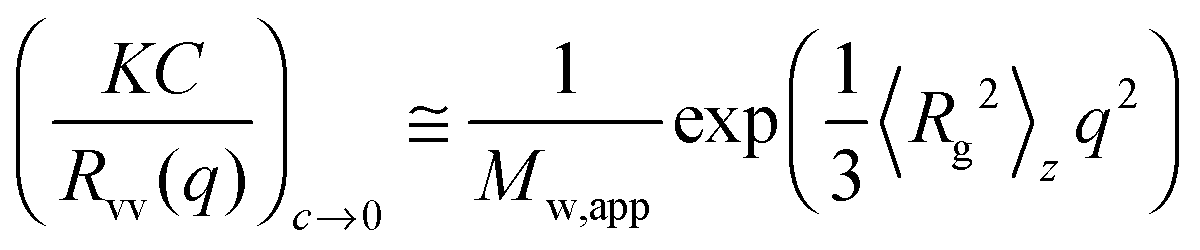PNIPAmx–PPO36–PNIPAmx thermo-sensitive triblock copolymers: chain conformation and adsorption behavior on a hydrophobic gold surface
Received
9th October 2015
, Accepted 16th November 2015
First published on 20th November 2015
Abstract
The chain conformations and adsorption behaviors of four thermo-sensitive poly(N-isopropylacrylamide)x–poly(propylene oxide)36–poly(N-isopropylacrylamide)x (PNIPAmx–PPO36–PNIPAmx) triblock copolymers with x values of 15, 33, 75, and 117 in dilute aqueous solutions were investigated by combined techniques of micro-differential scanning calorimetry (micro-DSC), static and dynamic light scattering (SLS & DLS), and the quartz crystal microbalance (QCM). PNIPAm15–PPO36–PNIPAm15 only exhibited the lower critical solution temperature (LCST) of the PPO block, i.e. 25 °C, because the PNIPAm block with x = 15 was too short to maintain its own LCST. With middle lengths x of 33 and 75, the LCSTs of PPO and PNIPAm blocks were observed, respectively. For the longest PNIPAm block with x = 117, only LCST of PNIPAm block dominated, i.e. 32.3 °C. DLS results revealed that the four PNIPAmx–PPO36–PNIPAmx triblock copolymers formed “associate” structures in their dilute aqueous solutions at 20 °C, which was well below the LCSTs of the PPO and PNIPAm blocks. QCM results indicated that the adsorption time constant decreased with increasing adsorption temperature but tended to increase with increasing length x of the PNIPAm block. A complex adsorption behavior with large adsorption amounts was only observed at the corresponding LCST of the PNIPAm block for PNIPAmx–PPO36–PNIPAmx with longer PNIPAm blocks with x = 33, 75, and 117. Furthermore, the adsorbed PNIPAmx–PPO36–PNIPAmx layers obtained at 20 °C were rigid with less energy dissipation.
Introduction
A block copolymer consisting of blocks of different monomers makes it possible to combine several different properties in one molecule. The chain conformations, solution properties and the interactions between block copolymers and solid surfaces might be varied by changing the chemical composition, length of the block, concentration of the block copolymer solution as well as the environmental properties like temperature, pH, ionic strength, etc.1–9 Depending on the chain conformations and solution properties, solutions of block copolymers might have potential applications as micelles for pay-load and -release, compatibilizers, or emulsifiers, etc.10,11 The chain conformations and the block length of block copolymers might strongly affect their adsorption behaviors on solid surfaces, which are relevant to many applications in physical, chemical and biological fields, including polymer coatings, self-assembly nanostructures, biomedicine, and so on. Many efforts have been carried out to investigate and understand the chain conformations and solution properties of block copolymers.12–18 The adsorption processes of polymers including homopolymers, diblock and triblock as well as multiblock copolymers on the solid substrates and the interactions between polymers and solid substrates were also experimentally and theoretically studied.19–29 The chain conformations, solution properties and adsorption behaviors of environmental responsive polymers have particularly attracted a lot of attention because the environmental stimuli such as temperature, pH, or ionic strength could alter their chain conformations and hence the adsorption behaviors. “Smart” surface coatings could be created by the adsorption of such environmental responsive polymers.
Typically, the adsorption behaviors of a thermo-responsive polymer, poly(N-isopropylacryamide) (PNIPAm), on solid substrates under various conditions have been extensively studied and correlated with the chain conformation of PNIPAm in aqueous solution.30–33 Liu et al.25 investigated the adsorption behaviors of a pentablock terpolymer, poly(2-dimethylaminoethyl methacrylate)–poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide)–poly(2-dimethylaminoethyl methacrylate) (PDMAEMA24–PEO132–PPO50–PEO132–PDMAEMA24) on various solid substrates, which were correlated with the effects of electrostatic screening, polymer hydrodynamic size, and solvency. Du et al.18,29,34,35 systematically studied the chain conformations, solution properties and adsorption behaviors of a series of double thermo-sensitive pentablock terpolymers, poly(N-isopropylacrylamide)–poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide)–poly(N-isopropylacrylamide) (PNIPAm–PEO–PPO–PEO–PNIPAm) with various relative block lengths of PNIPAm, PEO and PPO blocks, including PNIPAm110–PEO100–PPO65–PEO100–PNIPAm110 (PNIPAm110–F127–PNIPAm110) and PNIPAmx–PEO20–PPO70–PEO20–PNIPAmx (PNIPAmx–P123–PNIPAmx) with x values of 10, 63, and 97. PNIPAm110–F127–PNIPAm110 exhibited two lower critical solution temperatures (LCSTs) at 31 °C and 34 °C in dilute aqueous solution, which were attributed to the thermal phase transition of the PPO block and the PNIPAm block, respectively.18 Similarly, two LCSTs were also observed for PNIPAmx–P123–PNIPAmx pentablock terpolymers in dilute aqueous solutions.35 It was found that the adsorption behaviors and mechanisms of PNIPAm–PEO–PPO–PEO–PNIPAm pentablock terpolymers on hydrophobic gold surfaces were indeed strongly dependent on the chain conformations in aqueous solution at various temperatures and the gold substrates with different surface wetting and chemical properties. Especially, a complex adsorption behavior with large adsorption amounts was observed at the corresponding LCST of the PNIPAm block for the adsorption of PNIPAm110–F127–PNIPAm110 and PNIPAm97–P123–PNIPAm97 on hydrophobic gold substrates, which was attributed to the presence of the PNIPAm block and the premicellar associative adsorption right at the LCST of PNIPAm block.29,34 The adsorption behaviors of PEO100–PPO65–PEO100 (F127) at various temperatures were also investigated.34 However, no complex adsorption behavior was observed for F127.
To further understand and differentiate the effects of chain conformations and the length of the PNIPAm block on such complex adsorption behavior observed for PNIPAm–PEO–PPO–PEO–PNIPAm pentablock terpolymers, a series of thermo-sensitive PNIPAmx–PPO36–PNIPAmx triblock copolymers with x values of 15, 33, 75, and 117 were synthesized in the present work via atom transfer radical polymerization (ATRP). Only PPO and PNIPAm blocks were thus involved in these triblock copolymers. The chain conformations, solution and adsorption behaviors of PNIPAmx–PPO36–PNIPAmx in dilute aqueous solutions were investigated by micro-differential scanning calorimetry (micro-DSC), static and dynamic laser light scattering (SLS & DLS), and the quartz crystal microbalance (QCM). The effects of the PNIPAm block length x on the chain conformations, thermal transition and adsorption behaviors of PNIPAmx–PPO36–PNIPAmx were discussed. It was found that the PNIPAm block should necessarily reach a certain block length in order to maintain its own LCST and the presence of the PNIPAm block is not a sufficient condition for the observation of complex adsorption behavior. If the PNIPAm block exhibited its own LCST, the complex adsorption behavior was then observed at the corresponding LCST of the PNIPAm block for PNIPAmx–PPO36–PNIPAmx triblock copolymers.
Experimental section
Chemical and materials
N-Isopropylacrylamide (NIPAm, 98%) was purchased from Tokyo Chemical Industry (TCI) and purified by recrystallization from n-hexane. Polypropylene oxide (HO–PPO36–OH, Mn = 2000 g mol−1, Mw/Mn = 1.05) and tris(2-dimethylaminoethyl)amine (Me6TREN, 99%+) were purchased from Alfa Aesar and used as received. 2-Propanol was obtained from Sinopharm Chemical Reagent Co., Ltd and dried with CaH2 overnight, distilled under ambient pressure before use. CuBr (98%, Strem Chemicals Inc.) was purified by sequent washing with glacial acetic acid and absolute ethanol, and then dried under vacuum. 2-Bromoisobutyryl bromide (98%) was purchased from J&K Chemical Ltd and used as received. All other reagents were used as received.
Synthesis of ATRP macroinitiator Br–PPO36–Br
Bromo end functionalized Br–PPO36–Br was synthesized according to a reported method under modified conditions.36 Briefly, HO–PPO36–OH (10 mmol, 20 g) was first dissolved in 250 mL anhydrous toluene in a round-bottom flask. Secondly, triethylamine (30 mmol, 4.1 mL) was added and the solution was cooled to 0 °C using an ice bath. 2-Bromoisobutyryl bromide (30 mmol, 3.71 mL) was then added dropwise from a syringe over 30 min under stirring, and the reaction mixture was further stirred for 48 h at room temperature. The insoluble amine salt was then removed by filtration, and the resulting solution was extracted with 5% aqueous HCl three times and then with 5% aqueous NaHCO3 three times, and washed with distilled water. The resulting solution was further dried with MgSO4 and filtered again to give a clear solution, which was then dried by rotary evaporation to remove the solvent. The 1H NMR spectrum of Br–PPO36–Br indicated that the degree of esterification was approximately 100%.
