
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The donor OH stretching–libration dynamics of hydrogen-bonded methanol dimers in cryogenic matrices†
M.
Heger
 a,
J.
Andersen
b,
M. A.
Suhm
a and
R.
Wugt Larsen
*b
a,
J.
Andersen
b,
M. A.
Suhm
a and
R.
Wugt Larsen
*b
aInstitut für Physikalische Chemie, Universität Göttingen, Tammannstr. 6, D-37077 Göttingen, Germany
bDepartment of Chemistry, Technical University of Denmark, Kemitorvet 206, DK-2800 Kgs. Lyngby, Denmark. E-mail: rewl@kemi.dtu.dk
First published on 14th January 2016
Abstract
FTIR spectra of the methanol dimer trapped in neon matrices are presented. The fundamental, overtone and combination bands involving the donor OH libration and stretching motions were observed in order to extract relevant anharmonicity constants. We find a stretching–libration coupling constant of +43(5) cm−1 and a diagonal librational anharmonicity constant of −71(5) cm−1. The spectra are compared to a number of VPT2 calculations and a torsionally localized monomer model in order to enhance previous explanations of the observable OH stretching red-shift upon dimerization.
1 Introduction
For the past few decades, the methanol dimer has seen intensive spectroscopic characterization as a provider of dynamic details of the simplest aliphatic OH⋯O hydrogen bond.1–8 Specifically, interest has been directed to the OH stretching motions since they provide sensitive probes for the hydrogen-bonding interaction between the two constituent methanol molecules, and the 111 cm−1 red shift in the jet-cooled gas phase has recently been augmented with information on its diagonal anharmonicity.7 Together with a high-level harmonic reference for this band, it was inferred that the overall corrections from couplings to other vibrational modes should be on the order of 30 cm−1.7,9 It can be expected that most of this “off-diagonal” anharmonic correction stems from the librational motion, i.e. the torsion of the donor OH group around the central C–O bond which becomes hindered in the hydrogen bond. The term “libration” is used for this large-amplitude intermolecular hydrogen bond motion to distinguish this mode from the much less perturbed torsion of the acceptor OH group upon complexation. The recent observation of the OH libration fundamental excitation at 560 cm−1 by matrix- and jet-FTIR spectroscopy lends credibility to a series of quantum chemical predictions which reproduce the observable band positions of the donor OH stretching and libration bands and further predict a 60 cm−1 stretching–libration coupling element.7 This would coincidentally explain almost all of the total off-diagonal shift, because each coupling element enters the correction with a factor of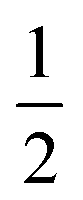 .10 Overall, the emergent picture has been largely consistent so far, but lacks stringent experimental corroboration of anharmonicity data for the important stretching–libration coupling.
.10 Overall, the emergent picture has been largely consistent so far, but lacks stringent experimental corroboration of anharmonicity data for the important stretching–libration coupling.
The easiest way to achieve this is to observe the according combination band at ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) s,l = s + l + xs,l (where s and l refer to the stretching and librational modes, respectively, and x denotes anharmonicity constants). Since the fundamental transitions s and l have already been characterized for the methanol dimer,7,8 the combination band remains as the only missing link for this analysis. The usual practice of backing the experiments with predictions from quantum chemical methods can of course be helpful in providing first estimates for the band positions and coupling constants; these can then in turn be corroborated or falsified once their true values have been extracted from the observations. However, realistic estimates from quantum chemistry for the methanol dimer may be hard to come by, given that many popular theoretical methods tend to notoriously misjudge the energetics of the hydrogen-bonded OH oscillator at least at the harmonic level.9,11 Chances are that the case is even more difficult when including the librational motion due to its much shallower potential. Furthermore, the anharmonic treatments undertaken here and previously are based on a second-order perturbational scheme12–16 in its implementation by Barone10,12 in which the harmonic wavenumber assumes the role of the zeroth-order reference. As with any perturbational approach, one should be wary if the perturbation is of considerable magnitude; seeing that the best harmonic librational prediction of 660 cm−1 suggests an anharmonic perturbation of this mode on the order of −100 cm−1, the previously found agreement with experiment may be serendipitous or misleading.8 It is thus indispensable to obtain direct experimental values for the coupling constant in order to test the predictions.
