonium-2-benzoquinone + OH (C4) products are formed with an exothermicity of 38.9 kcal mol−1 (13.0 kcal mol−1 less than in the neutral case). As shown by Fig. S10 in the ESI,† the reaction enthalpy for charge-tagged o-BQ + OH is reduced by separation of the charge tag and ring structure via inclusion of methylene linkages, indicating that differences shown in Fig. 6 are (in part) due to a through-space charge effect. Still, intermediates and transition states for both cases shown in Fig. 6 are well below the energy of the reactants and, therefore, OH-elimination is expected to be facile. This mechanism is consistent with the appearance of o-BQ (m/z 108) in neutral flow-tube experiments and ammonium-tagged o-BQ (m/z 124) in the distonic radical cation experiments.
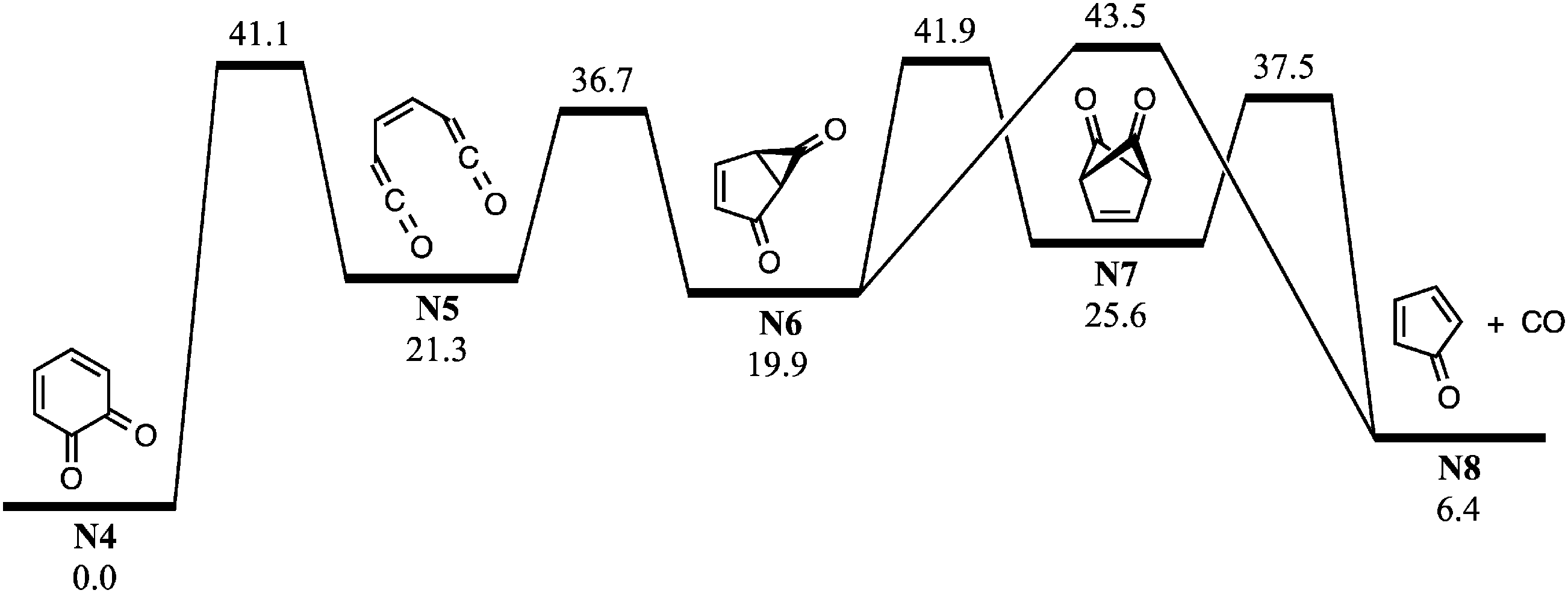 | ||
| Fig. 7 Potential energy schematic for CO loss from o-BQ (N4) along the singlet C6H4O2 surface. G3X-K 0 K enthalpies are provided in kcal mol−1 relative to o-BQ. | ||
Fig. S11 (ESI†) shows the potential energy scheme for CO elimination from ammonium-tagged o-BQ. The mechanisms shown in Fig. S11a (ESI†) feature barriers that exceed the entrance channel (5-ammonium-2-hydroxyphenyl + O2) by 2.8 kcal mol−1viaTSC6a → C8 and 6.7 kcal mol−1viaTSC6b → C8. In Fig. S11b (ESI†), however, the highest barrier is 35.0 kcal mol−1viaTSC4 → C5 (3.8 kcal mol−1 below 5-ammonium-2-hydroxyphenyl + O2). The decomposition reactions shown in Fig. S11 (ESI†) are less likely to proceed due to the reduced exothermicity of the charge-tagged o-BQ + OH and barriers to decarbonylation approaching the entrance channel limit. Collisional activation of the charge-tagged o-BQ intermediate should provide the activation energy required to generate charge-tagged CPO + CO, consistent with a loss of 28 Da from CID of m/z 