Synthesis of triblock copolymer PNIPAmx–PPO36–PNIPAmx
A standard ATRP procedure was applied to synthesize triblock copolymer PNIPAmx–PPO36–PNIPAmx. Br–PPO36–Br (0.1 mmol, 0.23 g), 6 mL 2-propanol, NIPAm (40 mmol, 4.53 g) and Me6TREN (0.2 mmol, 62 μL) were added to a 50 mL dry reaction tube. The mixture was degassed with three freezing–evacuation–thawing cycles before CuBr (0.2 mmol, 28.8 mg) was added. The mixture was allowed to stir for forming the copper complex, and then placed in an oil bath at 50 °C for polymerization. After given polymerization time, polymerization was terminated by freezing with liquid nitrogen. The reaction mixture was purified by dialysis against distilled water to remove the copper catalyst, solvent, and unreacted monomer and then freeze dried to give the final purified triblock copolymer PNIPAmx–PPO36–PNIPAmx.
Instrumentation and characterization
The molecular weight and molecular weight distribution of triblock copolymer PNIPAmx–PPO36–PNIPAmx were determined by using a Waters 1515 GPC (Waters Corporation) with DMF/0.05 M LiBr as the eluent and monodisperse poly(methyl methacrylate) (PMMA) as the calibration standard. The number average molecular weight, Mn, was also calculated from 1H-NMR of the triblock copolymer, which was performed by using a 400 MHz Varian Mercury Plus NMR instrument with CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard.
Micro-differential scanning calorimetry (micro-DSC) measurements were performed by using a VP DSC (MicroCal) in Hefei National Laboratory for Physical Sciences at Microscale, University of Science and Technology of China. The triblock copolymer PNIPAmx–PPO36–PNIPAmx was dissolved in de-ionized water to give a concentration of 5 mg mL−1. The heating rate was 1.0 °C min−1.
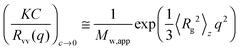
The static and dynamic light scattering (SLS & DLS) of the aqueous solutions of PNIPAmx–PPO36–PNIPAmx were carried out by using a commercial spectrometer ALV-CGS-3 Light Scattering Electronics and Multiple Tau Digital Correlator in Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. The wavelength of laser light λ was 632.8 nm. All of the solutions (0.5 mg mL−1) were filtered through 0.45 μm Millipore PVDF filters into the dust-free light scattering cells before measurements. The sample vial was then placed in the sample holder with a precise temperature control. The sample solution was equilibrated for 15 min at each set of temperatures before measurement. The average hydrodynamic radius 〈Rh〉 and the distribution of 〈Rh〉 from the DLS were calculated by using cumulant analysis and the CONTIN routine. The range of scattering angle θ used for SLS was from 45° to 135° with a step of 5°. The z-average root-mean-square radius of gyration, i.e. 〈Rg〉, of the triblock copolymer in a dilute aqueous solution can be determined from the angular dependence of the excess scattered intensity, i.e. the Rayleigh ratio Rvv(q), by using the Guinier equation:37,38
| |  | (1) |
where
K = 4π
n2(d
n/d
c)
2/(
NAλ4) and
q = (4π
n/
λ)sin(
θ/2), with d
n/d
c,
NA,
λ,
n and
θ being the specific refractive index increment of the solution, the Avogadro number, the wavelength of the laser light in a vacuum, the refractive index of the solvent, the scattering angle, respectively. d
n/d
c values of the triblock copolymers in aqueous solutions were determined by using a differential refractometer.
The adsorption behaviors of PNIPAmx–PPO36–PNIPAmx triblock copolymers on solid surfaces were investigated by using a quartz crystal microbalance (QCM, Resonant Probes GmbH, Goslar, Germany). AT-cut quartz crystals with a fundamental resonant frequency f0 of 5 MHz and gold electrodes were used (Hangzhou Dongwei Inc., Hangzhou, China). The gold electrodes of quartz crystals were modified by self-assembling a monolayer of 1-dodecanethiol (HS(CH2)11CH3) to give well-defined hydrophobic surfaces according to the method reported previously.29 The static water contact angles of the 1-dodecanethiol modified gold surfaces were ca. 108 ± 2°. The 1-dodecanethiol-modified quartz crystals were stored in ethanol before use. The triblock copolymers PNIPAmx–PPO36–PNIPAmx were dissolved in de-ionized water to give a concentration of 0.5 mg mL−1 at least one day before the QCM adsorption measurements. For QCM measurement, the 1-dodecanethiol-modified quartz crystal was taken and dried with nitrogen. The 1-dodecanethiol-modified quartz crystal was then installed in the QCM’s liquid cell with temperature control and a stable baseline was established in the flow of de-ionized water with a flow rate of 50 μL min−1. The de-ionized water was then replaced with the aqueous solution of PNIPAmx–PPO36–PNIPAmx. The adsorption of PNIPAmx–PPO36–PNIPAmx was monitored until equilibrium and adsorption was clearly observed unless otherwise stated. Afterwards, the quartz crystal was rinsed with de-ionized water for 15 minutes by replacing the copolymer solution with de-ionized water. The frequency shift (ΔF3/3) and half-band-half-width shift (ΔΓ3/3) of the quartz crystal were recorded vs. time at the 3rd overtone because of the sufficient energy trapping at the 3rd overtone of the quartz crystal operated in liquid.39 After the QCM measurement, the quartz crystal with the adsorbed polymer layer was taken out of the liquid cell and dried with nitrogen.
Results and discussion
Synthesis of triblock copolymer PNIPAmx–PPO36–PNIPAmx
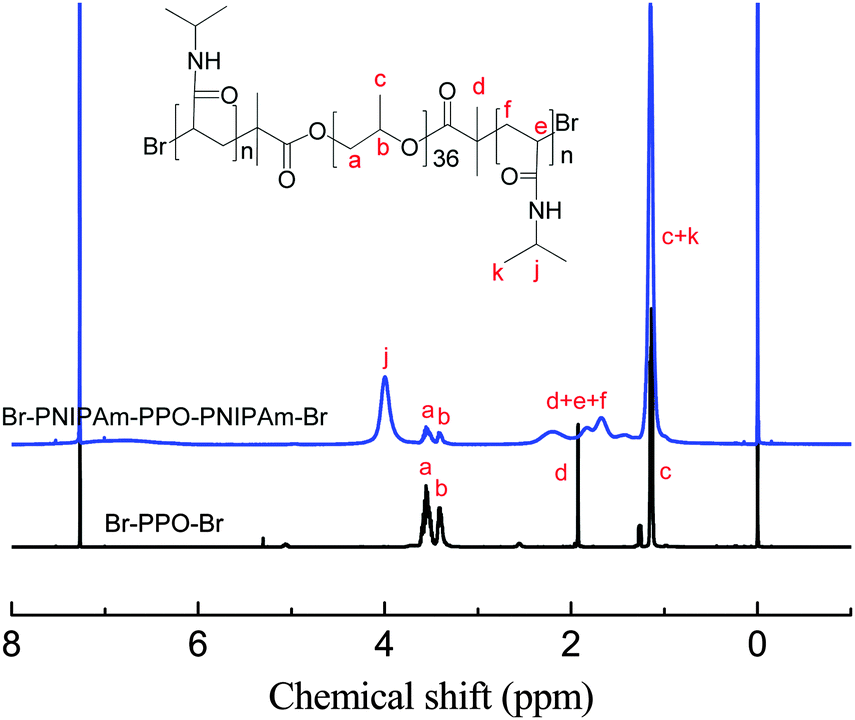
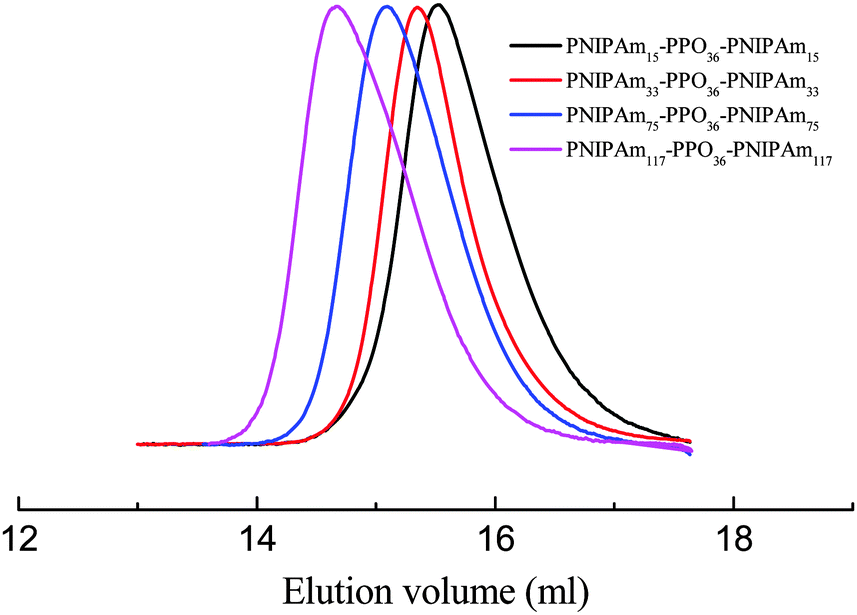
A standard ATRP procedure was applied to synthesize triblock copolymer PNIPAmx–PPO36–PNIPAmx. The macroinitiator Br–PPO36–Br was obtained by modifying the hydroxyl end-groups of HO–PPO36–OH with 2-bromoisobutyryl bromide, which was confirmed by the 1H-NMR spectrum shown in Fig. 1. A series of triblock copolymers, PNIPAmx–PPO36–PNIPAmx, were then synthesized via the ATRP procedure with macroinitiator Br–PPO36–Br and various feeding amounts of monomer NIPAm. The typical 1H-NMR spectrum of PNIPAmx–PPO36–PNIPAmx is shown in Fig. 1. The block length x of the PNIPAm block was determined from the ratio of the integrated intensities of the peaks j and a + b, which were attributed to the PNIPAm block and the PPO block, respectively. Fig. 2 shows the GPC traces of PNIPAmx–PPO36–PNIPAmx. Four triblock copolymers, PNIPAmx–PPO36–PNIPAmx with x of 15, 33, 75, and 117, respectively, and narrow molecular weight distribution were successfully obtained. The number average molecular weight Mn and the polydispersity Mw/Mn of PNIPAmx–PPO36–PNIPAmx are summarized in Table 1.