s,l = s + l + xs,l (where s and l refer to the stretching and librational modes, respectively, and x denotes anharmonicity constants). Since the fundamental transitions s and l have already been characterized for the methanol dimer,7,8 the combination band remains as the only missing link for this analysis. The usual practice of backing the experiments with predictions from quantum chemical methods can of course be helpful in providing first estimates for the band positions and coupling constants; these can then in turn be corroborated or falsified once their true values have been extracted from the observations. However, realistic estimates from quantum chemistry for the methanol dimer may be hard to come by, given that many popular theoretical methods tend to notoriously misjudge the energetics of the hydrogen-bonded OH oscillator at least at the harmonic level.9,11 Chances are that the case is even more difficult when including the librational motion due to its much shallower potential. Furthermore, the anharmonic treatments undertaken here and previously are based on a second-order perturbational scheme12–16 in its implementation by Barone10,12 in which the harmonic wavenumber assumes the role of the zeroth-order reference. As with any perturbational approach, one should be wary if the perturbation is of considerable magnitude; seeing that the best harmonic librational prediction of 660 cm−1 suggests an anharmonic perturbation of this mode on the order of −100 cm−1, the previously found agreement with experiment may be serendipitous or misleading.8 It is thus indispensable to obtain direct experimental values for the coupling constant in order to test the predictions.
Perchard and coworkers have extensively studied methanol embedded in nitrogen, argon and neon matrices,4,6,17 but without placing a particular focus on the stretching–libration couplings. Furthermore, only monomer transitions were assigned in the neon matrices where perturbations from the matrix environment are the smallest. We thus set out to explore the important xs,l anharmonicity constant by means of the less intense OH stretching–libration combination bands of the methanol dimer by this sensitive matrix isolation approach. Further data for the deuterated isotopologues are presented in the ESI.†
2 Experimental details
A pre-cooled (77 K) gas flow of neon (L'Air Liquide, 99.999%) was deposited with a flow rate of 0.02 mol h−1 on a gold-plated oxygen-free high thermal conductivity (OFHC) copper mirror at 3.6 K inside an immersion helium cryostat (IHC-3). This cryostat has been modified for matrix isolation spectroscopy18,19 and is mounted to the Bruker IFS 120 FTIR spectrometer installed on the infrared beam-line at the MAX IV facility hosted by Lund University. The pre-cooling of neon significantly reduces the heat load on the copper mirror and enables a total deposition time of about 1 hour per experiment. The pre-cooled neon gas flow was subsequently doped with “freeze–pump–thaw” purified methanol (Sigma Aldrich, 99.9%) and isotopically substituted methanol-d1 (Sigma Aldrich, 99.0% D), methanol-d3 (Sigma Aldrich, 99.0% D) and methanol-d4 (Sigma Aldrich, 99.0% D) samples with mixing ratios of 1–2‰. In the experiments for methanol-d1 and methanol-d4, the inner surfaces of the entire inlet system and mounted sample flasks were heavily deuterated with the enriched sample prior to the deposition procedure. A combination of resistive heaters and feedback electronics was employed to maintain a stable mirror temperature at 2.8 ± 0.1 K before and after the matrix deposition. The outer shroud of the cryostat was equipped with a CsI window to provide a viewing port and combined mid-infrared and near-infrared single-beam sample spectra were collected using a Bruker IFS 120 FTIR spectrometer employing tungsten and globar lamps as radiation sources. Liquid nitrogen cooled HgCdTe (broadband) and InSb detectors combined with Ge/KBr and CaF2 beam splitters were employed for the 600–5000 cm−1 and 1700–11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 spectral region, respectively. In all experiments, the doped neon matrices were subsequently annealed to 9 K to promote the diffusion of methanol and further formation of a dimer in the soft matrices. A spectral resolution of 0.5 cm−1 was chosen as the best compromise between a high signal-to-noise ratio and a sufficiently high spectral resolution to resolve the observed sub-band spectral features assigned to the methanol dimer (see Section 4.1). The single-beam background spectra were collected using the evacuated warm cryostat.
500 cm−1 spectral region, respectively. In all experiments, the doped neon matrices were subsequently annealed to 9 K to promote the diffusion of methanol and further formation of a dimer in the soft matrices. A spectral resolution of 0.5 cm−1 was chosen as the best compromise between a high signal-to-noise ratio and a sufficiently high spectral resolution to resolve the observed sub-band spectral features assigned to the methanol dimer (see Section 4.1). The single-beam background spectra were collected using the evacuated warm cryostat.