124 ions shown in Fig. 4c. The appearance of a small m/z 96 ion peak in Fig. 4b (<1%), prior to isolation of the m/z 124 ion, may result from decomposition of the high-energy portion of the nascent m/z 124 ion ensemble. It is likely that further exploration of o-BQ decomposition is required to reveal additional competitive pathways resulting in CPO + CO.
 | ||
| Fig. 8 Potential energy schematic comparing the OH-cis and OH-trans reaction pathways to the hydroxyoxepinoxyl and 6-carboxy-1-oxo-hex-2,4-dienyl intermediates along the neutral o-hydroxyphenyl + O2 reaction surface. The barrier to TSN3 → N4 (leading to o-BQ) and reaction enthalpy for the o-hydroxyphenoxyl radical (N40) are included for comparison. G3X-K energies are reported in kcal mol−1 relative to o-hydroxyphenyl + O2. | ||
Rearrangement of o-hydroxyphenylperoxyl (N2) toward 7-hydroxyoxepinoxyl (N23) and 6-carboxy-1-oxo-hex-2,4-dienyl radicals (N21) via dioxirane-hydroxycyclohexadienyl intermediates are described in Fig. 8. The reactions of these intermediates represent plausible unimolecular pathways to both m/z 80 and 108 ions in the ALS experiments. Reactions toward the 7-hydroxyoxepinoxyl radical proceeds through TSN2 → N22 18.1 kcal mol−1 above the barrier to o-BQ (TSN3 → N4 in Fig. 6) with an exothermicity of 102.3 kcal mol−1. Formation of the 6-carboxy-1-oxo-hex-2,4-dienyl radical (N21) occurs viaTSN2 → N20 at 11.6 kcal mol−1 above TSN3 → N4. Reactions of charged-tagged o-hydroxyphenylperoxyl toward hydroxyoxepinoxyl and 6-carboxyoxohexdienyl, shown in Fig. S13 (ESI†), generally parallel those described by Fig. 8. The rate limiting steps toward the two hydroxyoxepinoxyl (TS3c) and carboxyoxohexdienyl radicals (TS1c) are 22.2 and 5.1 kcal mol−1, respectively, above the barrier to charge-tagged o-BQ (TS C3 → C4, Fig. 6).