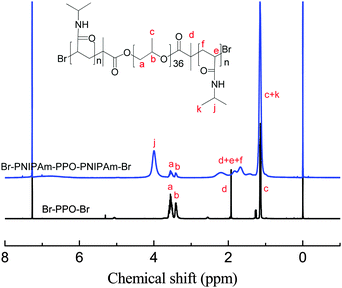 |
| | Fig. 1
1H-NMR spectra of macroinitiator Br–PPO36–Br and triblock copolymer PNIPAm117–PPO36–PNIPAm117. | |
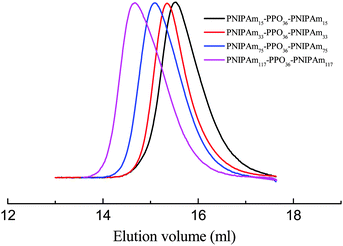 |
| | Fig. 2 GPC traces of the four triblock copolymers, PNIPAmx–PPO36–PNIPAmx. | |
Table 1 Some physical parameters of PNIPAmx–PPO36–PNIPAmx triblock copolymers
| Sample |
M
n
(×103 g mol−1) |
M
n
(×103 g mol−1) |
M
w/Mnb |
LCST (°C) |
Conversion of NIPAma (%) |
| I |
II |
|
Determined from 1H-NMR.
Determined from GPC.
|
| PNIPAm15–PPO36–PNIPAm15 |
7.4 |
5.7 |
1.26 |
25 |
— |
30 |
| PNIPAm33–PPO36–PNIPAm33 |
10.0 |
9.76 |
1.24 |
27.8 |
30 |
44 |
| PNIPAm75–PPO36–PNIPAm75 |
11.3 |
19.3 |
1.24 |
31.2 |
32.6 |
50 |
| PNIPAm117–PPO36–PNIPAm117 |
42.2 |
28.7 |
1.26 |
— |
32.3 |
58.5 |
Micro-DSC measurements of PNIPAmx–PPO36–PNIPAmx in dilute aqueous solutions
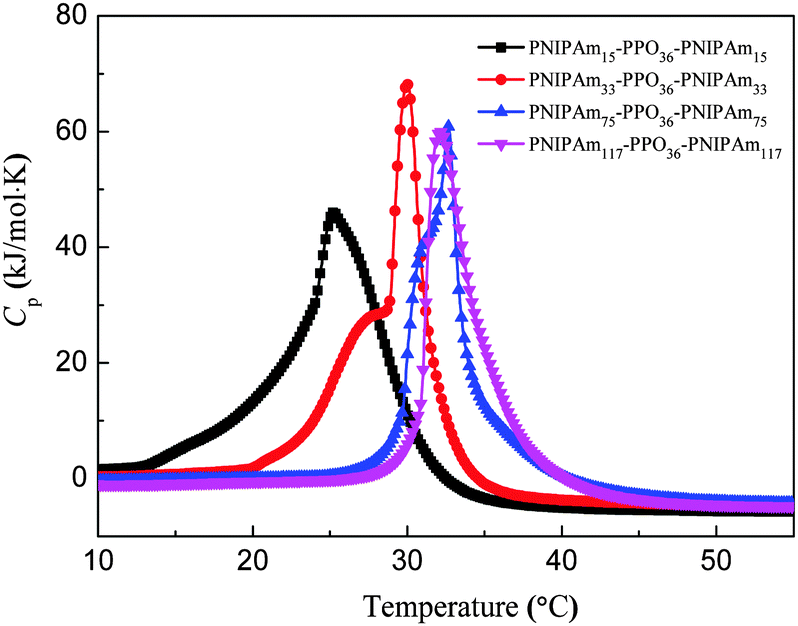
The micro-DSC data curves of PNIPAmx–PPO36–PNIPAmx triblock copolymers in dilute aqueous solutions with the concentration of 5 mg mL−1 are shown in Fig. 3. It can be seen that the thermal behavior of PNIPAmx–PPO36–PNIPAmx triblock copolymers in dilute aqueous solutions strongly depended on the length x of the PNIPAm block. With a shortest PNIPAm block length of 15, PNIPAm15–PPO36–PNIPAm15 exhibited a broad thermal transition with a peak center at 25 °C. However, PNIPAm33–PPO36–PNIPAm33 showed a bimodal transition with a shoulder endothermic peak centered at 27.8 °C and a strong sharp endothermic peak centered at 30 °C. PNIPAm75–PPO36–PNIPAm75 also showed a bimodal transition but with higher transition temperature, i.e. a shoulder endothermic peak centered at 31.2 °C and a strong sharp endothermic peak centered at 32.6 °C. Further increasing the length x of the PNIPAm block upto 117, PNIPAm117–PPO36–PNIPAm117 exhibited only one narrow thermal transition with a peak center at 32.3 °C. It has been reported that the linear homopolymers, PPO and PNIPAm, exhibited lower critical solution temperatures (LCST) at around 16 °C and 32–34 °C in aqueous solution, respectively.40–43 For PNIPAm15–PPO36–PNIPAm15, the PNIPAm block is too short to maintain its own LCST so that only one thermal transition was observed. Furthermore, the PNIPAm block is hydrophilic with temperature lower than its LCST, i.e. 32 °C. Therefore, the presence of hydrophilic PNIPAm blocks would broaden the thermal transition of the PPO block and shift the LCST of the PPO block to higher temperature, i.e. 25 °C, for PNIPAm15–PPO36–PNIPAm15. With increasing x to 33, the PNIPAm block is long enough to maintain its own LCST. As a result, PNIPAm33–PPO36–PNIPAm33 exhibited two LCSTs of 27.8 °C and 30 °C, which could be attributed to the LCSTs of the PPO block and the PNIPAm block, respectively. In this case, the LCST of the PPO block shifted to a higher temperature of 27.8 °C due to the presence of hydrophilic PNIPAm blocks. However, for the PNIPAm block, the PPO block is hydrophobic so that the LCST of PNIPAm blocks shifted to a lower temperature of 30 °C. Hasan et al. also observed two LCSTs for PPO–PNIPAm diblock copolymers in aqueous solutions.40 Similarly, for PNIPAm75–PPO36–PNIPAm75, the LCST of the PPO block further shifted to a higher temperature of 31.2 °C, whereas the LCST of PNIPAm blocks was 32.6 °C, which was close to that of linear homopolymer PNIPAm and higher than that of PNIPAm blocks in PNIPAm33–PPO36–PNIPAm33. Possibly, when the length of the PNIPAm block was much longer than that of the PPO block, the effect of the PPO block on the LCST of the PNIPAm block was weak. This possibility was supported by the evidence that for PNIPAm117–PPO36–PNIPAm117 with the longest PNIPAm block length of 117, the LCST of PNIPAm blocks dominated, i.e. 32.3 °C, and only one thermal transition was observed (cf.Fig. 3). Similarly, the LCST of the PNIPAm block of PNIPAm117–PPO36–PNIPAm117 was also close to that of linear hompolymer PNIPAm. Two LCSTs were also observed for PNIPAm110–PEO100–PPO65–PEO100–PNIPAm110 (PNIPAm110–F127–PNIPAm110) and PNIPAmx–PEO20–PPO70–PEO20–PNIPAmx (PNIPAmx–P123–PNIPAmx) pentablock terpolymers, which were attributed to the LCSTs of the PPO block and the PNIPAm block, respectively.18,35 Similarly, it was found that the LCST of the PPO block of PNIPAmx–P123–PNIPAmx was strongly dependent on the length x of the PNIPAm block. The LCST of the PPO block shifted to higher temperature with increasing x for PNIPAmx–P123–PNIPAmx. However, in the case of PNIPAmx–P123–PNIPAmx, the LCST of the PNIPAm block was less affected by the length of the PNIPAm block.
