
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and dynamics of the aliphatic cholesterol side chain in membranes as studied by 2H NMR spectroscopy and molecular dynamics simulation†
Alexander
Vogel
a,
Holger A.
Scheidt
a,
Dong Jae
Baek
bc,
Robert
Bittman‡
c and
Daniel
Huster
*a
aInstitute of Medical Physics and Biophysics, University of Leipzig, Härtelstr. 16-18, 04275 Leipzig, Germany. E-mail: daniel.huster@medizin.uni-leipzig.de; Fax: +49 (0)341 97 15709; Tel: +49 (0)341 97 15701
bCollege of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Jeonnam, Republic of Korea
cDepartment of Chemistry and Biochemistry, Queens College of the City University of New York, Flushing, NY 11367-1597, USA
First published on 7th January 2016
Abstract
Cholesterol is an evolutionarily highly optimized molecule particularly known for its ability to condense the phospholipids in cellular membranes. Until recently, the accompanying increase in the chain order of the surrounding phospholipids was attributed to the planar and rigid tetracyclic ring structure of cholesterol. However, detailed investigations of cholesterol's aliphatic side chain demonstrated that this side chain is responsible for approximately half of the condensation effect. Therefore, we investigated the structure and dynamics of the aliphatic side chain of cholesterol using 2H solid-state nuclear magnetic resonance (NMR) spectroscopy and microsecond timescale all-atom molecular dynamics (MD) simulations in four different model membranes: POPC, DPPC, PSM, and POPC/PSM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol/mol) and at three different temperatures: 5 °C, 37 °C, and 50 °C. A cholesterol variant, in which 11 hydrogens of the aliphatic side chain were exchanged for deuterium, was used and the respective 2H NMR spectra confirmed the axially asymmetric rotational diffusion of cholesterol in DPPC and PSM. Furthermore, NMR spectra indicated that some hydrogens showed an unexpected magnetic inequivalency. This finding was confirmed by all-atom molecular dynamics simulations and detailed analysis revealed that the hydrogens of the methylene groups at C22, C23, and C24 are magnetically inequivalent. This inequivalency is caused by steric clashes of the aliphatic side chain with the ring structure of cholesterol as well as the branched C21 methyl group. These excluded volume effects result in reduced conformational flexibility of the aliphatic side chain of cholesterol and explain its high order (order parameter of 0.78 for chain motions) and large contribution to the condensation effect. Additionally, the motional pattern of the side chain becomes highly anisotropic such that it shows larger fluctuations perpendicular to the ring plane of cholesterol with a biaxiality of the distribution of 0.046. Overall, our results shed light on the mechanism how the aliphatic side chain is able to contribute about half of the condensation effect of cholesterol.
:1 mol/mol) and at three different temperatures: 5 °C, 37 °C, and 50 °C. A cholesterol variant, in which 11 hydrogens of the aliphatic side chain were exchanged for deuterium, was used and the respective 2H NMR spectra confirmed the axially asymmetric rotational diffusion of cholesterol in DPPC and PSM. Furthermore, NMR spectra indicated that some hydrogens showed an unexpected magnetic inequivalency. This finding was confirmed by all-atom molecular dynamics simulations and detailed analysis revealed that the hydrogens of the methylene groups at C22, C23, and C24 are magnetically inequivalent. This inequivalency is caused by steric clashes of the aliphatic side chain with the ring structure of cholesterol as well as the branched C21 methyl group. These excluded volume effects result in reduced conformational flexibility of the aliphatic side chain of cholesterol and explain its high order (order parameter of 0.78 for chain motions) and large contribution to the condensation effect. Additionally, the motional pattern of the side chain becomes highly anisotropic such that it shows larger fluctuations perpendicular to the ring plane of cholesterol with a biaxiality of the distribution of 0.046. Overall, our results shed light on the mechanism how the aliphatic side chain is able to contribute about half of the condensation effect of cholesterol.
Introduction
Cholesterol is one of the most abundant bulk lipids in cellular membranes and plays the key role in the phase behavior and lateral organization of the plasma membrane.1 Specific interactions between cholesterol and sphingolipids are believed to represent the driving force for the domain structure of membranes consisting of cholesterol-depleted liquid-disordered (ld) and cholesterol-enriched liquid-ordered (lo) domains.2–4 However, as of now, there is no complete understanding of what brings about the complicated dynamic domain structure of the plasma membrane and of the role cholesterol plays in these dynamic domain forming processes.5–8Cholesterol preferentially interacts with saturated lipid chains.9–11 Attractive van der Waals interactions between the smooth α-face of cholesterol's planar and rigid ring structure and the lipid chains have been assumed to be responsible for this effect,12,13 which leads to a drastic increase in the lipid packing density that is called condensation.14 However, a recent report demonstrated that the iso-branched aliphatic side chain of cholesterol also plays an important role in the interaction with phospholipid chains and surprisingly accounts for 40–60% of the condensation effect.15 This result was also confirmed by recent molecular dynamics (MD) simulations.16 Sterols bearing an unbranched side chain also promote lipid condensation, but to a lesser extent than the native branched iso-alkyl chain of cholesterol.17
There is relatively little published work on the structural and dynamical aspects of the aliphatic side chain of cholesterol as opposed to the large body of literature on the order and dynamics of phospholipid chains.18–20 In particular, quadrupolar 2H and dipolar 1H–13C NMR order parameters have advanced our understanding of the influence of cholesterol on lipid packing, lateral organization, and domain formation in model membrane systems.11,21–23 In a first investigation of the cholesterol side chain, it was found that the two chemically identical methyl groups C26 and C27 (see Fig. 1 for the Nomenclature) did not provide equivalent 2H NMR signals.24 This was further confirmed by Dufourc et al. for the C24 methylene group.25 For the methylene segments of the cholesterol side chain, much higher order parameters were found compared to phospholipid acyl chains, also the aforementioned inequivalence between the two protons of the same methylene group has only been observed in phospholipid chains for the C2 segments. As a consequence of a very elaborated analysis of these experimental findings, the cholesterol side chain was pronounced to be “as ordered as the condensed ring structure”.25 However, the very low quadrupolar splittings of the terminal methyl groups of C26 and C27, observed in 2H NMR spectra, have not been understood in the framework of this model.25
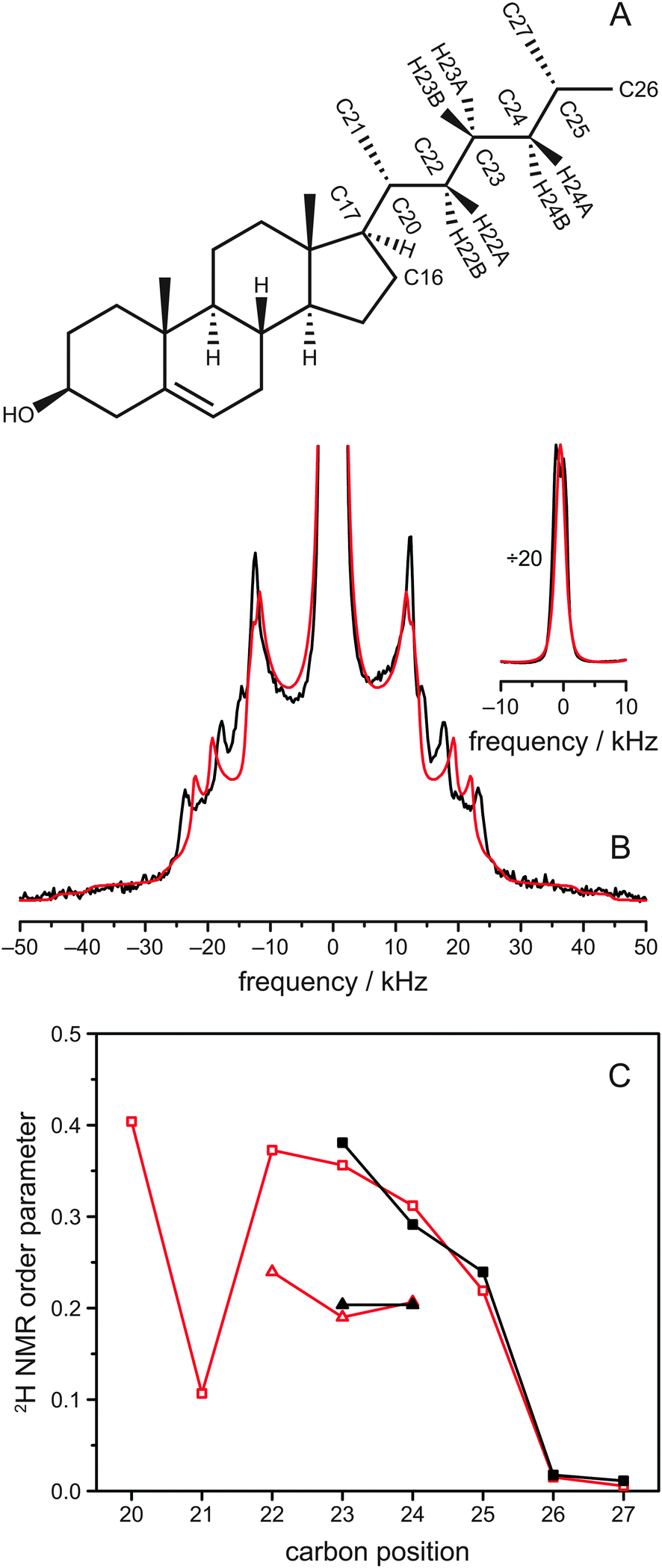 | ||