In the case where either hydroxyoxepinoxyl or carboxyoxohexdienyl radicals are produced, their decomposition could possibly follow pathways described by Fig. S14 and S15 (ESI†). Likely products, by analogy to phenyl radical oxidation,16,61 include o-BQ + OH, CPO + HOCO, 3-hydroxy-2-benzoquinone + H, and 2-hydroxy-cyclopentadienone + HCO with barriers far below the reactants. The absence of peaks at m/z 96 and 124 within ALS experimental results (Fig. 1) and the high barriers to hydroxyoxepinoxyl and carboxyoxohexdienyl intermediates indicate that at most only a small fraction of the reaction flux follows these channels. Furthermore, preliminary RRKM modelling of the o-hydroxyphenylperoxyl radical (N2) decomposition, utilizing MultiWell,45 indicates H-migration and OH-loss to form o-BQ (N4, shown in Fig. 6) comprehensively outcompetes the pathways toward hydroxyoxepinoxyl and carboxyoxohexadienyl radicals (shown in Fig. 8, N2 toward TS1n and TS4n). The sums of states for salient transition states and corresponding rate coefficients are provided in Table S2 (ESI†). The oxepinoxyl pathways (viaTS1n and TS4n) experience comparatively tight transition states with state counts several orders of magnitude lower than any other along the o-BQ pathway. This is in accord with our previous statement that the prevailing mechanism is formation of o-BQ via an o-hydroxyperoxylphenoxyl radical intermediate (N3). The appearance of m/z 80 in ALS experiments is explained by o-BQ decomposition, supported by distonic experiments that show connectivity between the analogous charge-tagged species.
4. Conclusions
Product detection experiments conducted at the ALS synchrotron reveal that the o-hydroxyphenyl + O2 reaction produces two major products detected at m/z 80 and 108 that are consistent with CPO and o-BQ. We conclude that CPO forms as a secondary product from prompt decomposition of o-BQ and dissociative ionisation of o-BQ leads to some enhancement of the m/z 80 signal at photoionisation energies ≥9.8 eV. There are indications of a minor o-hydroxyphenoxyl + O(3P) pathway in the ALS experiments. To establish connections between the major reaction products, distonic radical analogue ammonium-tagged o-hydroxyphenyl + O2 reactions were studied using ion-trap mass spectrometry. Reactions of the 5-ammonium-2-hydroxyphenyl radical cation (m/z 109) with O2 produced product ions consistent with ammonium-tagged o-BQ produced via O2 addition, H-atom migration and subsequent OH loss. CID of the m/z 124 ions yielded a species assigned ammonium-tagged CPO produced by CO loss.Second order rate coefficients (k2nd) for 5-ammonium-2-hydroxyphenyl (m/z 109) + O2 were measured to have a 5% reaction efficiency. Additional kinetic measurements for O2 reactions with PD generated 5-ammonium-2-methylphenyl and 5-(N,N,N-trimethylammonium)-2-hydroxyphenyl radical cations and a previous investigation of trimethylammonium-tagged o-methylphenyl + O2 reaction kinetics17 demonstrate for this small set that the identity of the charged-tag and ortho-substituent does not significantly affect the reaction efficiency (ca. 5%).
Quantum chemical calculations are in accord with our experimental observations, where a 1,5-H shift in the o-hydroxyphenylperoxyl adduct and subsequent OH elimination is the minimum energy pathway for both o-hydroxylphenyl + O2 and the ammonium-tagged counterpart. Decomposition of the o-BQ toward CPO does encounter large barriers. However, the indication from preliminary kinetic modelling is that production of o-BQ is the dominant unimolecular pathway.
The prevailing mechanism for decomposition of the o-hydroxyphenylperoxyl radical produced by O2 addition is via 1,5-H migration and OH loss from the hydroperoxyphenoxyl radical intermediate to produce o-BQ. Its decomposition via ring opening, cyclisation, and CO elimination is the likely pathway to CPO. These proposed pathways to o-BQ and CPO serve as source of OH and CO species in reactive environments.