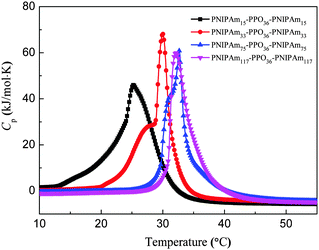 |
| | Fig. 3 Temperature dependence of the specific heat capacity (Cp) for PNIPAmx–PPO36–PNIPAmx in aqueous solutions. The concentration was 5 mg mL−1 and the heating rate was 1.0 °C min−1. | |
Chain conformations of PNIPAmx–PPO36–PNIPAmx in dilute aqueous solutions
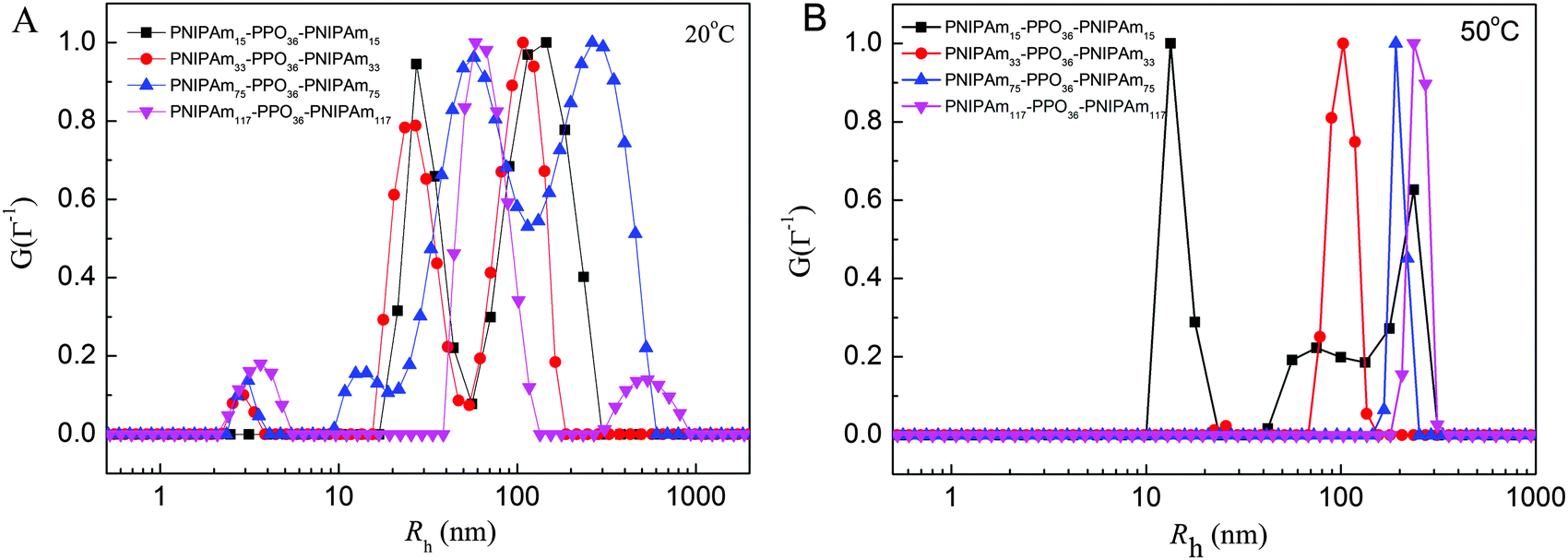
The chain conformations of PNIPAmx–PPO36–PNIPAmx triblock copolymers in dilute aqueous solutions at various temperatures, especially at the corresponding LCSTs, were studied by static and dynamic light scattering (SLS & DLS). Fig. 4 shows the distribution of the hydrodynamic radius 〈Rh〉 of PNIPAmx–PPO36–PNIPAmx in dilute aqueous solutions at 20 °C and 50 °C, as measured by DLS. Multimodal distribution was observed at 20 °C (Fig. 4A), which was well below the LCSTs of the PPO and PNIPAm blocks of PNIPAmx–PPO36–PNIPAmx triblock copolymers (Fig. 4A). Note that the LCSTs of PPO blocks for PNIPAm15–PPO36–PNIPAm15, PNIPAm33–PPO36–PNIPAm33 and PNIPAm75–PPO36–PNIPAm75 were 25 °C, 27.8 °C, and 31.2 °C, respectively (cf.Table 1 and Fig. 3). Because PPO and PNIPAm blocks were soluble in aqueous solution at 20 °C, the observation of large 〈Rh〉 indicated the formation of “associate” structures” for PNIPAmx–PPO36–PNIPAmx triblock copolymers. There was a dynamic equilibrium between single chain conformation and “associate” structures for PNIPAmx–PPO36–PNIPAmx triblock copolymers in aqueous solution at 20 °C.18 The “associate” structures have been observed for many PEO-containing block copolymers such as PEO-b-PNIPAm, PEO-b-poly(N,N-dimethylacrylamide) (PEO-b-PDMA), and PNIPAm–PEO–PPO–PEO–PNIPAm terpolymers in their dilute aqueous solutions at low temperature, at which each block was soluble in water.14,15,44,45 It was supposed that the formation of such “associate” structures could be attributed to the incompatibility between PNIPAm and PEO.44,46 Here, the “associate” structures were observed for PNIPAm–PPO–PNIPAm triblock copolymers in aqueous solutions. PPO and PNIPAm homopolymers are polar polymers and exhibit thermo-sensitive character. The dissolution of PNIPAm in aqueous solution at low temperature is attributed to the favorable enthalpy of amide–water hydrogen bonding, which stabilizes the swollen state of PNIPAm by overcoming the unfavorable entropy of water solvation of the isopropyl hydrophobic groups.45,47 With increasing the temperature above the LCST of PNIPAm, the amide–water hydrogen bonding is broken and bounding water is released, leading to the collapse of PNIPAm chains. The dissolution of PPO in aqueous solution at low temperature could be also attributed to the hydrogen bonding of oxygen atoms and water. It is thus possible that hydrogen bonding might be formed between oxygen atoms of the PPO block and the amide group of the PNIPAm block, leading to the formation of “associate” structures for PNIPAm–PPO–PNIPAm triblock copolymers in aqueous solution at low temperature. At temperature (50 °C) well above the LCSTs of the PPO and PNIPAm blocks, PNIPAmx–PPO36–PNIPAmx formed uniform compact nanoparticles in aqueous solutions except PNIPAm15–PPO36–PNIPAm15 with the shortest PNIPAm block (Fig. 4B). For PNIPAm15–PPO36–PNIPAm15 in aqueous solution, multimodal distribution of the hydrodynamic radius was observed from 20 °C to 50 °C, suggesting the formation of “associate” structures at low temperature and aggregates at high temperature. The hydrodynamic radii 〈Rh〉 of nanoparticles formed by PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117 in aqueous solutions at 50 °C were 102 nm, 192 nm, and 237 nm, respectively, which increased with the length x of the PNIPAm block. Although the PPO and PNIPAm blocks were hydrophobic at 50 °C, the as-formed nanoparticles of PNIPAm–PPO–PNIPAm triblock copolymers were stable and well dispersed in aqueous solutions. No precipitation was observed at 50 °C during the experimental time scale. The radii of gyration 〈Rg〉s of the nanoparticles formed at 50 °C were also determined by static light scattering to be 83 nm, 146 nm, and 125 nm, for PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117, respectively. The corresponding values of 〈Rg〉/〈Rh〉 were then calculated to be 0.8137, 0.7604, and 0.5274 for PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117 nanoparticles, respectively.
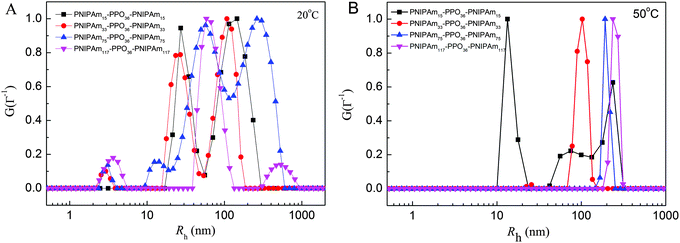 |
| | Fig. 4 The distribution of the hydrodynamic radius 〈Rh〉 measured by DLS at a scattering angle of 90° for PNIPAmx–PPO36–PNIPAmx triblock copolymers in aqueous solutions with the concentration of 0.5 mg mL−1 at temperatures of (A) 20 °C and (B) 50 °C. | |
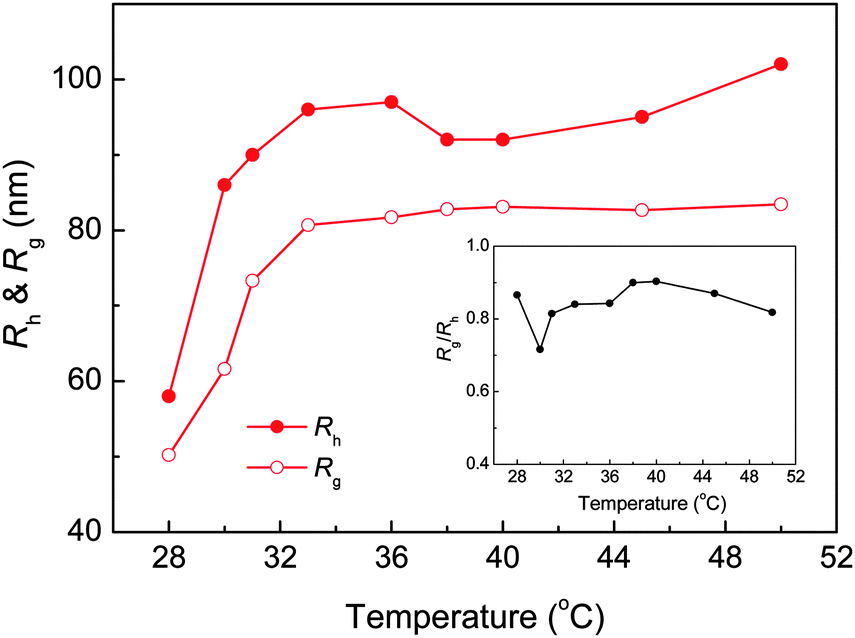
The evolution of 〈Rh〉 and 〈Rg〉 as a function of solution temperature for PNIPAm33–PPO36–PNIPAm33 in aqueous solution was further investigated by DLS & SLS, as shown in Fig. 5. Note that the values of 〈Rh〉 and 〈Rg〉 could not be determined below the LCST of the PPO block due to the formation of “associate structures” and multimodal size distribution. For PNIPAm33–PPO36–PNIPAm33, the LCSTs of PPO and PNIPAm blocks were 27.8 °C and 30 °C, respectively. With increasing temperature to ca. 28 °C, the PPO block became insoluble in aqueous solution and micelles with the hydrophobic PPO block as the core and the hydrophilic PNIPAm block as the corona were formed. The smallest values of 〈Rg〉 and 〈Rh〉 were obtained at 28 °C. Further increasing the temperature to 30 °C, PNIPAm blocks were no longer well dissolved in aqueous solution and collapsed, which hence increased the micelle sizes. With temperature well above the LCST of PNIPAm blocks, stable uniform nanoparticles were formed, leading to the constant value of 〈Rg〉 and 〈Rh〉. The inset of Fig. 5 shows the corresponding values of 〈Rg〉/〈Rh〉 ratios. Note that the value of 〈Rg〉/〈Rh〉 can be used to describe the conformation of a polymer chain or the architecture of polymer aggregates in solution or the cross-linking density distribution of the microgels.48–50 For example, for linear and flexible polymer chains, 〈Rg〉/〈Rh〉 is around 1.5, while for uniform hard spheres, 〈Rg〉/〈Rh〉 is 0.778. For the PNIPAm microgels with a heterogeneous cross-linking network, 〈Rg〉/〈Rh〉 was found to be around 0.55–0.6 at room temperature.48–50 It can be seen that the 〈Rg〉/〈Rh〉 value of PNIPAm33–PPO36–PNIPAm33 nanoparticles formed at elevated temperatures was close to that of the hard sphere, i.e. 0.778.