| Fig. 1 (A) Chemical structure of cholesterol including the relevant nomenclature. In cholesterol-d11, hydrogens were replaced by deuterium at positions C23, C24, C25, C26, and C27. (B) 2H NMR spectra of POPC/cholesterol-d11 (3:1 mol/mol) at 37 °C. The experimentally obtained 2H NMR spectrum is shown in black, while the spectrum calculated from the MD simulation is drawn in red. The inset shows the same spectrum vertically divided by a factor of 20 to show the intense Pake doublets from the two terminal CH3 groups. (C) Corresponding 2H NMR order parameters of the aliphatic cholesterol side chain. Experimental data is shown in black for all deuterated carbon positions of cholesterol-d11, while the order parameters from MD simulation, which are shown in red, were calculated for the whole chain. Data for C22, C23, and C24 shows two values because A (triangles) and B (squares) hydrogens are not magnetically equivalent at these positions. In the MD simulation data, the three hydrogens of the CH3 groups at C21, C26, and C27 are averaged, because their order parameters differ by less than 0.002. The first 500 ns were considered as equilibration of the MD simulation and omitted for data evaluation. | ||
In addition to 2H NMR studies, 13C NMR separated local field experiments under magic-angle spinning conditions have proven very useful to study the order parameters of molecular segments of biomolecules26,27 and in particular lipids and cholesterol.23,28–30 In a recent paper, the order parameters of cholesterol were also determined by this method, though at lower resolution than in the 2H NMR studies.28 Experimental results were also compared to a 100 ns MD simulation. The simulated 1H–13C order parameters showed good agreement with the previously published 2H NMR data. Also, simulation and experiment showed reasonable agreement for the ring structure of cholesterol but not for the side chain, where the MD results were too small by a factor of ∼2, possibly due to the united atom GROMOS force field31 or the short simulation time.
Clearly, a better understanding of the putatively rigid cholesterol side chain with the highly mobile phospholipid chains will advance our understanding of the lateral organization of the lipid molecules in the membrane and their relevance for the organization of biological membranes. Here, we focus on the structural and dynamical behaviour of the aliphatic cholesterol side chain by analyzing the 2H NMR spectra of a chain deuterated cholesterol-d11 molecule32 in POPC, DPPC, PSM, as well as in a model lipid raft mixture and four all-atom MD simulations, each of more than 1.5 μs length.
Using these methods we could show that some conformations of the side chain result in steric clashes with the ring structure of cholesterol and therefore are forbidden. This can be interpreted as a reduction of the volume that is accessible to the side chain and leads to high chain order and anisotropic chain motions.
Materials and methods
Materials
The lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and N-palmitoyl-D-erythro-sphingosylphosphorylcholine (PSM) were purchased from Avanti Polar Lipids, Inc. (Alabaster, Al) and used without further purification. Cholesterol-d11 was synthesized as described in literature.32Sample preparation
For the 2H NMR measurements, cholesterol-d11 was mixed with the individual phospholipids in chloroform at a molar ratio of 1:3 (25 mol% cholesterol-d11). In addition, a lipid raft mixture of POPC/PSM/cholesterol-d11 at a molar ratio of 37.5/37.5/25 mol/mol/mol was prepared. After the removal of the chloroform using a vacuum evaporator, the samples were redissolved in cyclohexane and lyophilized. Samples were hydrated with 40 wt% deuterium-depleted H2O and equilibrated by ten freeze-thaw cycles and gentle centrifugation.
2H NMR spectroscopy
The static 2H NMR spectra were recorded on a Bruker Avance I 750 MHz NMR spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) at a resonance frequency of 115.1 MHz for 2H using a single-channel probe with a 5 mm coil. The spectra were acquired with a phase-cycled quadrupolar echo sequence.33 The typical length of the 90° pulses was around 3 μs, the relaxation delay was 1 s.Molecular dynamics simulations
Four all-atom MD simulations were carried out: two simulations of a POPC/cholesterol at 37 °C and 50 °C, one simulation of DPPC/cholesterol at 50 °C, and one simulation of PSM/cholesterol at 50 °C. The most detailed data analysis was conducted on the POPC/cholesterol simulation at 37 °C. Each simulation consisted of 150 phospholipids (POPC, DPPC, PSM), 50 cholesterols, and TIP3 water and was run for ∼1550 ns. Setup of the membranes was conducted according to published procedures.34 Particular attention was paid to avoid any molecule from being threaded through one of the cholesterol rings. The program NAMD35 was employed for the simulation under conditions of normal pressure (1.013 bar), using the most recent CHARMM all-H C36 lipid force field36 and the modified C36c force field37 for cholesterol. This allowed to run the simulation with flexible surface area and therefore enabled the system to adapt to the condensation effect of cholesterol (no additional force was applied in the plane of the membrane). The PSM force field values were taken from literature.38 The smooth particle mesh Ewald algorithm was used to compute the electrostatic forces39 and the SHAKE algorithm was used to keep rigid all bonds involving hydrogen atoms, allowing a 2 fs time step.40Results
2H solid-state NMR spectroscopy represents a very suitable tool to obtain detailed information on the structure and dynamics of the aliphatic side chain of cholesterol.24,25 In such spectra, a Pake doublet is observed for each deuterium in the molecule, where the magnitude of the splitting of the doublet is determined by the possible orientations of the C–2H bond with respect to the membrane normal and directly proportional to the motional amplitude of this bond.41,42 Sophisticated models have been developed to interpret these quadrupolar splittings in terms of orientation and dynamics of the deuterated molecule.25,43–45Here, we have used a specifically deuterated cholesterol-d11, in which the hydrogen atoms attached to C23, C24, C25, C26 and C27 were replaced by deuterium, acting as probes in our samples (Fig. 1A).32 This cholesterol variant was mixed with various phospholipids to investigate the influence of the lipid environment on the aliphatic cholesterol side chain. The 2H NMR spectrum of the sample of POPC/cholesterol-d11 (molar ratio 75:25) at 37 °C is shown in black in Fig. 1B and consists of a superposition of several Pake doublets. There is a very intense Pake doublet with very small quadrupolar splitting, which can be assigned to the deuterons of the methyl groups at C26 and C27 due to the fast rotation of the CH3 groups in agreement with literature.25 In addition to this spectral component, four more Pake patterns with larger quadrupolar splittings are clearly visible, which originate from the five deuterons at C23, C24, and C25. As shown before, the two deuterons of the C24 segment of cholesterol are not equivalent.25 Therefore, to obtain a total of four Pake patterns, we expected the two deuterium atoms on C23 to be equivalent and give rise to one Pake doublet with double intensity. This doublet should have the largest quadrupolar splitting (i.e., highest order) because it was shown that the deuterons on C22 are equivalent and of higher order than on C24,25 and we expected C23 to be of intermediate order. In our NMR spectrum, however, the innermost Pake doublet (aside from the CH3 doublets) showed double intensity.