Acknowledgements
The authors are grateful for the financial support of the Australian Research Council through the Discovery programs (SJB: DP140101237; AJT and GdS: DP 130100862; GdS: FT130101304) and Centre of Excellence For Free Radical Chemistry and Biotechnology (CE0561607). The authors also acknowledge the generous allocation of computing resources by the NCI National Facility (Canberra, Australia) under Merit Allocation Scheme. BBK is supported by the National Aeronautics and Space Administration (NNH13AV43I). JDS, DLO, and CAT are supported by the Division of Chemical Sciences, Geosciences, and Biosciences, the Office of Basic Energy Sciences, the U.S. Department of Energy. Sandia is a multi-program laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the National Nuclear Security Administration under contract DE-AC04-94-AL85000. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility at the Lawrence Berkeley National Laboratory. MBP would like to acknowledge Dr. Phillip J. Tracey for discussions on distonic radical cation experiments.References
- G. Gellerstedt, J. Li, I. Eide, M. Kleinert and T. Barth, Chemical Structures Present in Biofuel Obtained from Lignin, Energy Fuels, 2008, 22, 4240–4244 CrossRef CAS.
- Y. Huang, H. Sakamoto, S. Kudo, K. Norinaga and J.-i. Hayashi, Pyrolysis of Lignite with Internal Recycling and Conversion of Oil, Energy Fuels, 2014, 28, 7285–7293 CrossRef CAS.
- W. W. Focke, I. van der Westhuizen, A. B. L. Grobler, K. T. Nshoane, J. K. Reddy and A. S. Luyt, The Effect of Synthetic Antioxidants on the Oxidative Stability of Biodiesel, Fuel, 2012, 94, 227–233 CrossRef CAS.
- N. A. Santos, A. M. T. M. Cordeiro, S. S. Damasceno, R. T. Aguiar, R. Rosenhaim, J. R. Carvalho Filho, I. M. G. Santos, A. S. Maia and A. G. Souza, Commercial Antioxidants and Thermal Stability Evaluations, Fuel, 2012, 97, 638–643 CrossRef CAS.
- S. Schober and M. Mittelbach, The Impact of Antioxidants on Biodiesel Oxidation Stability, Eur. J. Lipid Sci. Technol., 2004, 106, 382–389 CrossRef CAS.
- B. D. Batts and A. Z. Fathoni, A Literature Review on Fuel Stability Studies with Particular Emphasis on Diesel Oil, Energy Fuels, 1991, 5, 2–21 CrossRef CAS.
- S. Lomnicki, H. Truong and B. Dellinger, Mechanisms of Product Formation from the Pyrolytic Thermal Degradation of Catechol, Chemosphere, 2008, 73, 629–633 CrossRef CAS PubMed.
- E. Bjergbakke, A. Sillesen and P. Pagsberg, UV Spectrum and Kinetics of Hydroxycyclohexadienyl Radicals, J. Phys. Chem., 1996, 100, 5729–5736 CrossRef CAS.
- C. Venkat, K. Brezinsky and I. Glassman, High Temperature Oxidation of Aromatic Hydrocarbons, Proc. Combust. Inst., 1982, 19, 143–152 CrossRef.
- G. da Silva, M. R. Hamdan and J. W. Bozzelli, Oxidation of the Benzyl Radical: Mechanism, Thermochemistry, and Kinetics for the Reactions of Benzyl Hydroperoxide, J. Chem. Theory Comput., 2009, 5, 3185–3194 CrossRef CAS PubMed.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, M. R. Nimlos, H.-H. Carstensen and G. Barney Ellison, Unimolecular Thermal Decomposition of Phenol and D5-Phenol: Direct Observation of Cyclopentadiene Formation Via Cyclohexadienone, J. Chem. Phys., 2012, 136, 044309 CrossRef PubMed.
- Y. Z. He, W. G. Mallard and W. Tsang, Kinetics of Hydrogen and Hydroxyl Radical Attack on Phenol at High Temperatures, J. Phys. Chem., 1988, 92, 2196–2201 CrossRef CAS.
- T. Berndt and O. Boge, Gas-Phase Reaction of OH Radicals with Phenol, Phys. Chem. Chem. Phys., 2003, 5, 342–350 RSC.
- R. Atkinson, Kinetics and Mechanisms of the Gas-Phase Reactions of the Hydroxyl Radical with Organic Compounds, J. Phys. Chem. Ref. Data, 1989, Monograph No. 1, 246 Search PubMed.