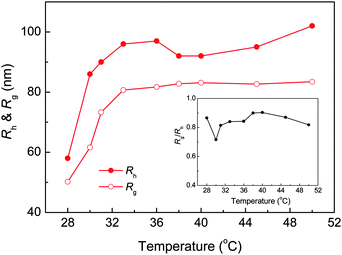 |
| | Fig. 5 The evolution of 〈Rh〉 and 〈Rg〉 of PNIPAm33–PPO36–PNIPAm33 in aqueous solution as a function of temperature. The concentration of PNIPAm33–PPO36–PNIPAm33 was 0.5 mg mL−1. The inset shows the corresponding value of the 〈Rg〉/〈Rh〉 ratio. | |
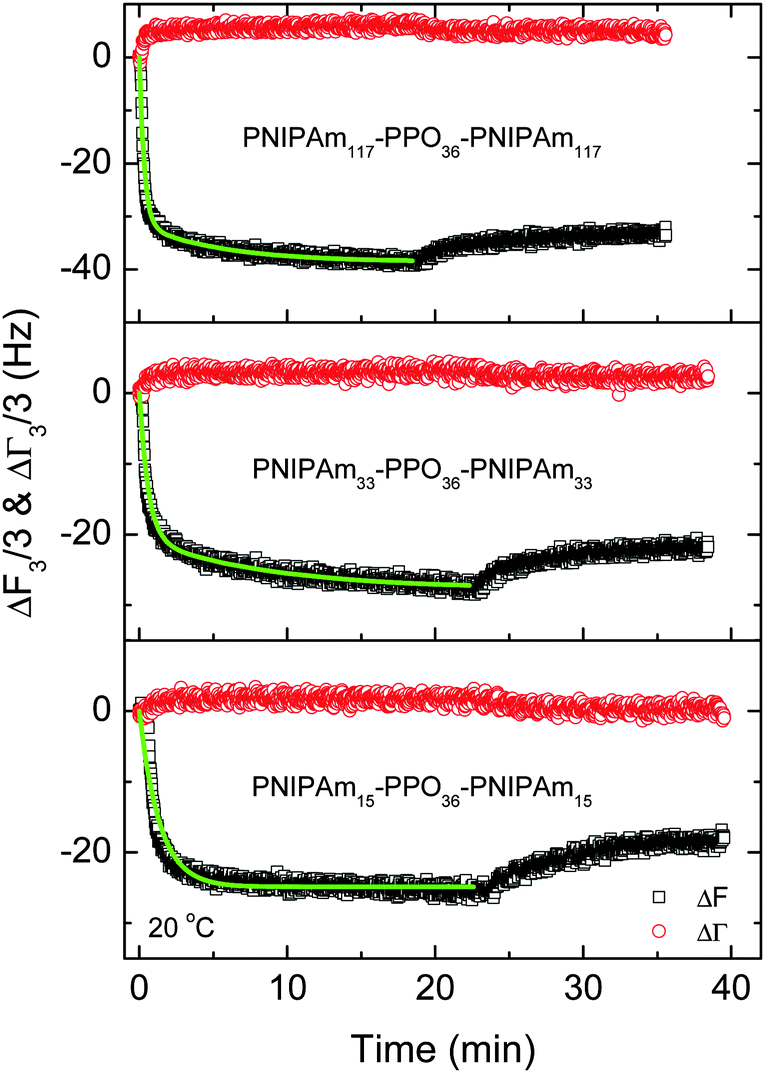
Adsorption behaviors of PNIPAmx–PPO36–PNIPAmx triblock copolymers on solid surfaces
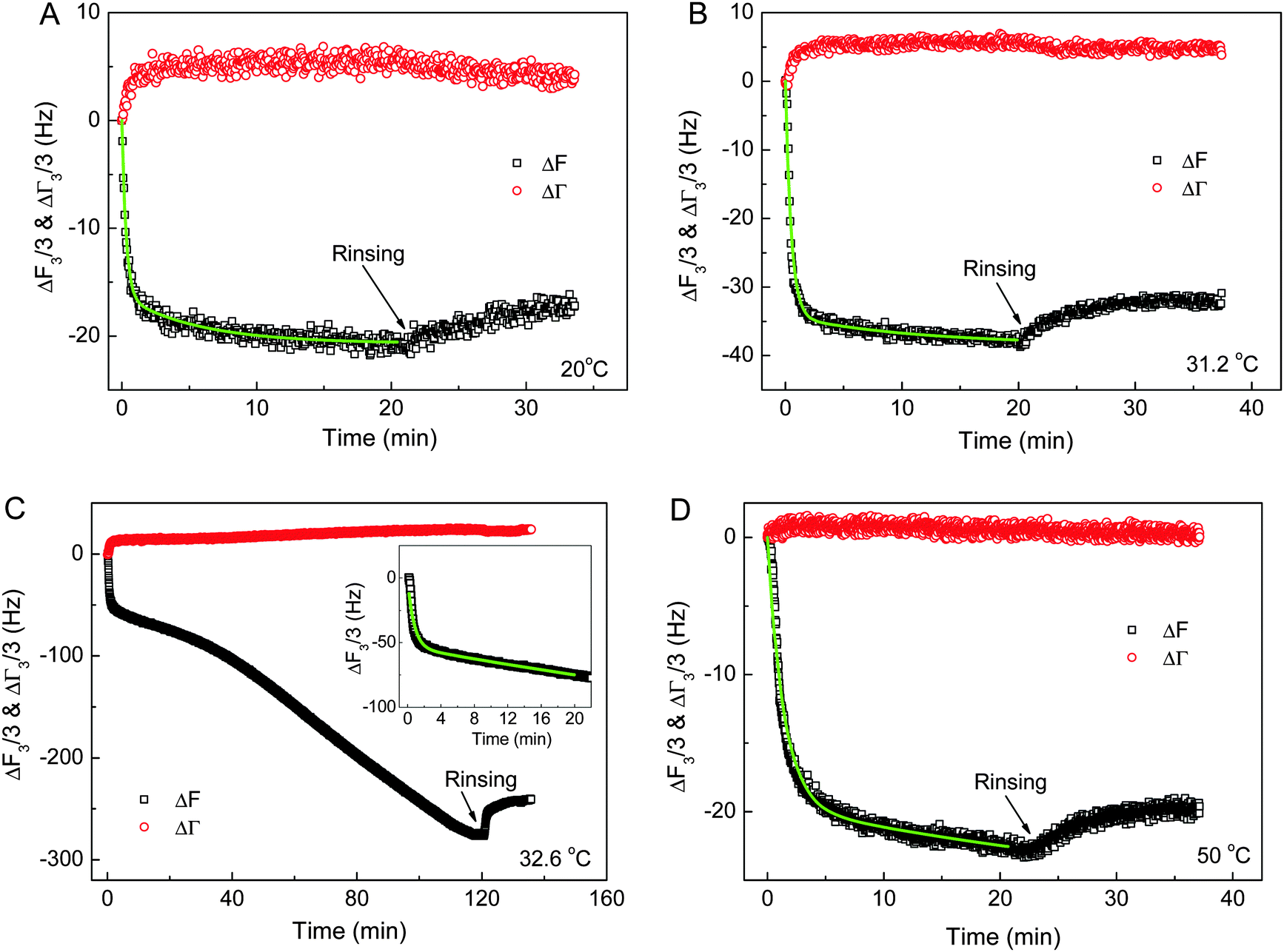
The effects of the chain conformations, solution temperature and block length x of PNIPAm on the adsorption behaviors of PNIPAmx–PPO36–PNIPAmx triblock copolymers on the hydrophobic dodecanethiol (HS(CH2)11CH3) modified gold electrode were then investigated by using the QCM technique. Fig. 6 shows the frequency (ΔF3/3) and the half-band-half-width shifts (ΔΓ3/3) of the quartz crystals during the adsorption of PNIPAm75–PPO36–PNIPAm75 on the hydrophobic dodecanethiol-modified gold surface at various temperatures, respectively. Note that the LCSTs of PPO and PNIPAm blocks were ca. 31.5 °C and 32.6 °C for PNIPAm75–PPO36–PNIPAm75 in dilute aqueous solution, respectively. It can be seen that the adsorption of PNIPAm75–PPO36–PNIPAm75 reached equilibrium within 20 min at temperatures of 20 °C, 31.2 °C, and 50 °C. However, continuous adsorption of PNIPAm75–PPO36–PNIPAm75 was observed at 32.6 °C, which was the LCST of the PNIPAm block. This continuous adsorption was arbitrarily stopped at 120 min by replacing the polymer solution with distilled water (Fig. 6C). We will come back to this point later. The adsorption kinetics of PNIPAm75–PPO36–PNIPAm75 could be described with a two-step Langmuir adsorption model given as:18,29,34| | | ΔF(t) ∝ A(t) = A1(1 − e−k1t) + A2(1 − e−k2t) | (2) |
where k1 and k2 were the apparent adsorption time constants to characterize the fast and slow adsorption processes, respectively. A1 and A2 presented the corresponding surface coverage contributed by the fast and slow adsorption processes, respectively. The green lines in Fig. 6 showed that the experimental data were well fitted with eqn (2). Note that for the adsorption at 32.6 °C, only the data obtained in the first 20 minutes were fitted with eqn (2). The obtained fitting parameters are summarized in Table 2. From Table 2, it could be seen that the adsorption rate decreased with increasing adsorption temperature. The apparent adsorption time constants k1 decreased from 3.25 min−1 at 20 °C to 0.71 min−1 at 50 °C. It is understandable because the chain conformation of PNIPAm75–PPO36–PNIPAm75 changes with increasing solution temperature. The hydrophilicity and solubility of PPO and PNIPAm blocks will decrease with increasing temperature and finally become hydrophobic at a temperature above their LCSTs. A similar phenomenon was observed for the adsorption of the PNIPAm homopolymer on the gold surface,51 of which the adsorption time constant decreased with increasing adsorption temperature. The micelles or possible aggregate structures formed at higher temperature would result in a longer translational diffusion time constant than that of the linear chain at lower temperature, leading to the smaller adsorption rate of the polymer onto the gold surface. Furthermore, small half-band-half-width shifts (ΔΓ3/3) were observed for the adsorption of PNIPAm75–PPO36–PNIPAm75 at 20 °C, 31.2 °C, and 50 °C, indicating that the adsorbed polymer layers were rigid and resulted in less energy dissipation.