3 Computational details
Quantum chemical calculations have previously been carried out for the methanol monomer and dimer at the MP2, B2PLYP-D3BJ and B3LYP-D3BJ levels of theory using the cc-pVTZ (“VTZ”) basis sets.8 We revisit these calculations herein to judge their robustness and extend the study to the deuterated isotopologues (see ESI†). All calculations were carried out using the Gaussian 09 package,20 including Grimme's D3 dispersion for the DFT calculations.21 Tight optimization convergence criteria and, for DFT calculations, ultrafine integration grids were used throughout (opt = tight and int = ultrafine keywords, respectively). Anharmonic treatments were then carried out using the perturbational VPT2 method10,12 implemented in this software package.Due to potential incompatibilities of the VPT2 implementation in Gaussian09 with Becke–Johnson damping, the results presented herein have been obtained with zero-damping.22 We have also explored the numerical sensitivity of low-frequency DFT VPT2 predictions to minute dimer structure variations, suggesting larger error bars in some of our earlier results than implied before. In particular, the off-diagonal librational anharmonicity constants calculated previously for the methanol dimer8 will be slightly impacted by this, as we demonstrate in the ESI;† similar variations will exist in the off-diagonal coupling terms to the OH stretching wavenumbers in methanol–ethene,9 but the impact on our previous results is likely to be small.
4 Results
4.1 Neon matrix spectral data
The recorded absorption spectra of methanol are shown in Fig. 1 for the OH stretching fundamental, overtone and stretching–libration combination bands as well as the libration overtone region. The spectra and calculations for the deuterated isotopologues are shown in the ESI.† Throughout, black and red traces show pre- and post-annealing spectra, respectively, and blue traces (annotated “diff.”) show their difference. Due to optical saturation, the OH stretching fundamental band lacks direct intensity information, and we omit its annealing difference trace. All experiments were repeated at different methanol concentrations, and the dimer assignments we put forward below are based on the observed concentration dependence and annealing trends.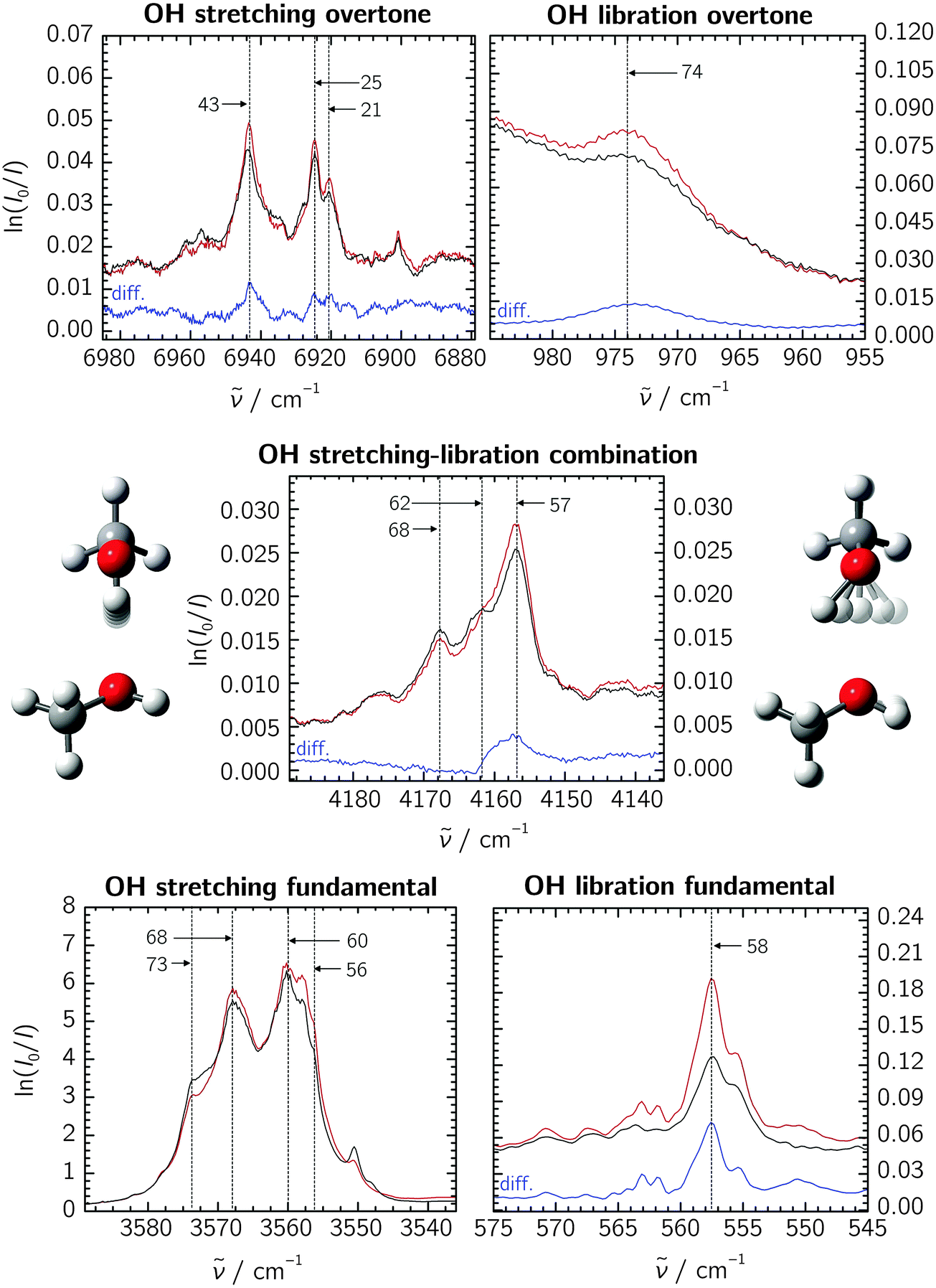 | ||
| Fig. 1 Neon matrix spectra of the methanol dimer in the relevant spectral regions. The black and red traces show pre- and post-annealing spectra, respectively; blue traces show the difference spectra (“diff.”, omitted for the OH stretching fundamental due to saturation). Annotated are the last two digits of assigned sub-band positions. The OH libration fundamental spectrum is reproduced from a previous study at a lower methanol concentration.8 The absorbance scale is defined by the natural logarithm (napierian absorbance) for all the spectral regions. | ||