- D. J. Robichaud, A. M. Scheer, C. Mukarakate, T. K. Ormond, G. T. Buckingham, G. B. Ellison and M. R. Nimlos, Unimolecular Thermal Decomposition of Dimethoxybenzenes, J. Chem. Phys., 2014, 140, 234302 CrossRef PubMed.
- G. da Silva, C.-C. Chen and J. W. Bozzelli, Toluene Combustion: Reaction Paths, Thermochemical Properties, and Kinetic Analysis for the Methylphenyl Radical + O2 Reaction, J. Phys. Chem. A, 2007, 111, 8663–8676 CrossRef CAS PubMed.
- M. B. Prendergast, P. A. Cooper, B. B. Kirk, G. da Silva, S. J. Blanksby and A. J. Trevitt, Hydroxyl Radical Formation in the Gas Phase Oxidation of Distonic 2-Methylphenyl Radical Cations, Phys. Chem. Chem. Phys., 2013, 15, 20577–20584 RSC.
- D. C. DeJongh, R. Y. Van Fossen and C. F. Bourgeois, Comparative Studies of Electron-Impact and Thermolytic Fragmentation. II. o-Phenylene Sulfite, Tetrahedron Lett., 1967, 8, 271–276 CrossRef.
- D. DeJongh and D. Brent, Mass Spectra and Pyrolyses of o-Phenylene Carbonate and Related Compounds, J. Org. Chem., 1970, 35, 4204–4208 CrossRef CAS.
- O. L. Chapman and C. L. McIntosh, Cyclopentadienone, J. Chem. Soc. D, 1971, 770–771 RSC.
- T. K. Ormond, A. M. Scheer, M. R. Nimlos, D. J. Robichaud, T. P. Troy, M. Ahmed, J. W. Daily, T. L. Nguyen, J. F. Stanton and G. B. Ellison, Pyrolysis of Cyclopentadienone: Mechanistic Insights from a Direct Measurement of Product Branching Ratios, J. Phys. Chem. A, 2015, 119, 7222–7234 CrossRef CAS PubMed.
- B. N. Moore, S. J. Blanksby and R. R. Julian, Ion-Molecule Reactions Reveal Facile Radical Migration in Peptides, Chem. Commun., 2009, 5015–5017 RSC.
- D. J. M. Ray, A. Redfearn and D. J. Waddington, Gas-Phase Oxidation of Alkenes: Decomposition of Hydroxy-Substituted Peroxyl Radicals, J. Chem. Soc., Perkin Trans. 2, 1973, 540–543 RSC.
- D. J. M. Ray and D. J. Waddington, Gas Phase Oxidation of Alkenes—Part II. The Oxidation of 2-Methylbutene-2 and 2,3-Dimethylbutene-2, Combust. Flame, 1973, 20, 327–334 CrossRef CAS.
- O. Welz, J. Zádor, J. D. Savee, L. Sheps, D. L. Osborn and C. A. Taatjes, Low-Temperature Combustion Chemistry of N-Butanol: Principal Oxidation Pathways of Hydroxybutyl Radicals, J. Phys. Chem. A, 2013, 117, 11983–12001 CrossRef CAS PubMed.
- P. E. Williams, B. J. Jankiewicz, L. Yang and H. I. Kenttämaa, Properties and Reactivity of Gaseous Distonic Radical Ions with Aryl Radical Sites, Chem. Rev., 2013, 113, 6949–6985 CrossRef CAS PubMed.
- B. B. Kirk, D. G. Harman, H. I. Kenttämaa, A. J. Trevitt and S. J. Blanksby, Isolation and Characterization of Charge-Tagged Phenylperoxyl Radicals in the Gas Phase: Direct Evidence for Products and Pathways in Low Temperature Benzene Oxidation, Phys. Chem. Chem. Phys., 2012, 14, 16719–16730 RSC.