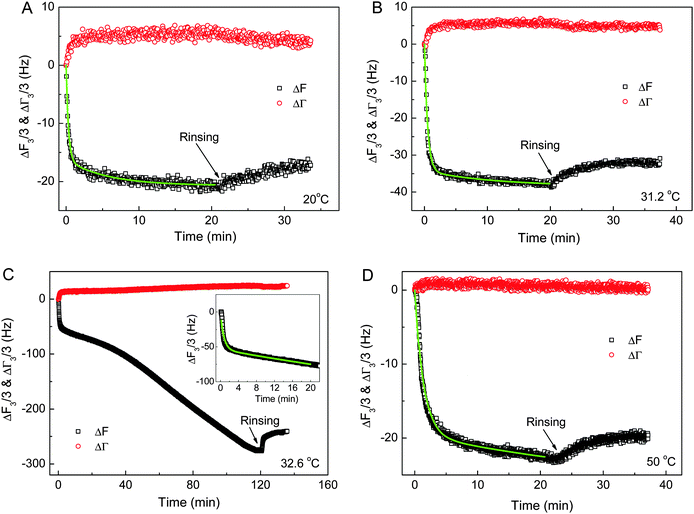 |
| | Fig. 6 The frequency shifts (ΔF3/3) and half-band-half-width shifts (ΔΓ3/3) of the quartz crystals during the adsorption of PNIPAm75–PPO36–PNIPAm75 on hydrophobic dodecanethiol-modified gold surfaces at various temperatures. (A) 20 °C, (B) 31.2 °C, (C) 32.6 °C, and (D) 50 °C. The inset of (C) shows the corresponding ΔF3/3 at the first 20 minutes. The green lines are the fits with eqn (2). The concentration of PNIPAm75–PPO36–PNIPAm75 aqueous solution was 0.5 mg mL−1. | |
Table 2 The fitting parameters of eqn (2) for the adsorption kinetics of PNIPAm75–PPO36–PNIPAm75 on the hydrophobic dodecanethiol-modified gold surface at various temperatures
|
T (°C) |
Eqn (2)
|
| ΔF1 (Hz) |
k
1 (min−1) |
ΔF2 (Hz) |
k
2 (min−1) |
R
2
|
|
Only the data obtained in the first 20 minutes were fitted with eqn (2).
|
| 20 |
−16.2 ± 0.1 |
3.25 ± 0.08 |
−4.4 ± 0.1 |
0.18 ± 0.01 |
0.95 |
| 31.2 |
−34.2 ± 0.2 |
2.00 ± 0.03 |
−4.2 ± 0.2 |
0.09 ± 0.02 |
0.97 |
| 32.6a |
−53.6 ± 0.4 |
1.30 ± 0.03 |
−114.3 ± 56.2 |
0.01 ± 0.01 |
0.98 |
| 50 |
−19.5 ± 0.2 |
0.71 ± 0.01 |
−8.1 ± 4.113 |
0.02 ± 0.02 |
0.98 |
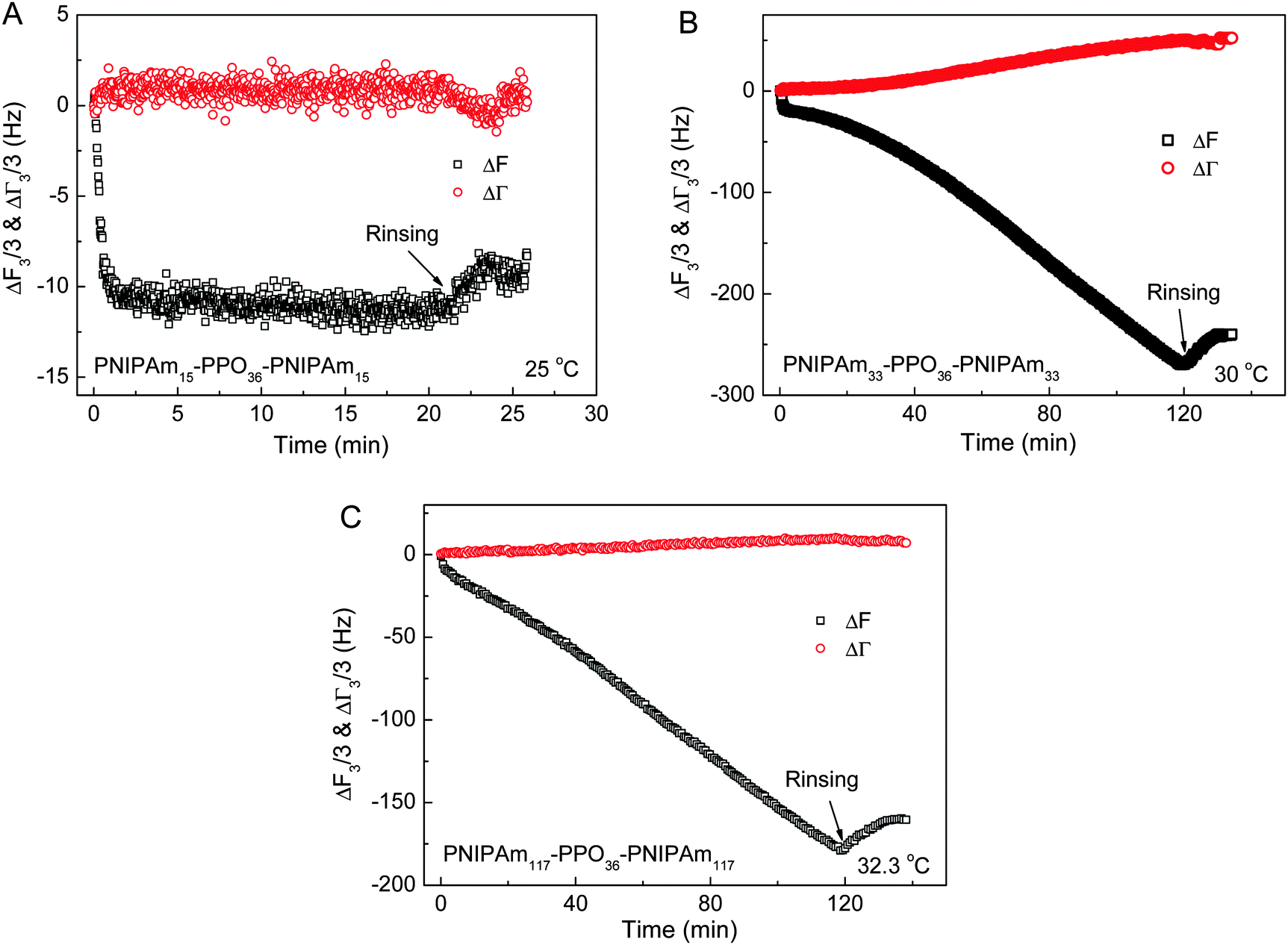
Fig. 6C shows the continuous adsorption and large amounts of adsorption for PNIPAm75–PPO36–PNIPAm75 at the LCST of the PNIPAm block, i.e. 32.6 °C. The frequency shift (ΔF3/3) was more than −270 Hz for 120 min adsorption. Such complex adsorption behavior and large amounts of adsorption have been previously observed for PNIPAm110–PEO100–PPO65–PEO100–PNIPAm110 and PNIPAmx–PEO20–PPO70–PEO20–PNIPAmx pentablock terpolymers with various lengths x of the PNIPAm block on the gold substrates at the temperature equal to the corresponding LCST of PNIPAm blocks. For example, complex adsorption behaviors were observed for PNIPAm110–PEO100–PPO65–PEO100–PNIPAm110 and PNIPAm97–PEO20–PPO70–PEO20–PNIPAm97 at their corresponding LCST of PNIPAm blocks, i.e. 34 °C and 34.7 °C, respectively.29,34 However, normal adsorption behavior was observed for the PEO100–PPO65–PEO100 triblock copolymer and the adsorption reached equilibrium within 20 min regardless of the temperatures investigated, even at the LCST of the PPO block.34 Such complex adsorption phenomena were attributed to the presence of the PNIPAm block. The large amounts of adsorption were explained by the premicellar associative adsorption, which occurred just prior to solution micellization.25,29,34,52 In the present work, it was further found that the length x of the PNIPAm block plays the key role in the adsorption behaviors of PNIPAmx–PPO36–PNIPAmx triblock copolymers at their corresponding LCSTs. Fig. 7 shows the frequency shifts (ΔF3/3) and half-band-half-width shifts (ΔΓ3/3) of the quartz crystals as a function of adsorption time for the adsorption of PNIPAm15–PPO36–PNIPAm15 at 25 °C, PNIPAm33–PPO36–PNIPAm33 at 30 °C, and PNIPAm117–PPO36–PNIPAm117 at 32.3 °C. Normal adsorption behavior was observed for the adsorption of PNIPAm15–PPO36–PNIPAm15 at 25 °C. For PNIPAm15–PPO36–PNIPAm15, the PNIPAm block was too short to maintain its own LCST so that only the LCST of the PPO block at 25 °C was observed (cf.Fig. 3). Fig. 7A shows that the adsorption of PNIPAm15–PPO36–PNIPAm15 on the hydrophobic gold surface at the LCST of the PPO block, i.e. 25 °C, reached equilibrium within 20 min, which was similar with the results obtained at 20 and 50 °C. For PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117 in aqueous solutions, the PNIPAm block is long enough to maintain its own LCST. The LCSTs of PNIPAm blocks were 30 °C, 32.6 °C, and 32.3 °C, respectively, for PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117. Complex adsorption behavior and large amounts of adsorption were thus observed for the adsorption of PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117 at their corresponding LCSTs of PNIPAm blocks (cf.Fig. 6C, 7B and C). These results suggested that the presence of the PNIPAm block is not a sufficient condition for the observation of complex adsorption behavior and the PNIPAm block should necessarily reach a certain block length in order to maintain its own LCST. If the PNIPAm block exhibited its own LCST, the complex adsorption behavior was then observed at the LCST of the PNIPAm block for PNIPAmx–PPO36–PNIPAmx triblock copolymers.