Since it is not straightforward to assign the individual Pake doublets to the carbon positions in the aliphatic cholesterol side chain, we conducted an all-atom MD simulation of the same system. The simulation consisted of 150 POPC and 50 cholesterol molecules, which were simulated for ∼1.5 μs at 37 °C. From the simulation, the angles β of all C–H bonds in the aliphatic cholesterol side chain with respect to the membrane normal were extracted and subsequently converted into order parameters SCDvia
| SCD = 1/2(3cos2β − 1).45 |
:1 mol/mol) at several temperatures from experimental 2H NMR spectra and MD simulations. The first 500 ns were considered as equilibration of the MD simulation and omitted for data evaluation
| 5 °C | 37 °C | 50 °C | |||
|---|---|---|---|---|---|
| NMR | NMR | MD | NMR | MD | |
| a Not determined due to the lack of deuteration. | |||||
| H20 | n.d.a | n.d. | 0.4040 | n.d. | 0.3954 |
| H21A | n.d. | n.d. | 0.1058 | n.d. | 0.1035 |
| H21B | n.d. | n.d. | 0.1070 | n.d. | 0.1026 |
| H21C | n.d. | n.d. | 0.1072 | n.d. | 0.1035 |
| H22A | n.d. | n.d. | 0.2396 | n.d. | 0.2232 |
| H22B | n.d. | n.d. | 0.3726 | n.d. | 0.3591 |
| H23A | 0.2771 | 0.2036 | 0.1900 | 0.1804 | 0.1719 |
| H23B | 0.4256 | 0.3808 | 0.3562 | 0.3617 | 0.3402 |
| H24A | 0.2771 | 0.2036 | 0.2065 | 0.1804 | 0.1877 |
| H24B | 0.3497 | 0.2914 | 0.3120 | 0.2715 | 0.2938 |
| H25 | 0.3114 | 0.2395 | 0.2190 | 0.2140 | 0.1982 |
| H26A | 0.0224 | 0.0176 | 0.0155 | 0.0176 | 0.0136 |
| H26B | 0.0154 | 0.0142 | |||
| H26C | 0.0144 | 0.0148 | |||
| H27A | 0.0176 | 0.0112 | 0.0056 | 0.0096 | 0.0053 |
| H27B | 0.0050 | 0.0047 | |||
| H27C | 0.0059 | 0.0050 | |||
The observed 2H NMR quadrupolar splitting ΔνQ is directly related to the angle β between the C–2H bond and the membrane normal via
| ΔνQ = 3/4χQ(3cos2β − 1), |
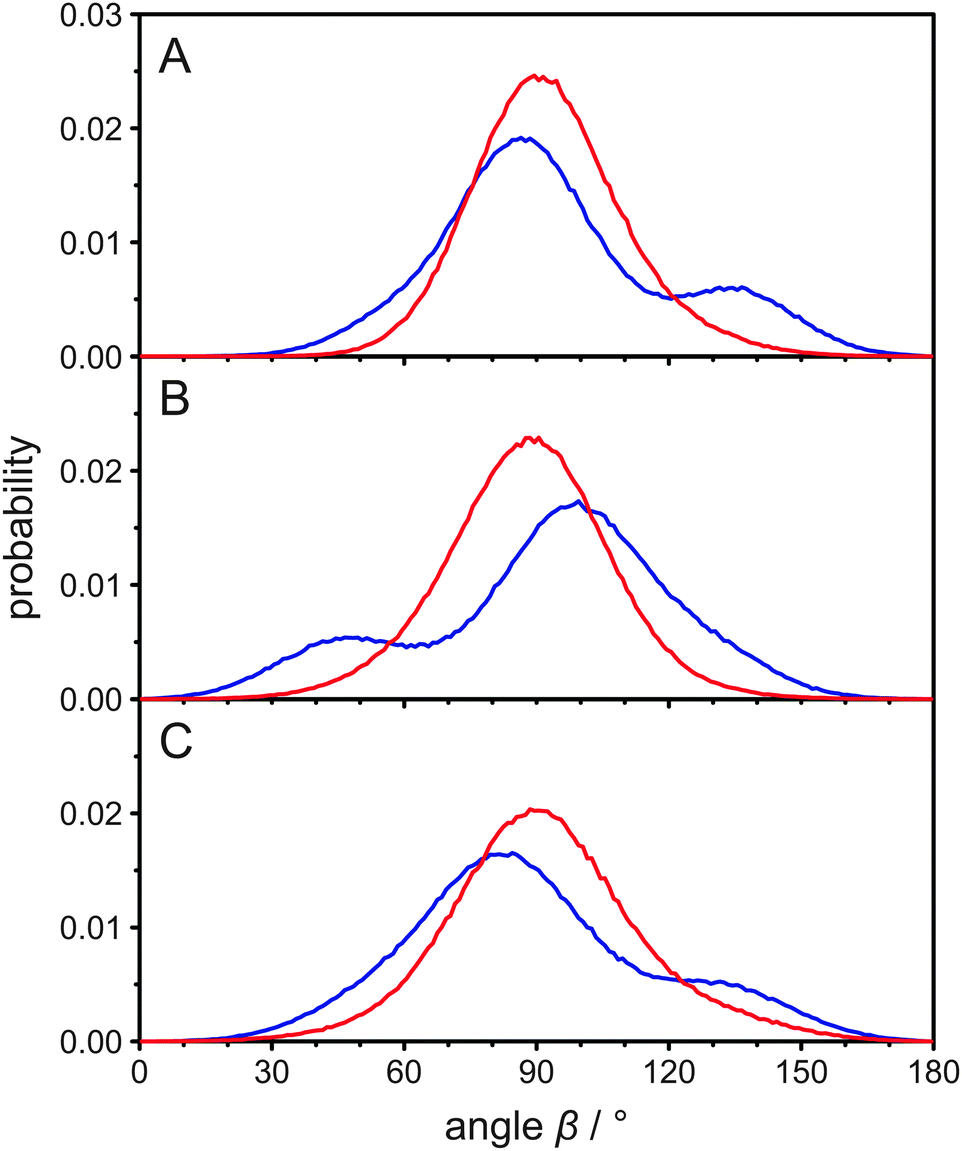 | ||
| Fig. 2 (A) Histograms of the angle β between the two carbon hydrogen bonds attached to C22 and the membrane normal as obtained from the MD simulation of POPC/cholesterol (3:1 mol/mol) at 37 °C. Panels (B) and (C) show the same histograms for C23 and C24, respectively. The data for hydrogen A is shown in blue and for hydrogen B in red. | ||
In the report by Dufourc et al.,25 it has been speculated that the two inequivalent deuterons at C24 have “different average orientations with respect to the axis of segmental motion”. However, from the MD simulation we calculated the mean angle β for both atoms attached to C24 and found they were both very similar (89.3° for the A deuteron and 92.2° for the B deuteron). This small alteration is not sufficient to account for the observed differences in the quadrupolar splittings. From the histograms of the angle β in Fig. 2C it is clear, however, that the explanation of the inequivalent deuterons in the cholesterol side chain is more complex. Clearly, it is not the average orientation of each bond vector that differs but rather their distribution. To investigate the orientations in some more detail, we also determined the mean rotational axis of cholesterol, which is on average relatively parallel to the membrane normal. Due to the cholesterol wobbling motion, the mean angle between this axis and the membrane normal is 17.1°, while the most probable angle is ∼13°. This agrees well with results from other MD simulations.28,37 We also determined the molecular order parameter of the cholesterol ring system, which is 0.839 in our MD simulation. This value agrees well with experimental results.25
Next, we calculated the mean angle between the C24–hydrogen bond and this rotational axis for both hydrogens and again, they were very similar (89.6° for the A hydrogen and 93.0° for the B hydrogen). The histograms of this angle and also for the groups of C22 and C23 are displayed in Fig. S2 (ESI†) and overall show better defined orientations than the histograms in Fig. 2. This is due to the fact that the wobbling motion of cholesterol is not relevant in Fig. S2 (ESI†), as the rotational axis of cholesterol is fixed within the molecule. Nevertheless, the general appearance is the same and the distributions differ significantly for hydrogens that are bound to the same carbon.
One possible explanation for the observed magnetic inequivalency of the C–H bonds at C22, C23, and C24 are steric clashes within the cholesterol molecule. Therefore, we calculated histograms of the dihedral angles between four consecutive carbons along the aliphatic cholesterol side chain from the MD simulation, which are shown as black lines in Fig. 3. A typical histogram that is expected for four consecutive carbons in a non-branched hydrocarbon chain is observed for the dihedral angle C22–C23–C24–C25 shown in Fig. 3C. The trans orientation (dihedral angle ≈ 180°) is most likely, while the two gauche orientations (dihedral angle ≈ ±60°) are less likely, but have identical probability. All other dihedral angles along the chain differ from this expected distribution. For the dihedral angles C17–C20–C22–C23 and C20–C22–C23–C24 the ≈−60° orientation is prohibited. We also extracted these dihedral angles from a total of 38 crystal structures46–49 and the corresponding histograms are shown as grey bars in Fig. 3. The angles that are forbidden in our MD simulations also did not occur in these crystal structures, while all other possible angles are found at least once in the crystal structures.