- D. L. Osborn, P. Zou, H. Johnsen, C. C. Hayden, C. A. Taatjes, V. D. Knyazev, S. W. North, D. S. Peterka, M. Ahmed and S. R. Leone, The Multiplexed Chemical Kinetic Photoionization Mass Spectrometer: A New Approach to Isomer-Resolved Chemical Kinetics, Rev. Sci. Instrum., 2008, 79, 104103 CrossRef PubMed.
- A. G. Suits, P. Heimann, X. Yang, M. Evans, C.-W. Hsu, K.-t. Lu, Y. T. Lee and A. H. Kung, A Differentially Pumped Harmonic Filter on the Chemical Dynamics Beamline at the Advanced Light Source, Rev. Sci. Instrum., 1995, 66, 4841–4844 CrossRef CAS.
- S. R. Leone, M. Ahmed and K. R. Wilson, Chemical Dynamics, Molecular Energetics, and Kinetics at the Synchrotron, Phys. Chem. Chem. Phys., 2010, 12, 6564–6578 RSC.
- D. P. Biddiscombe and J. F. Martin, Vapour Pressures of Phenol and the Cresols, Trans. Faraday Soc., 1958, 54, 1316–1322 RSC.
- Normalised collision energy is a term used by the instrument vendor as explained here: http://https://static.thermoscientific.com/images/D13507~.pdf. Zero, on this scale, means that no additional resonant excitation is applied.
- T.-Y. Kim, M. S. Thompson and J. P. Reilly, Peptide Photodissociation at 157 Nm in a Linear Ion Trap Mass Spectrometer, Rapid Commun. Mass Spectrom., 2005, 19, 1657–1665 CrossRef CAS PubMed.
- T. Ly and R. R. Julian, Residue-Specific Radical-Directed Dissociation of Whole Proteins in the Gas Phase, J. Am. Chem. Soc., 2008, 130, 351–358 CrossRef CAS PubMed.
- T. Ly, B. B. Kirk, P. I. Hettiarachchi, B. L. J. Poad, A. J. Trevitt, G. da Silva and S. J. Blanksby, Reactions of Simple and Peptidic Alpha-Carboxylate Radical Anions with Dioxygen in the Gas Phase, Phys. Chem. Chem. Phys., 2011, 13, 16314–16323 RSC.
- C. S. Hansen, B. B. Kirk, S. J. Blanksby, R. A. J. O'Hair and A. J. Trevitt, UV Photodissociation Action Spectroscopy of Haloanilinium Ions in a Linear Quadrupole Ion Trap Mass Spectrometer, J. Am. Soc. Mass Spectrom., 2013, 24, 932–940 CrossRef CAS PubMed.
- D. G. Harman and S. J. Blanksby, Investigation of the Gas Phase Reactivity of the 1-Adamantyl Radical Using a Distonic Radical Anion Approach, Org. Biomol. Chem., 2007, 5, 3495–3503 CAS.
- W. A. Donald, G. N. Khairallah and R. A. J. O'Hair, The Effective Temperature of Ions Stored in a Linear Quadrupole Ion Trap Mass Spectrometer, J. Am. Soc. Mass Spectrom., 2013, 24, 811–815 CrossRef CAS PubMed.
- P. Langevin, A Fundamental Formula of Kinetic Theory, Ann. Chim. Phys., 1905, 5, 245 CAS.
- G. da Silva, G3X-K Theory: A Composite Theoretical Method for Thermochemical Kinetics, Chem. Phys. Lett., 2013, 558, 109–113 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- J. Zheng, Y. Zhao and D. G. Truhlar, The DBH24/08 Database and Its Use to Assess Electronic Structure Model Chemistries for Chemical Reaction Barrier Heights, J. Chem. Theory Comput., 2009, 5, 808–821 CrossRef CAS PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, A Complete Basis Set Model Chemistry. VI. Use of Density Functional Geometries and Frequencies, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, A Complete Basis Set Model Chemistry. VII. Use of the Minimum Population Localization Method, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef CAS.