The spectra reveal some complex band patterns for the features assigned to the OH stretching fundamental of the methanol dimer.7 However, the combined concentration dependency and dedicated pre- and post-annealing measurements together with the direct comparison with previous jet observations5 unambiguously support the dimer origin of both the complex band patterns observed in the 3555–3575 cm−1 region for regular methanol and methanol-d3, and the 2625–2635 cm−1 region for methanol-d1 and methanol-d4 (see ESI†). The independent absorption spectra recorded previously for regular methanol embedded in both neon and para-H2 matrices have revealed similar spectral splittings, although smaller in the para-H2 host.7 A detailed physical interpretation of the observed spectral splittings is beyond the scope of the current contribution concerned with the determination of much larger anharmonicity constants. However, the vibrational spectrum of the methanol monomer embedded in neon and para-H2 has previously revealed some complex torsional–vibrational couplings with resulting A–E splittings of the ground-state and the different fundamental states.17,23 The usual A–A and E–E gas-phase selection rules do not seem to apply in the environments of para-H2 and neon, and each of the fundamental bands for the methanol monomer thus gives rise to four different sub-bands with splittings determined by the size of the individual torsional–vibrational couplings.17,23 The spectral splittings observed for the OH-stretching fundamental of the methanol dimer appear to follow a similar trend, although the exact pattern is less clear. CH3 deuteration qualitatively conserves this pattern, whereas OH deuteration does not (see ESI†). The pronounced isotope effects rule out the presence of different random trapping sites in the solid neon matrix cages, as does the fact that the spectral splittings are also observed for the methanol dimer embedded in much larger para-H2 matrix cages.24
Our attempts at identifying individual sub-features are marked by anchor lines in the spectra, annotated with the last two digits of the respective band origins. Accepting these features to stem from some yet unclear vibrational dynamics within the methanol dimer, we correlate them among the fundamental and overtone bands to extract the diagonal anharmonicity constants xs,s. The results are listed in Table 1, yielding an average value of −97 cm−1 for the methanol dimer. Agreement with previous Ne, para-H2 and jet experiments is very good,7 and we assume that the desired results for the stretching–libration coupling are similarly transferable to the gas-phase situation.
Concerning the stretching–libration combination band, we are tempted to correlate the three assigned methanol dimer features with those on the lower-wavenumber side of the stretching fundamental band. Extracting the coupling constant xs,l further requires the librational fundamental band position, which has previously been established as 558 cm−1.8 The resulting xs,l values for the individual sub-bands are given in Table 1, yielding an average of +43 cm−1. In addition to the stretching–libration combination band, we suggest an assignment of the broad transition at 974 cm−1 to the overtone of the librational motion (see Fig. 1). Together with the fundamental band at 558 cm−1,8 this yields a diagonal anharmonicity constant xl,l = −71 cm−1. Based on the extensive band splittings, certain mismatches between the individual correlated sub-features, and residual matrix shifts even in neon, we assume an error bar of ±5 cm−1 throughout for all experimentally determined anharmonicity constants.