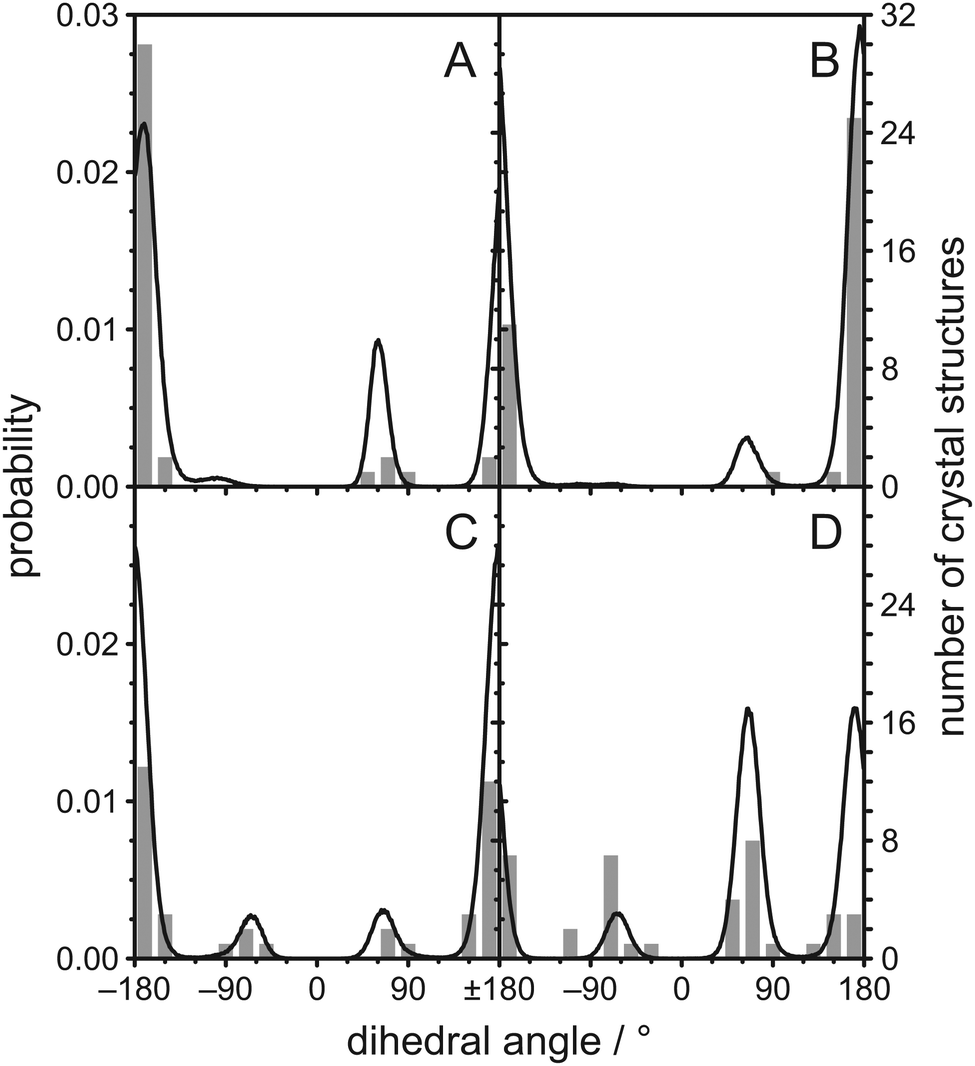 | ||
| Fig. 3 Histograms of dihedral angles in the aliphatic chain of cholesterol as obtained from the MD simulation of POPC/cholesterol (3:1 mol/mol) at 37 °C and from 38 crystal structures found in the literature46–49 for (A) C17–C20–C22–C23, (B) C20–C22–C23–C24, (C) C22–C23–C24–C25, and (D) C23–C24–C25–C26. Data from the MD simulation is shown as black lines while data from the crystal structures is shown as grey bars. | ||
To investigate what causes these −60° orientations to be prohibited, we calculated how the cholesterol structure would look like with exactly this orientation at the C17−C20−C22−C23 dihedral angle and all other dihedral angles in the chain in their most likely conformation (Fig. 4A). In this structure, the hydrogens H16B and H23B have a distance of 1.40 Å, which is significantly less than twice the hydrogen van der Waals radius of 1.09 Å.50 Furthermore, H16A and H23B have a distance of 1.74 Å. Contacts this close are not tolerated and prohibit the dihedral angle to reach −60°. The cholesterol structure with the dihedral angle C20–C22–C23–C24 in the −60° orientation is shown in Fig. 4B. In this structure, H21C (since the CH3 group can rotate freely it also could be H21A or H21B) and H24B have a distance of 1.03 Å and therefore display a significant steric clash. For C23–C24–C25–C26 the ≈−60° orientation has a reduced probability while the +60° and 180° orientations are virtually equally probable (Fig. 3D). To investigate what causes this distribution, the cholesterol structures with the dihedral angle C23–C24–C25–C26 at +60°, 180°, and −60° are shown in Fig. 4C–E, respectively. In the two similarly likely orientations of +60° and 180°, one of the two CH3 groups is in the trans configuration, while in the less likely −60° orientation both CH3 groups are in the gauche configuration. Obviously, it is favorable to have at least one of the bulky CH3 groups extended towards the membrane center. However, this distribution might change in an environment with very short lipid chains, where cholesterol might be forced to adapt to the hydrophobic thickness.51 In the crystal structures however, the +60°, −60°, and 180° orientations have similar probability, which supports our interpretation of a membrane mediated effect that cannot be observed in the crystal structures where no membrane is present.
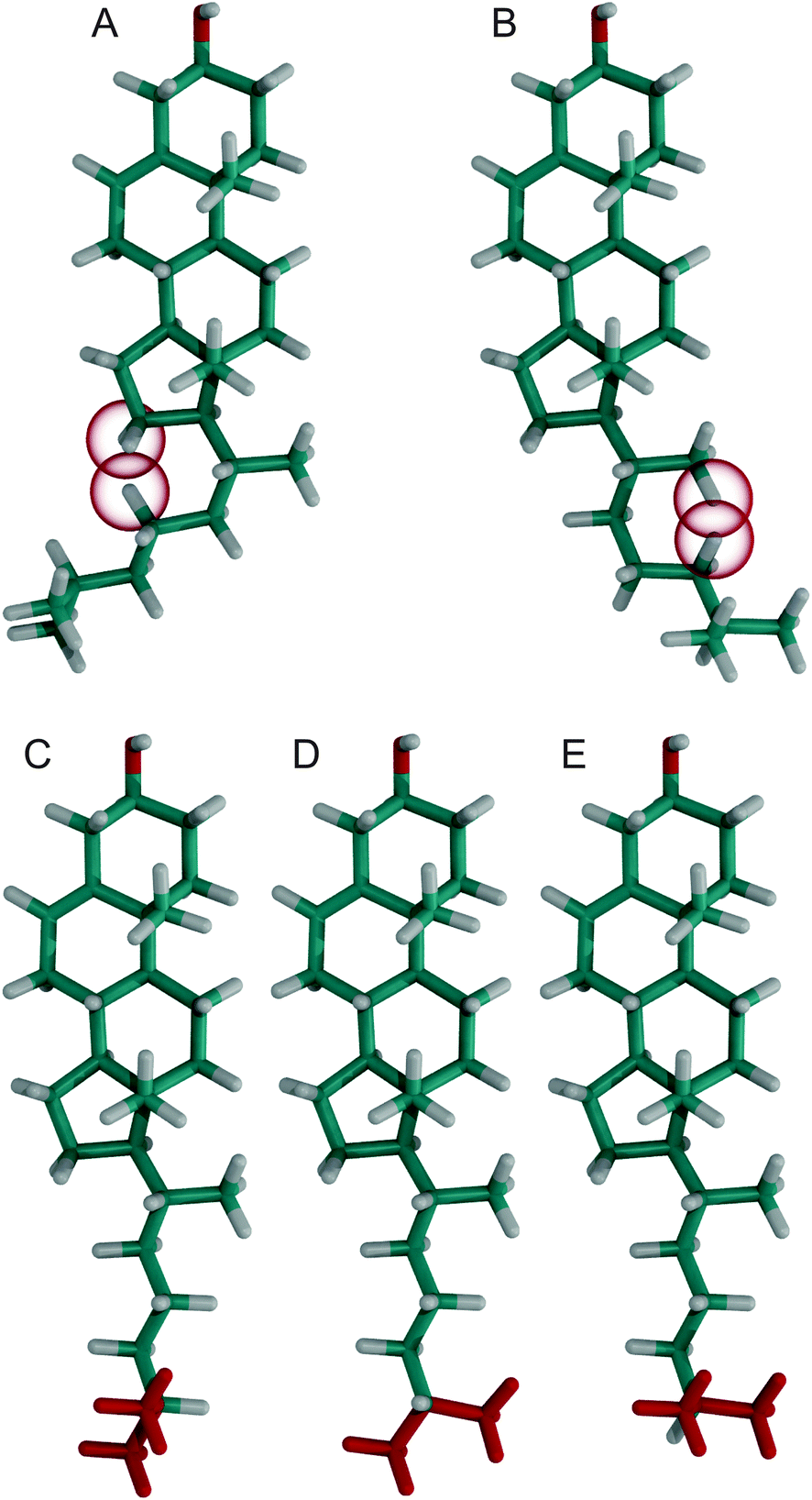 | ||
| Fig. 4 Cholesterol structures with various dihedral angles, where hydrogens that have the closest steric contact are highlighted by a red sphere with the van der Waals radius of hydrogen. All dihedral angles were in their most likely conformation with the exception of (A) C17–C20–C22–C23 at −60°, (B) C20–C22–C23–C24 at −60°, (C) C23–C24–C25–C26 at +60°, (D) C23–C24–C25–C26 at 180°, and (E) C23–C24–C25–C26 at −60° respectively. In panels (C)–(E) the two terminal CH3 groups were colored red for better visibility. | ||
To investigate how the steric clashes influence the distribution of the angles between the carbon hydrogen bonds and the rotational axis of cholesterol, the cholesterol structures with the dihedral angle C17–C20–C22–C23 in the 180°, +60°, and the prohibited −60° orientation are compared in Fig. 5. In this figure, the A hydrogens at C22, C23, and C24 are colored blue and in the two allowed dihedral angles they have very different orientations with respect to the rotational axis of cholesterol, which is shown in green. This explains why in the histogram (Fig. S2, ESI†) several maxima at different angles are observed. For the B hydrogens, however, in the two allowed dihedral angles the angle between the carbon hydrogen bond and the rotational axis of cholesterol is always close to 90°, explaining the single maximum around 90° in Fig. S2 (ESI†). The dihedral angle of −60° that would cause angles between the carbon hydrogen bond and the rotational axis of cholesterol other than 90° is prohibited by the steric clashes between H16B and H23B as well as H16A and H23B.