- (a) MultiWell-2013.1 Software, designed and maintained by John R. Barker with contributors N. F. Ortiz, J. M. Preses, L. L. Lohr, A. Maranzana, P. J. Stimac, T. Lam Nguyen, and T. J. Dhilip Kumar; University of Michigan, Ann Arbor, MI; http://aoss.engin.umich.edu/multiwell/; (b) J. R. Barker, Int. J. Chem. Kinet., 2001, 33, 232–245 CrossRef CAS; (c) J. R. Barker, Int. J. Chem. Kinet., 2009, 41, 748–763 CrossRef CAS.
- R. Schulz and A. Schweig, Elucidation of Thermal Reactions by Variable Temperature Photoelectron Spectral Detection of Reactive Intermediates. The UV Photoelectron Spectra of Transient Fulveneketene, Fulvenethioketene, A Ketoketene and Thiobenzpropiolactone, Tetrahedron Lett., 1979, 20, 59–62 CrossRef.
- H. Bock, T. Hirabayashi and S. Mohmand, Gasphasen-Reaktionen, 21. Thermische Erzeugung Von Alkyl- Und Halogenketenen, Chem. Ber., 1981, 114, 2595–2608 CrossRef CAS.
- S. G. Lias, R. D. Levin and S. A. Kafafi, in NIST Chemistry Webbook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2011 Search PubMed.
- C. A. Taatjes, D. L. Osborn, T. M. Selby, G. Meloni, A. J. Trevitt, E. Epifanovsky, A. I. Krylov, B. Sirjean, E. Dames and H. Wang, Products of the Benzene + O(3P) Reaction, J. Phys. Chem. A, 2010, 114, 3355–3370 CrossRef CAS PubMed.
- K. Fuke, H. Yoshiuchi, K. Kaya, Y. Achiba, K. Sato and K. Kimura, Multiphoton Ionization Photoelectron Spectroscopy and Two-Color Multiphoton Ionization Threshold Spectroscopy on the Hydrogen Bonded Phenol and 7-Azaindole in a Supersonic Jet, Chem. Phys. Lett., 1984, 108, 179–184 CrossRef CAS.
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin and W. G. Mallard, in NIST Chemistry Webbook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2011 Search PubMed.
- N. Akai, S. Kudoh, M. Takayanagi and M. Nakata, Photoreaction Mechanisms of 2-Bromophenols Studied by Low-Temperature Matrix-Isolation Infrared Spectroscopy and Density-Functional-Theory Calculation, Chem. Phys. Lett., 2002, 363, 591–597 CrossRef CAS.
- T. K. Ormond, P. Hemberger, T. P. Troy, M. Ahmed, J. F. Stanton and G. B. Ellison, The Ionisation Energy of Cyclopentadienone: A Photoelectron–Photoion Coincidence Study, Mol. Phys., 2015, 113, 2350–2358 CrossRef CAS.
- J. Yang, Photonionization Cross Section Database, Center for Advanced Combustion and Energy, 2011 Search PubMed.
- D. S. N. Parker, R. I. Kaiser, T. P. Troy, O. Kostko, M. Ahmed and A. M. Mebel, Toward the Oxidation of the Phenyl Radical and Prevention of PAH Formation in Combustion Systems, J. Phys. Chem. A, 2015, 119, 7145–7154 CrossRef CAS PubMed.
- V. K. Potapov and V. V. Sorokin, Photoionization and Ion-Molecule Reactions in Quinones and Alcohols, High Energy Chem., 1971, 5, 435 Search PubMed.
- M. H. Palmer, W. Moyes, M. Speirs and J. N. A. Ridyard, The Electronic Structure of Substituted Benzenes; Ab Initio Calculations and Photoelectron Spectra for Phenol, the Methyl- and Fluoro-Derivatives, and the Dihydroxybenzenes, J. Mol. Struct., 1979, 52, 293–307 CrossRef CAS.