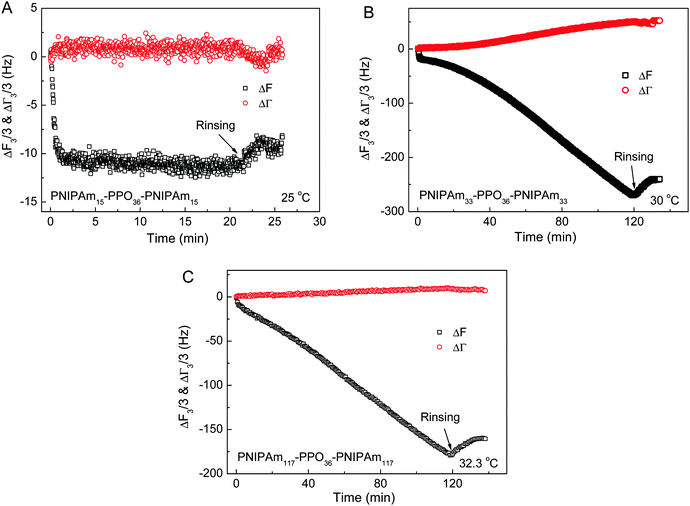 |
| | Fig. 7 The frequency shifts (ΔF3/3) and half-band-half-width shifts (ΔΓ3/3) of the quartz crystals as a function of adsorption time for the adsorption of (A) PNIPAm15–PPO36–PNIPAm15 at 25 °C, (B) PNIPAm33–PPO36–PNIPAm33 at 30 °C, and (C) PNIPAm117–PPO36–PNIPAm117 at 32.3 °C on hydrophobic dodecanethiol-modified gold surfaces. The concentration of each PNIPAmx–PPO36–PNIPAmx aqueous solution was 0.5 mg mL−1. | |
Fig. 8 shows the frequency shifts (ΔF3/3) and half-band-half-width shifts (ΔΓ3/3) of the quartz crystals as a function of adsorption time for the adsorption of PNIPAm15–PPO36–PNIPAm15, PNIPAm33–PPO36–PNIPAm33 and PNIPAm117–PPO36–PNIPAm117 at 20 °C on hydrophobic dodecanethiol-modified gold surfaces. Together with the data curve of Fig. 6A, it can be clearly seen that the adsorption kinetics and adsorption amounts of PNIPAmx–PPO36–PNIPAmx triblock copolymers at 20 °C were dependent on the length x of the PNIPAm block. The adsorption amounts slightly increased with increasing x except x = 75. The frequency shifts (ΔF3/3) at adsorption equilibrium were −26 Hz, −28 Hz, −21 Hz, and −38 Hz for PNIPAm15–PPO36–PNIPAm15, PNIPAm33–PPO36–PNIPAm33, PNIPAm75–PPO36–PNIPAm75, and PNIPAm117–PPO36–PNIPAm117, respectively. A minimum adsorption was observed for the adsorption of PNIPAm75–PPO36–PNIPAm75 on the dodecanethiol-modified gold surface at 20 °C. The adsorption kinetics could be well fitted with eqn (2) and the fitting parameters are listed in Table 3. With increasing length x of the PNIPAm block from 15 to 117, the apparent adsorption time constants k1 increased from 0.74 ± 0.01 min−1 to 3.06 ± 0.06 min−1. Note that for PNIPAm15–PPO36–PNIPAm15 with the shortest PNIPAm block, only one adsorption time constant was needed to describe its adsorption kinetics. These results were different with those previously observed for the adsorption of PNIPAmx–P123–PNIPAmx pentablock terpolymers onto the bare hydrophobic gold surfaces at 20 °C.29 It was observed that the apparent adsorption time constants k1 decreased with increasing length x of the PNIPAm block for PNIPAmx–P123–PNIPAmx. In the case of PNIPAmx–P123–PNIPAmx, it was found that the sizes of “associate” structures formed at 20 °C increased with increasing length x of the PNIPAm block.35 The large “associate” structures would diffuse slowly in the dilute solution, leading to a small adsorption rate on the solid surface.33,51 As a result, the adsorption time constant decreased with increasing length x of the PNIPAm block for PNIPAmx–P123–PNIPAmx at 20 °C. However, for the case of PNIPAmx–PPO36–PNIPAmx, the “associate” structures formed at 20 °C did not increase with length x, as shown in Fig. 4A. A multimodal size distribution was observed for the “associate” structures of PNIPAmx–PPO36–PNIPAmx in dilute aqueous solutions at 20 °C. Fig. 4A shows that the dominated peak of size distribution even shifts to a smaller size when increasing the length x of the PNIPAm block. This might be the possible reason why the apparent adsorption time constants k1 increased with increasing length x of the PNIPAm block for the adsorption of PNIPAmx–PPO36–PNIPAmx on the dodecanethiol-modified gold surface at 20 °C. In fact, a maximum adsorption rate constant of 3.25 ± 0.08 min−1 was observed for PNIPAm75–PPO36–PNIPAm75. With further increasing length x to 117, the adsorption rate constant decreased to 3.06 ± 0.06 min−1. Fig. 8 also shows that small half-band-half-width shifts (ΔΓ3/3) were observed for the adsorption of PNIPAm15–PPO36–PNIPAm15, PNIPAm33–PPO36–PNIPAm33 and PNIPAm117–PPO36–PNIPAm117 at 20 °C, which were consistent with that observed for PNIPAm75–PPO36–PNIPAm75 (cf.Fig. 4A). Such small ΔΓ3/3 indicated that the adsorbed PNIPAmx–PPO36–PNIPAmx layers were rigid regardless of the length x of the PNIPAm block.