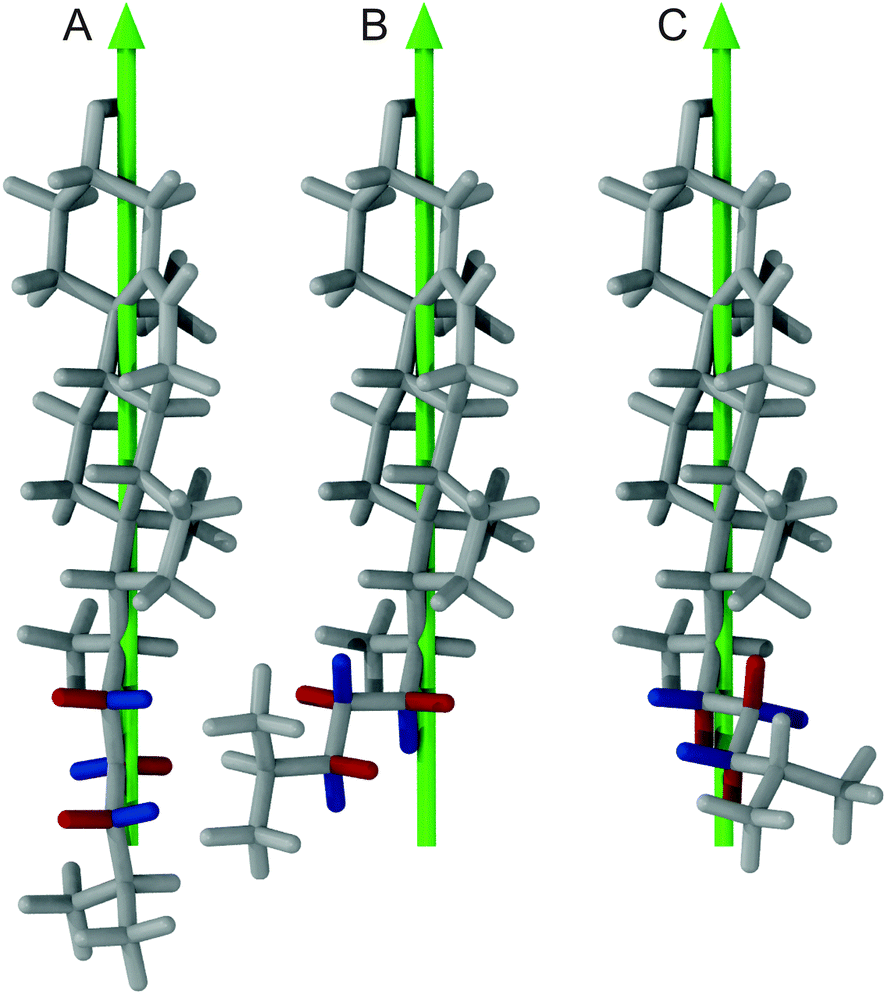 | ||
| Fig. 5 Cholesterol structures with the dihedral angle C17–C20–C22–C23 in (A) 180°, (B) +60°, and the prohibited (C) −60° orientation respectively. The green arrow indicates the main rotational axis of cholesterol while A hydrogens at C22, C23, and C24 are colored blue and B hydrogens are colored red. | ||
To further investigate the influence of the surrounding phospholipids on the cholesterol molecule and its aliphatic side chain, experiments as well as MD simulations with DPPC, PSM, and a POPC/PSM (1:1 mol/mol) mixture (no MD simulation for this system) were conducted. Such binary and ternary systems also represent the essential lipid species in raft mixtures. In Fig. 6, the experimental spectra (black lines) at various temperatures are summarized. All 2H NMR spectra with sufficient resolution were fitted (red lines) by a superposition of seven Pake doublets, where quadrupolar splitting, line width, chemical shift, and overall sample ellipticity were adjusted. Good fits were obtained for cholesterol-d11 in POPC and POPC/PSM, while fits in DPPC and PSM show clear deviations indicative of slight axially asymmetric motion25,41,42 even at 50 °C. From these fits, 2H NMR order parameters were obtained and assigned in the same sequence as the order parameters derived from the MD simulation of POPC/cholesterol at 37 °C. All order parameters are summarized in Fig. S3 and Table S1 (ESI†) (data for POPC at 37 °C is found in Fig. 1C and Table 1). Comparing the spectral shape and order parameter profile a high similarity is observed between POPC and the POPC/PSM mixture (the average order parameter is higher in the latter) at all temperatures. This indicates that the overall behavior of the aliphatic side chain is similar in both environments on the NMR timescale. In contrast, the 2H NMR spectrum of cholesterol-d11 in PSM looks quite different, although the order parameters are similar to those in the POPC/PSM mixture. The numerical fits of the spectra clearly show signs of slight axially asymmetric motion, which means that cholesterol might not achieve total rotational symmetry on the NMR timescale of several microseconds even at 50 °C. This indicates that cholesterol rotation is slower in PSM than in POPC/PSM. The same is observed for DPPC.