- Y. Pan, L. Zhang, T. Zhang, H. Guo, X. Hong and F. Qi, Photoionization Studies on Various Quinones by an Infrared Laser Desorption/Tunable VUV Photoionization Tof Mass Spectrometry, J. Mass Spectrom., 2008, 43, 1701–1710 CrossRef CAS PubMed.
- B. K. Carpenter, Computational Prediction of New Mechanisms for the Reactions of Vinyl and Phenyl Radicals with Molecular Oxygen, J. Am. Chem. Soc., 1993, 115, 9806–9807 CrossRef CAS.
- M. J. Fadden, C. Barckholtz and C. M. Hadad, Computational Study of the Unimolecular Decomposition Pathways of Phenylperoxy Radical, J. Phys. Chem. A, 2000, 104, 3004–3011 CrossRef CAS.
- M. J. Fadden and C. M. Hadad, Unimolecular Decomposition of the 2-Oxepinoxy Radical: A Key Seven-Membered Ring Intermediate in the Thermal Oxidation of Benzene, J. Phys. Chem. A, 2000, 104, 8121–8130 CrossRef CAS.
- I. V. Tokmakov, G.-S. Kim, V. V. Kislov, A. M. Mebel and M. C. Lin, The Reaction of Phenyl Radical with Molecular Oxygen: A G2M Study of the Potential Energy Surface, J. Phys. Chem. A, 2005, 109, 6114–6127 CrossRef CAS PubMed.
- B. B. Kirk, D. G. Harman and S. J. Blanksby, Direct Observation of the Gas Phase Reaction of the Cyclohexyl Radical with Dioxygen Using a Distonic Radical Ion Approach, J. Phys. Chem. A, 2010, 114, 1446–1456 CrossRef CAS PubMed.
- C. H. Li, G. N. Khairallah, A. K. Y. Lam, R. A. J. O'Hair, B. B. Kirk, S. J. Blanksby, G. da Silva and U. Wille, Reaction of Aromatic Peroxyl Radicals with Alkynes: A Mass Spectrometric and Computational Study Using the Distonic Radical Ion Approach, Chem. – Asian J., 2013, 8, 450–464 CrossRef CAS PubMed.
- G. da Silva and J. W. Bozzelli, Variational Analysis of the Phenyl + O2 and Phenoxy + O Reactions, J. Phys. Chem. A, 2008, 112, 3566–3575 Search PubMed.
- J. J. Casero and J. A. Joens, Thermochemistry of Gas-Phase Molecular Complexes of Fluorobenzene and Toluene with Oxygen, J. Phys. Chem. A, 1999, 103, 7136–7138 CrossRef CAS.
- M. L. Chabinyc, S. L. Craig, C. K. Regan and J. I. Brauman, Gas-Phase Ionic Reactions: Dynamics and Mechanism of Nucleophilic Displacements, Science, 1998, 279, 1882–1886 CrossRef CAS PubMed.
- S. Skokov, A. Kazakov and F. L. Dryer, presented in part at the Fourth Joint Meeting of the U.S. Sections of the Combustion Institute, Philadelphia, PA, March 20-23, 2005.
- C. Barckholtz, M. J. Fadden and C. M. Hadad, Computational Study of the Mechanisms for the Reaction of O2(3σg) with Aromatic Radicals, J. Phys. Chem. A, 1999, 103, 8108–8117 CrossRef CAS.
- R. P. Lindstedt and G. Skevis, Detailed Kinetic Modeling of Premixed Benzene Flames, Combust. Flame, 1994, 99, 551–561 CrossRef CAS.
- N. Sebbar, H. Bockhorn and J. Bozzelli, Thermodynamic Properties of the Species Resulting from the Phenyl Radical with O2 Reaction System, Int. J. Chem. Kinet., 2008, 40, 583–604 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02953h |
| This journal is © the Owner Societies 2016 |