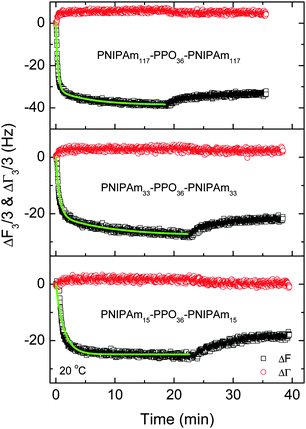 |
| | Fig. 8 The frequency shifts (ΔF3/3) and half-band-half-width shifts (ΔΓ3/3) of the quartz crystals as a function of adsorption time for the adsorption of PNIPAm15–PPO36–PNIPAm15, PNIPAm33–PPO36–PNIPAm33 and PNIPAm117–PPO36–PNIPAm117 at 20 °C on hydrophobic dodecanethiol-modified gold surfaces. The concentration of each PNIPAmx–PPO36–PNIPAmx aqueous solution was 0.5 mg mL−1. | |
Table 3 The fitting parameters of eqn (2) for the adsorption kinetics of PNIPAmx–PPO36–PNIPAmx on the hydrophobic dodecanethiol-modified gold surface at 20 °C
| |
Eqn (2)
|
| ΔF1 (Hz) |
k
1 (min−1) |
ΔF2 (Hz) |
k
2 (min−1) |
R
2
|
| PNIPAm15–PPO36–PNIPAm15 |
−24.9 ± 0.1 |
0.74 ± 0.01 |
— |
— |
0.93 |
| PNIPAm33–PPO36–PNIPAm33 |
−20.5 ± 0.2 |
1.69 ± 0.04 |
−7.3 ± 0.1 |
0.12 ± 0.01 |
0.97 |
| PNIPAm75–PPO36–PNIPAm75 |
−16.2 ± 0.1 |
3.25 ± 0.08 |
−4.4 ± 0.1 |
0.18 ± 0.01 |
0.95 |
| PNIPAm117–PPO36–PNIPAm117 |
−31.5 ± 0.2 |
3.06 ± 0.06 |
−7.1 ± 0.2 |
0.18 ± 0.01 |
0.97 |
Conclusions
Four thermo-sensitive PNIPAmx–PPO36–PNIPAmx triblock copolymers with x values of 15, 33, 75, and 117 were obtained. The length x strongly affected the thermal transition of PNIPAmx–PPO36–PNIPAmx in dilute aqueous solutions. The PNIPAm block with x = 15 was too short to maintain its own LCST so that only the LCST of the PPO block was observed, i.e. 25 °C. For the longest PNIPAm block with x = 117, only the LCST of the PNIPAm block dominated, i.e. 32.3 °C. With middle length x, PNIPAm33–PPO36–PNIPAm33 and PNIPAm75–PPO36–PNIPAm75 exhibited two LCSTs, which were corresponding to LCSTs of PPO and PNIPAm blocks, respectively. All the four PNIPAmx–PPO36–PNIPAmx triblock copolymers formed “associate” structures in the dilute aqueous solutions at 20 °C, which was well below the LCSTs of the PPO and PNIPAm blocks. The adsorption behaviors of PNIPAmx–PPO36–PNIPAmx triblock copolymers on dodecanethiol-modified gold surfaces were strongly dependent on the length x of the PNIPAm block and the corresponding chain conformation in dilute aqueous solutions at various temperatures. For PNIPAm75–PPO36–PNIPAm75, the adsorption time constant decreased with increasing adsorption temperature. However, the adsorption time constant increased with increasing length x of the PNIPAm block. The presence of the PNIPAm block is not a sufficient condition for the observation of complex adsorption behavior. When the PNIPAm block was long enough to maintain its own LCST, the complex adsorption behavior with large adsorption amounts was thus observed at the corresponding LCST of the PNIPAm block for PNIPAmx–PPO36–PNIPAmx with x = 33, 75, and 117. Furthermore, the adsorbed PNIPAmx–PPO36–PNIPAmx layers obtained at 20 °C were rigid regardless of the value of x, resulting in less energy dissipation.
Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21274129 and 21322406), the Fundamental Research Funds for the Central Universities (2014XZZX003-21), the third level of 2013 Zhejiang Province 151 Talent Project, and Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences for financial support.
References
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- S. Forster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- S. A. Jenekhe and X. L. Chen, Science, 1999, 283, 372–375 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- L. Hong, F. Zhu, J. Li, T. Ngai, Z. Xie and C. Wu, Macromolecules, 2008, 41, 2219–2227 CrossRef CAS.
- Y. Zhou, K. Jiang, Q. Song and S. Liu, Langmuir, 2007, 23, 13076–13084 CrossRef CAS PubMed.
- J. S. Lintuvuori and M. R. Wilson, Phys. Chem. Chem. Phys., 2009, 11, 2116–2125 RSC.
- V. Hugouvieux, M. A. V. Axelos and M. Kolb, Macromolecules, 2009, 42, 392–400 CrossRef CAS.
- B. Du, A. Mei, K. Yin, Q. Zhang, J. Xu and Z. Fan, Macromolecules, 2009, 42, 8477–8484 CrossRef CAS.
- B. Du, A. Mei, Y. Yang, Q. Zhang, Q. Wang, J. Xu and Z. Fan, Polymer, 2010, 51, 3493–3502 CrossRef CAS.
- A. X. Mei, X. L. Guo, Y. W. Ding, X. H. Zhang, J. T. Xu, Z. Q. Fan and B. Y. Du, Macromolecules, 2010, 43, 7312–7320 CrossRef CAS.
- Y. Yan, X. Zhou, J. Ji, L. Yan and G. Zhang, J. Phys. Chem. B, 2006, 110, 21055–21059 CrossRef CAS PubMed.
- E. J. Park, D. D. Draper and N. T. Flynn, Langmuir, 2007, 23, 7083–7089 CrossRef CAS PubMed.
- I. G. Sedeva, D. Fornasiero, J. Ralston and D. A. Beattie, Langmuir, 2009, 25, 4514–4521 CrossRef CAS PubMed.
- A. Naderi, J. Iruthayaraj, T. Pettersson, R. Makuška and P. M. Claesson, Langmuir, 2008, 24, 6676–6682 CrossRef CAS PubMed.
- G. Zhang, Macromolecules, 2004, 37, 6553–6557 CrossRef CAS.
- S. Bhattacharya, H. P. Hsu, A. Milchev, V. G. Rostiashvili and T. A. Vilgis, Macromolecules, 2008, 41, 2920–2930 CrossRef CAS.
- X. Liu, A.-H. Vesterinen, J. Genzer, J. V. Seppälä and O. J. Rojas, Langmuir, 2011, 27, 9769–9780 CrossRef CAS PubMed.
- A. Corsi, A. Milchev, V. G. Rostiashvili and T. A. Vilgis, J. Chem. Phys., 2005, 122, 094907 CrossRef PubMed.
- Y. A. Kriksin, P. G. Khalatur and A. R. Khokhlov, J. Chem. Phys., 2005, 122, 114703 CrossRef PubMed.
- K. Sumithra, M. Brandau and E. Straube, J. Chem. Phys., 2009, 130, 234901 CrossRef CAS PubMed.
- T. Q. Chen, Y. P. Lu, T. Y. Chen, X. H. Zhang and B. Y. Du, Phys. Chem. Chem. Phys., 2014, 16, 5536–5544 RSC.
- G. M. Liu, H. Cheng, L. F. Yan and G. Z. Zhang, J. Phys. Chem. B, 2005, 109, 22603–22607 CrossRef CAS PubMed.
- G. M. Liu and G. Z. Zhang, J. Phys. Chem. B, 2005, 109, 743–747 CrossRef CAS PubMed.
- M. A. Plunkett, Z. Wang, M. W. Rutland and D. Johannsmann, Langmuir, 2003, 19, 6837–6844 CrossRef CAS.
- K. Wu, B. Wu, P. Wang, Y. Hou, G. Zhang and D.-M. Zhu, J. Phys. Chem. B, 2007, 111, 8723–8727 CrossRef CAS PubMed.
- Y. P. Lu, X. H. Zhang, Z. Q. Fan and B. Y. Du, Polymer, 2012, 53, 3791–3801 CrossRef CAS.
- Y. P. Lu, T. Q. Chen, A. X. Mei, T. Y. Chen, Y. W. Ding, X. H. Zhang, J. T. Xu, Z. Q. Fan and B. Y. Du, Phys. Chem. Chem. Phys., 2013, 15, 8276–8286 RSC.
- C. M. Li, N. J. Buurma, I. Haq, C. Turner, S. P. Armes, V. Castelletto, I. W. Hamley and A. L. Lewis, Langmuir, 2005, 21, 11026–11033 CrossRef CAS PubMed.
- J. Ye, J. Xu, J. Hu, X. Wang, G. Zhang, S. Liu and C. Wu, Macromolecules, 2008, 41, 4416–4422 CrossRef CAS.
- J. Ye, Y. Hou, G. Zhang and C. Wu, Langmuir, 2008, 24, 2727–2731 CrossRef CAS PubMed.
- D. Johannsmann, Phys. Chem. Chem. Phys., 2008, 10, 4516–4534 RSC.
- E. Hasan, M. Zhang, A. H. E. Muller and C. B. Tsvetanov, J. Macromol. Sci., Part A: Pure Appl.Chem., 2004, A41, 467–486 CrossRef CAS.
- P. Alexandridis, T. Nivaggioli, J. F. Holzwarth and T. A. Hatton, Abstr. Pap., Jt. Conf. – Chem. Inst. Can. Am. Chem. Soc., 1994, 207, 234 Search PubMed.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed.
- J. Yan, W. Ji, E. Chen, Z. Li and D. Liang, Macromolecules, 2008, 41, 4908–4913 CrossRef CAS.
- Z. Ahmed, E. A. Gooding, K. V. Pimenov, L. Wang and S. A. Asher, J. Phys. Chem. B, 2009, 113, 4248–4256 CrossRef CAS PubMed.
- F. Ke, X. Mo, R. Yang, Y. Wang and D. Liang, Macromolecules, 2009, 42, 5339–5344 CrossRef CAS.
- F. Meersman, J. Wang, Y. Wu and K. Heremans, Macromolecules, 2005, 38, 8923–8928 CrossRef CAS.
- H. Senff and W. Richtering, J. Chem. Phys., 1999, 111, 1705–1711 CrossRef CAS.
- D. Kuckling, C. D. Vo, H. J. P. Adler, A. Völkel and H. Cölfen, Macromolecules, 2006, 39, 1585–1591 CrossRef CAS.
- H. Chen, J. Li, Y. Ding, G. Zhang, Q. Zhang and C. Wu, Macromolecules, 2005, 38, 4403–4408 CrossRef CAS.
- D.-M. Zhu, K. Wu, B. Wu, P. Wang, J. Fang, Y. Hou and G. Zhang, J. Phys. Chem. C, 2007, 111, 18679–18686 CAS.
- M. Malmsten, P. Linse and T. Cosgrove, Macromolecules, 1992, 25, 2474–2481 CrossRef CAS.
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.