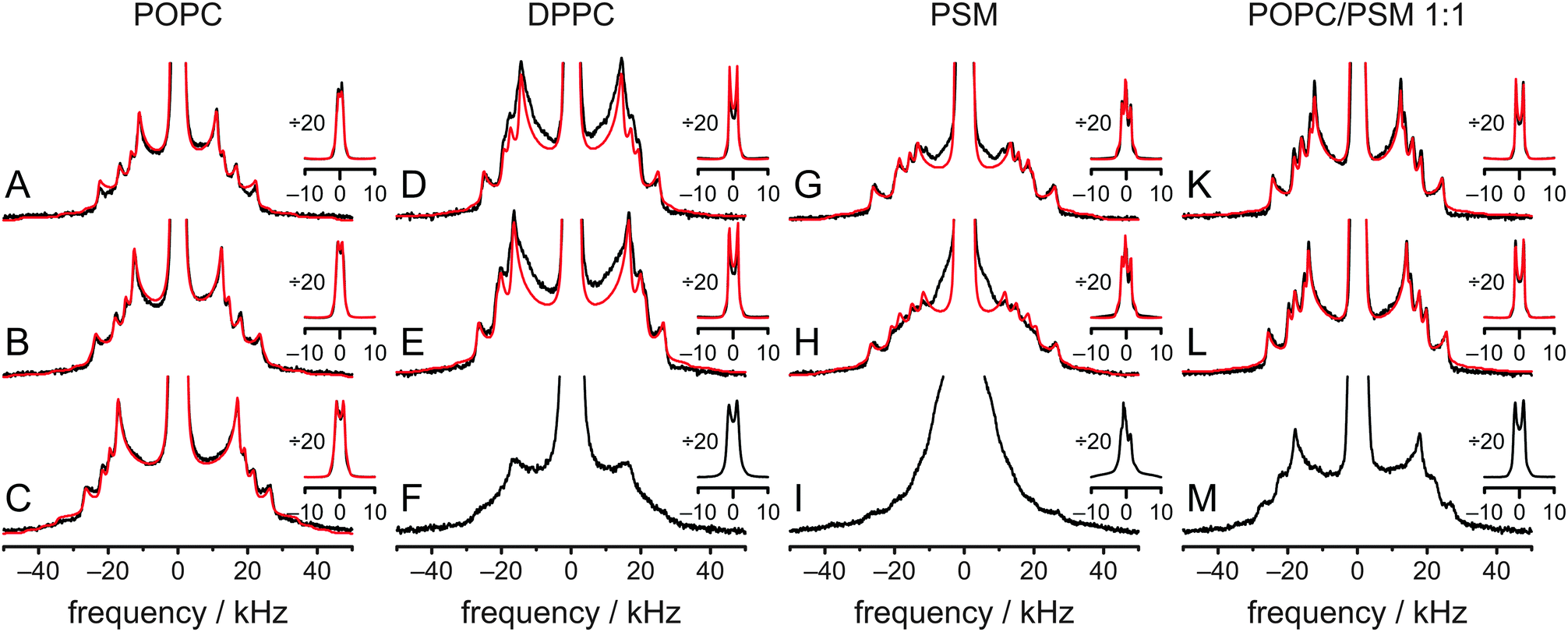 | ||
| Fig. 6
2H NMR spectra of cholesterol-d11 (25 mol% relative to all other lipids) in various environments at 50 °C (A, D, G and K), 37 °C (B, E, H and L), and 5 °C (C, F, I and M). NMR spectra were measured in POPC (A–C), DPPC (D–F), PSM (G–I) and POPC/PSM (1:1 mol/mol) (K–M). The inset shows the same spectra vertically divided by a factor of 20 to show the intense Pake doublets from the two terminal CH3 groups. Measured spectra are shown in black, while numerical fits are red. | ||
The order parameters also were extracted from MD simulations of cholesterol in POPC, DPPC, and PSM at 50 °C. Overall, the agreement for the two MD simulations in DPPC and PSM was not as good as for the POPC simulations at 37 °C and 50 °C, but still much better than reported previously.28 In the POPC MD simulations, the average order parameter was slightly smaller by 4.3% (37 °C) or 3.1% (50 °C) than the experiment. In contrast, it was somewhat bigger by 16.3% and 8.7% in the DPPC and PSM MD simulations, respectively. From the MD order parameters at 50 °C, 2H NMR spectra were calculated and a direct comparison to the spectra obtained from experiment is found in Fig. S4 (ESI†) (the comparison for POPC at 37 °C is found in Fig. 1B). Again, differences are small for POPC and larger for DPPC and PSM.
Discussion
It is well known that cholesterol is an evolutionarily highly optimized molecule. Its most striking effect in membranes is the cholesterol induced condensation of the surrounding phospholipids and the accompanying increase in molecular order and membrane stiffness and a concomitant reduction in membrane permeability.15,52 No matter which modifications of the cholesterol structure are tested, none of them can significantly improve this condensation effect.53 In fact, many modifications lead to dramatic reductions of this effect even if the modification was very small, such as changing a single double bond within the ring structure.54–56 Recently, however, it has been shown that rather surprisingly, approximately half of the condensation effect is caused by the aliphatic side chain of cholesterol.15 Furthermore, the condensation effect depends on the chain length and is highest for the natural chain length of cholesterol.15 In addition, the condensation effect is reduced in case there is no branched methyl group at the chain end.17In this article, details of the structure and dynamics of the aliphatic side chain of cholesterol and the molecular origin of the observed effects were investigated in four different membrane environments using 2H NMR spectroscopy and MD simulations. The general features of all experimental spectra are similar, indicating that the overall behavior of the aliphatic side chain of cholesterol is comparable in all environments. At 37 °C, the average order parameter is lowest for POPC (0.128) and highest for DPPC (0.160). Average order parameters in PSM and POPC/PSM are relatively similar (0.149 and 0.147, respectively). The lineshapes of the respective 2H NMR spectra, however, are rather different. In POPC and POPC/PSM, the NMR spectra are rather similar and indicate axially symmetric motions of cholesterol, while in DPPC and PSM some line broadening, possibly indicative of asymmetric behavior even at 50 °C is observed, which probably originates from incomplete rotational averaging on the NMR timescale of several microseconds. This indicates that the presence of POPC accelerates cholesterol rotational motion in PSM. At 5 °C, individual Pake doublets cannot be discriminated anymore in DPPC and PSM (see Fig. 6), while the 2H NMR spectra in POPC and POPC/PSM still resolve more details.
To analyze the NMR spectra in more detail, several MD simulations were conducted and overall, their agreement with experiment was very reasonable. It was exceptionally good with less than 5% deviation for the two POPC MD simulations at different temperatures, while previous MD simulations of a very similar system differed by a factor of ∼2.28 The large difference in the aliphatic side chain region of the previous MD simulation (despite of the otherwise good agreement) can be explained by the observed steric clashes as described in this article. The previous simulations used a force field with united atoms in the aliphatic side chain, which therefore could not capture the steric clashes of the involved hydrogens. Unfortunately, the steric clashes are separated by two dihedral angles (refer Fig. 4A and B). If they were separated by just one dihedral angle, the steric clashes could be incorporated into the dihedral angle force field parameters. Therefore, cholesterol force fields with united atoms in the aliphatic side chains should be used with caution.
In our study, agreement with experimental data was still reasonable for the MD simulations of cholesterol in DPPC and PSM with 16.3% and 8.7% deviation, respectively. Nevertheless, this was significantly worse than in the simulations of the POPC/cholesterol mixture. Since the agreement was good in POPC, force field inaccuracies are unlikely. However, the NMR spectra of these samples already show signs of slight axially asymmetric motion on a timescale of several microseconds (Fig. 6). Therefore, the origin of this discrepancy most likely is incomplete sampling despite the very long 1.5 μs MD simulation time. Possibly, rapid MD sampling techniques57–59 would yield better agreement. This interpretation is supported by the relatively good agreement between experiment and simulation for the more mobile CH3 groups, where sampling issues are diminished.
Therefore, the MD simulation of cholesterol in POPC at 37 °C, where agreement was excellent, was analyzed in detail. The most interesting observation in this study was that the hydrogens at C22, C23, and C24 are not magnetically equivalent and that this is caused by steric clashes of atoms within the cholesterol molecule. These clashes prevent the occurrence of certain conformations. This reduced conformational flexibility is also reflected in the order parameters that were calculated for the C–D bonds in the aliphatic cholesterol side chain. These order parameters are relatively high compared to those in the methylene and methyl segments in the lipids, which are similarly deeply embedded in the bilayer11 and comparable to those in the rigid ring determined previously.25 It is surprising that experimental investigations showed the hydrogens on C22 to be equivalent, while those on C24 were not equivalent.25 The steric clashes we found should lead to inequalities at both positions and our MD results are not consistent with this experimental observation, which we cannot explain at the moment.
Conclusions
The observed steric clashes shed light on the question why the aliphatic side chain of cholesterol contributes significantly to the condensation effect of the molecule in membranes. It is located close to the methylene and methyl segments in the lipids, which exhibit the lowest order and, therefore, can be ordered to the highest extent. There, it occupies conformational space and thereby limits the conformational flexibility of the lipid acyl chains. The relatively high order that is observed for the aliphatic cholesterol side chain magnifies this effect. The prohibited conformations reduce the volume accessible to the aliphatic side chain, which reduces the number of possible conformations. In turn, this also reduces the possible conformations that the surrounding lipid acyl chains can adapt.Interestingly, the prohibited conformations are those, in which the aliphatic side chain would stay in the plane of the cholesterol ring structure (see Fig. 4A and B). Therefore, we analyzed the positions of the C25 group when looking along the main rotational axis of cholesterol, as shown in Fig. 7. It is immediately obvious that the aliphatic side chain has a much higher tendency to move perpendicular to the ring system instead of staying within the plane of the rings. For a more detailed analysis, the chain axis was defined as the vector from C17 to C25 and the order parameter matrix for this axis calculated in the internal frame of cholesterol via
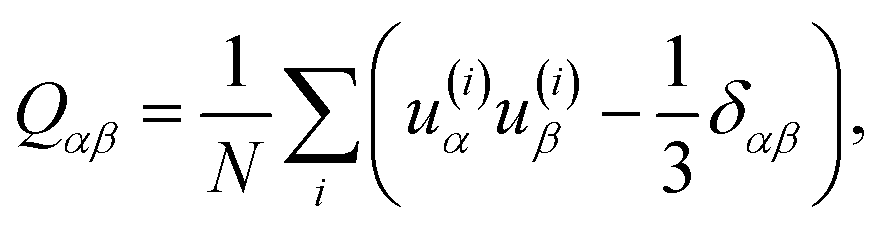
The resulting order parameter S was 0.78 with a biaxiality B of 0.046 (for the MD simulation of cholesterol in POPC at 50 °C those values were 0.76 and 0.051, respectively). These values of biaxiality are comparable to values observed for nematic liquid crystals with biaxial behavior.60 Therefore, the forbidden conformations not only increase the order of the aliphatic side chain, but also lead to a very anisotropic mobility pattern. This possibly modulates cholesterol lipid interactions, but the implications of this behavior are not understood yet and should be investigated in the future.
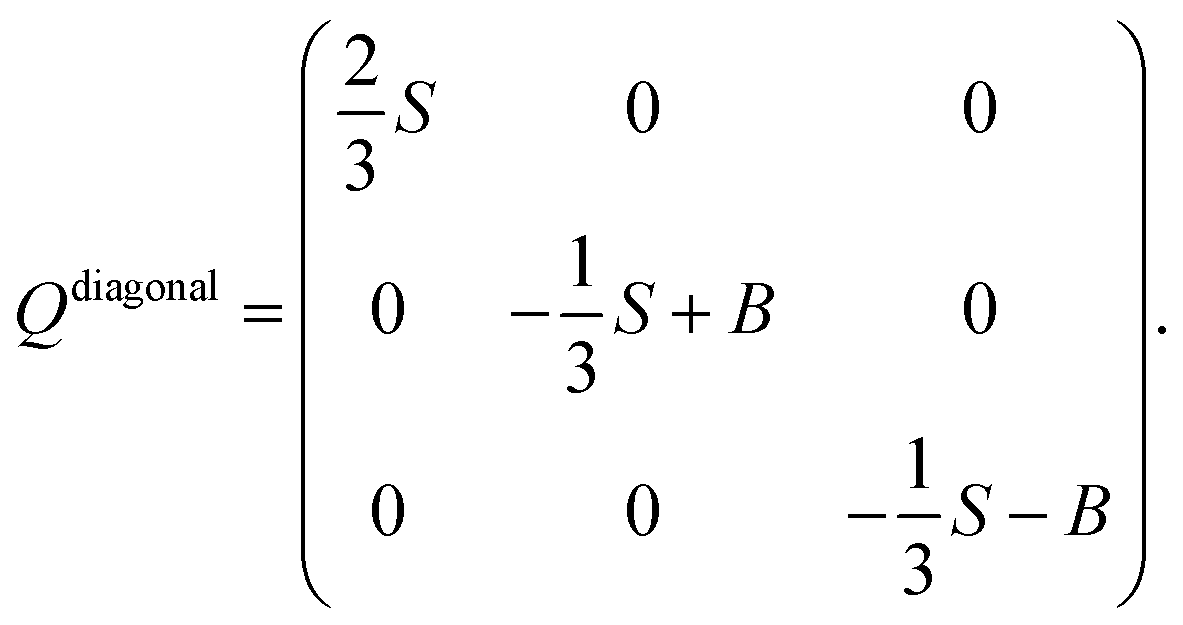
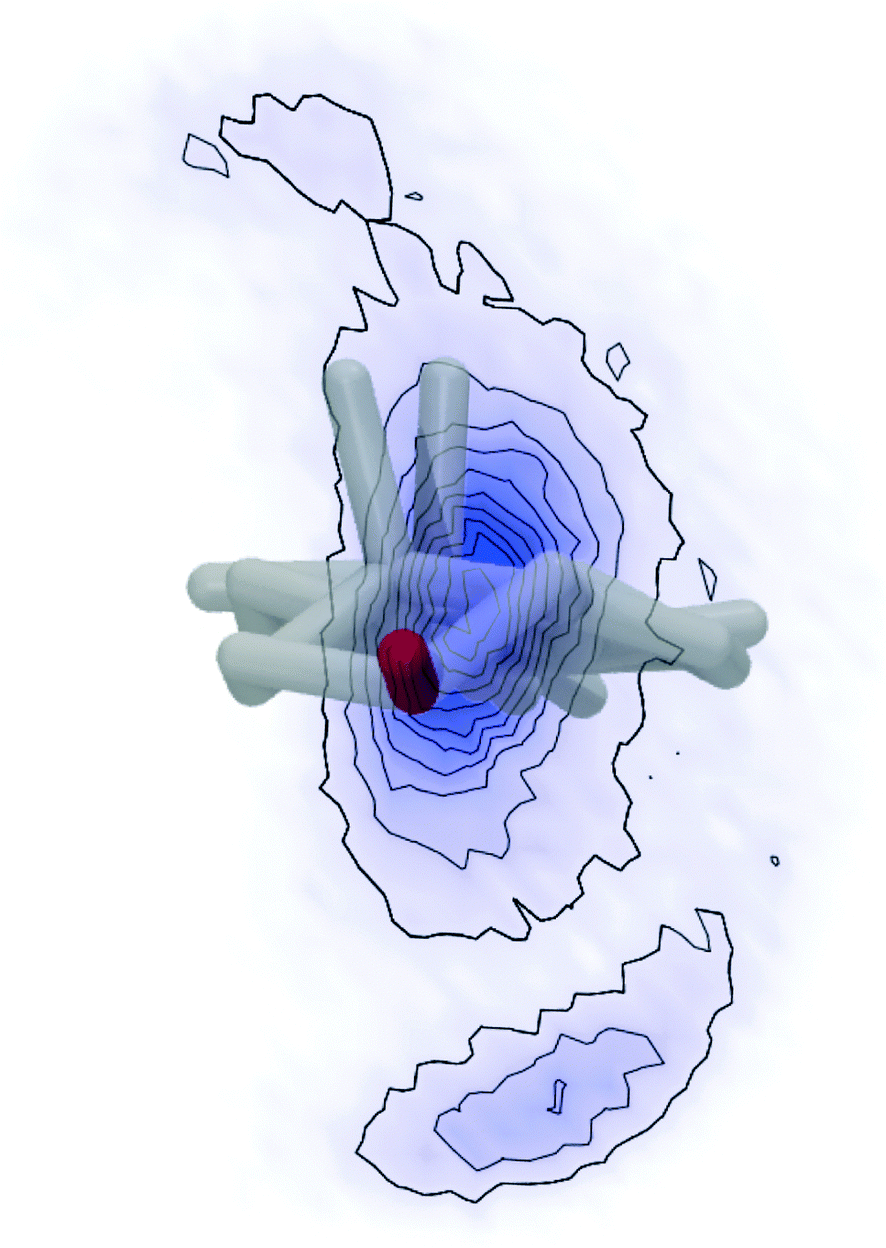 | ||
| Fig. 7 Distribution of the C25 position when looking at cholesterol from the top (looking at the OH group (red) towards the aliphatic side chain along the main rotational axis of cholesterol). Hydrogens were omitted from the cholesterol structure for clarity and the two CH3 groups that stick out of the plane of the cholesterol rings can be seen pointing upwards. The probability distribution of the C25 position is indicated by blue color, where more intense color corresponds to higher probability. | ||
Acknowledgements
The authors would like to thank Prof. Dr Norbert Sträter for useful comments on cholesterol structures. The study was supported by the Deutsche Forschungsgemeinschaft (SFB 1052, B6).References
- M. G. van, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112 CrossRef PubMed.
- K. Jacobson, O. G. Mouritsen and R. G. W. Anderson, Nat. Cell Biol., 2007, 9, 7 CrossRef PubMed.
- M. L. Frazier, J. R. Wright, A. Pokorny and P. F. Almeida, Biophys. J., 2007, 92, 2422 CrossRef PubMed.
- A. Tsamaloukas, H. Szadkowska and H. Heerklotz, J. Phys.: Condens. Matter, 2006, 18, S1125 CrossRef PubMed.
- K. Simons and E. Ikonen, Science, 2000, 290, 1721 CrossRef PubMed.
- D. A. Brown and E. London, Annu. Rev. Cell Dev. Biol., 1998, 14, 111 CrossRef PubMed.
- S. Munro, Cell, 2003, 115, 377 CrossRef PubMed.
- N. Shimokawa, M. Nagata and M. Takagi, Phys. Chem. Chem. Phys., 2015, 17, 20882 RSC.
- P. F. Almeida, A. Pokorny and A. Hinderliter, Biochim. Biophys. Acta, 2005, 1720, 1 CrossRef PubMed.
- D. Huster, K. Arnold and K. Gawrisch, Biochemistry, 1998, 37, 17299 CrossRef PubMed.
- A. Bunge, P. Müller, M. Stöckl, A. Herrmann and D. Huster, Biophys. J., 2008, 94, 2680 CrossRef PubMed.
- M. A. Davies, H. F. Schuster, J. W. Brauner and R. Mendelsohn, Biochemistry, 1990, 29, 4368 CrossRef CAS.
- G. Orädd, V. Shahedi and G. Lindblom, Biochim. Biophys. Acta, 2009, 1788, 1762 CrossRef PubMed.
- E. Oldfield, M. Meadows, D. Rice and R. Jacobs, Biochemistry, 1978, 17, 2727 CrossRef CAS PubMed.
- H. A. Scheidt, T. Meyer, J. Nikolaus, D. J. Baek, I. Haralampiev, L. Thomas, R. Bittman, A. Herrmann, P. Müller and D. Huster, Angew. Chem., Int. Ed., 2013, 52, 12848 CrossRef CAS PubMed.
- J. R. Robalo, J. P. Prates Ramalho, D. Huster and L. M. Loura, Phys. Chem. Chem. Phys., 2015, 17, 22736 RSC.
- T. Meyer, D. J. Baek, R. Bittman, I. Haralampiev, P. Müller, A. Herrmann, D. Huster and H. A. Scheidt, Chem. Phys. Lipids, 2014, 184, 1 CrossRef CAS PubMed.
- D. Huster, Biochim. Biophys. Acta, 2014, 1841, 1146 CrossRef CAS PubMed.
- M. F. Brown, Biochemistry, 2012, 51, 9782 CrossRef CAS PubMed.
- A. Leftin and M. F. Brown, Biochim. Biophys. Acta, 2011, 1808, 818 CrossRef CAS PubMed.
- T. Bartels, R. S. Lankalapalli, R. Bittman, K. Beyer and M. F. Brown, J. Am. Chem. Soc., 2008, 130, 14521 CrossRef CAS PubMed.
- S. L. Veatch, O. Soubias, S. L. Keller and K. Gawrisch, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17650 CrossRef CAS PubMed.
- A. Leftin, T. R. Molugu, C. Job, K. Beyer and M. F. Brown, Biophys. J., 2014, 107, 2274 CrossRef CAS PubMed.
- M. G. Taylor, T. Akiyama, H. Saito and I. C. Smith, Chem. Phys. Lipids, 1982, 31, 359 CrossRef CAS PubMed.
- E. J. Dufourc, E. J. Parish, S. Chitrakorn and C. P. Smith, Biochemistry, 1984, 23, 6062 CrossRef CAS.
- P. Schmidt, L. Thomas, P. Müller, H. A. Scheidt and D. Huster, Chemistry, 2014, 20, 4986 CrossRef CAS.
- L. Thomas, J. Kahr, P. Schmidt, U. Krug, H. A. Scheidt and D. Huster, J. Biomol. NMR, 2015, 61, 347 CrossRef CAS PubMed.
- T. M. Ferreira, F. Coreta-Gomes, O. H. Ollila, M. J. Moreno, W. L. Vaz and D. Topgaard, Phys. Chem. Chem. Phys., 2013, 15, 1976 RSC.
- K. Gawrisch, N. V. Eldho and I. V. Polozov, Chem. Phys. Lipids, 2002, 116, 135 CrossRef CAS PubMed.
- K. Mors, C. Roos, F. Scholz, J. Wachtveitl, V. Dotsch, F. Bernhard and C. Glaubitz, Biochim. Biophys. Acta, 2013, 1828, 1222 CrossRef PubMed.
- M. Holtje, T. Forster, B. Brandt, T. Engels, R. W. von and H. D. Holtje, Biochim. Biophys. Acta, 2001, 1511, 156 CrossRef CAS.
- D. J. Baek and R. Bittman, Chem. Phys. Lipids, 2013, 175–176, 99 CrossRef CAS PubMed.
- J. H. Davis, K. R. Jeffrey, M. Bloom, M. I. Valic and T. P. Higgs, Chem. Phys. Lett., 1976, 42, 390 CrossRef CAS.
- S. Jo, T. Kim and W. Im, PLoS One, 2007, 2, e880 Search PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830 CrossRef CAS PubMed.
- J. B. Lim, B. Rogaski and J. B. Klauda, J. Phys. Chem. B, 2012, 116, 203 CrossRef CAS PubMed.
- R. M. Venable, A. J. Sodt, B. Rogaski, H. Rui, E. Hatcher, A. D. MacKerell, Jr., R. W. Pastor and J. B. Klauda, Biophys. J., 2014, 107, 134 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577 CrossRef CAS.
- W. van Gunsteren and H. J. C. Berendsen, Mol. Phys., 1977, 34, 1311 CrossRef CAS.
- J. H. Davis, Biochim. Biophys. Acta, 1983, 737, 117 CrossRef CAS.
- J. Seelig, Q. Rev. Biophys., 1977, 10, 353 CrossRef CAS PubMed.
- H. I. Petrache, S. W. Dodd and M. F. Brown, Biophys. J., 2000, 79, 3172 CrossRef CAS PubMed.
- M. F. Brown, S. Lope-Piedrafita, G. V. Martinez, H. I. Petrache, in Modern Magnetic Resonance, ed. G. Webb, Springer, Heidelberg, 2006, pp. 241–252 Search PubMed.
- A. Vogel, H. A. Scheidt, S. E. Feller, J. Metso, R. M. Badeau, M. J. Tikkanen, K. Wahala, M. Jauhiainen and D. Huster, Biophys. J., 2014, 107, 114 CrossRef CAS PubMed.
- H. S. Shieh and C. E. Nordmann, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2002, 58, 079 Search PubMed.
- R. J. Galloway, S. A. Raza, R. D. Young and I. D. H. Oswald, Cryst. Growth Des., 2012, 12, 231 CAS.
- H. S. Shieh, L. G. Hoard and C. E. Nordmann, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1538 CrossRef.
- L. Y. Hsu, J. W. Kampf and C. E. Nordman, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 260 Search PubMed.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384 CrossRef CAS.
- W. G. Wu and L. M. Chi, J. Am. Chem. Soc., 1991, 113, 4683 CrossRef CAS.
- O. G. Mouritsen and M. J. Zuckermann, Lipids, 2004, 39, 1101 CrossRef CAS PubMed.
- T. Rog, M. Pasenkiewicz-Gierula, I. Vattulainen and M. Karttunen, Biophys. J., 2007, 92, 3346 CrossRef CAS PubMed.
- H. A. Scheidt, P. Müller, A. Herrmann and D. Huster, J. Biol. Chem., 2003, 278, 45563 CrossRef CAS PubMed.
- D. Huster, H. A. Scheidt, K. Arnold, A. Herrmann and P. Müller, Biophys. J., 2005, 88, 1838 CrossRef CAS PubMed.
- S. Milles, T. Meyer, H. A. Scheidt, R. Schwarzer, L. Thomas, M. Marek, L. Szente, R. Bittman, A. Herrmann, P. T. Gunther, D. Huster and P. Müller, Biochim. Biophys. Acta, 2013, 1828, 1822 CrossRef CAS PubMed.
- A. Vogel, G. Reuther, M. B. Roark, K. T. Tan, H. Waldmann, S. E. Feller and D. Huster, Biochim. Biophys. Acta, 2010, 1798, 275 CrossRef CAS PubMed.
- Y. M. Rhee and V. S. Pande, Biophys. J., 2003, 84, 775 CrossRef CAS PubMed.
- D. Hamelberg, J. Mongan and J. A. McCammon, J. Chem. Phys., 2004, 120, 11919 CrossRef CAS PubMed.
- K. Severing and K. Saalwächter, in Thermotropic Liquid Crystals, ed.A. Ramamoorthy, Springer, Berlin, 2007, ch. 5, pp. 141–170 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05084g |
| ‡ Prof. Dr Robert Bittman passed away on 1 October 2014. |
| This journal is © the Owner Societies 2016 |