The analyses for the deuterated isotopologues are analogous to the methanol case and shall not be outlined here in greater detail, not least because our assignments are less complete and the annealing effects are much smaller for the CD3 species. The spectra and data are given in the ESI.† For the dimer of methanol-d3, the diagonal anharmonicity constant of −99(5) cm−1 is in good agreement with the methanol results. Likewise, a rich band structure is again observed between 4175 and 4157 cm−1, and correlating the stretching and libration fundamental bands yields a coupling constant of +42(5) cm−1. This structure however vanishes in methanol-d1 and methanol-d4 upon deuteration of the hydroxy group, and we refrain from correlating individual bands. Placing their combination bands at 3075 and 3076 cm−1, respectively, suggests xs,l coupling elements on the order of 25(5) cm−1 in light of the general expanse of the fundamental band structures. Furthermore, we were unable to identify the librational overtones in the CD3 species due to overlapping strong monomer features. For methanol-d1, we assign a band at 766 cm−1 to the librational overtone, and together with the 420 cm−1 fundamental,8 we obtain a diagonal anharmonicity constant of xl,l = −37(5) cm−1, about half that of the non-deuterated methanol dimer.
4.2 Quantum chemical predictions of anharmonic coupling constants
Estimates for xs,s and xs,l in the methanol dimer have been obtained before at various levels of theory.7,9 It was established that the former anharmonicity constant is reproduced quite well by all methods, with deviations of a few cm−1. For the stretching–libration coupling constant, MP2 and DFT calculations yielded consistent predictions of +59 cm−1.7,8 This consistency contrasts with substantially larger variations in the predicted vibrational transitions which are correlated by this coupling constant. The variation does not only involve different theoretical levels, but also subtly different structure optimizations at exactly the same level, when DFT methods are employed (see Section 3 of the ESI†). A particularly striking indicator for this is the harmonically second-lowest intermolecular vibration, representing the hindered rotation of the acceptor methanol molecule around an axis approximately parallel to the C–O bond (Ia in the free monomer). The fundamental and overtone bands of this mode fluctuate over a large wavenumber range across all calculations, often reaching imaginary values and unreasonable anharmonicity corrections. Similar variations, albeit less drastic, persist for the librational band, and uncertainties in the fundamental band position directly impact the stretching–libration band as well.Based on a series of DFT calculations on the methanol dimer (see Section 3 in the ESI†), we assume uncertainties of at least 10 cm−1 for the position of the libration fundamental, 5 cm−1 for the donor OH stretching fundamental, and thus up to 15 cm−1 for the stretching–libration combination band. At the same time, these instabilities are practically absent in MP2, which leads us to suspect difficulties in the numerical integration in DFT. These numerical instabilities add to the methodical inaccuracy of the perturbational approach for such large-amplitude vibrations, which is valid for any electronic structure level. However, we concentrate here on the important xs,s, xs,l and xl,l anharmonicity constants, which turn out to be robust on all employed levels of theory (see Table 1). The results show a consistent overestimation of xs,l across all methods by some 35(5)%, while xl,l is underestimated by about the same relative amount.
4.3 Analysis of the donor OH-stretching dimerization red-shift
Having established an experimental value for the stretching–libration coupling, we can further update our previous analyses of the observable OH stretching red shift in the methanol dimer, −Δs.9 Since we require also anharmonicity information for the monomer, we henceforth introduce superscripts “D” and “M” for the dimer and the monomer, respectively. The OH stretching wavenumber for the dimer is given by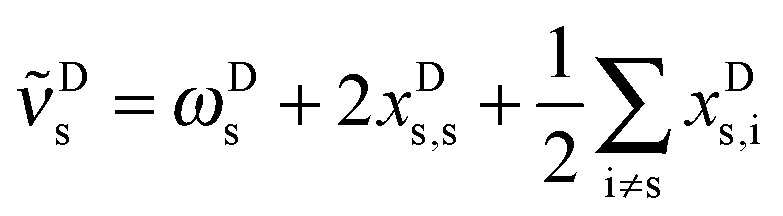 | (1) |
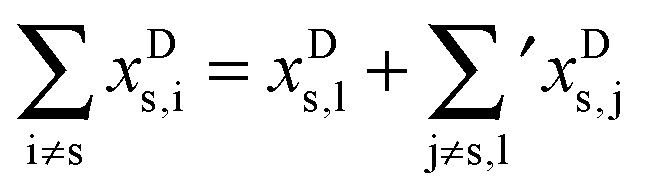 | (2) |
s requires to take the difference of all terms, where we imply the different summation ranges in a common term: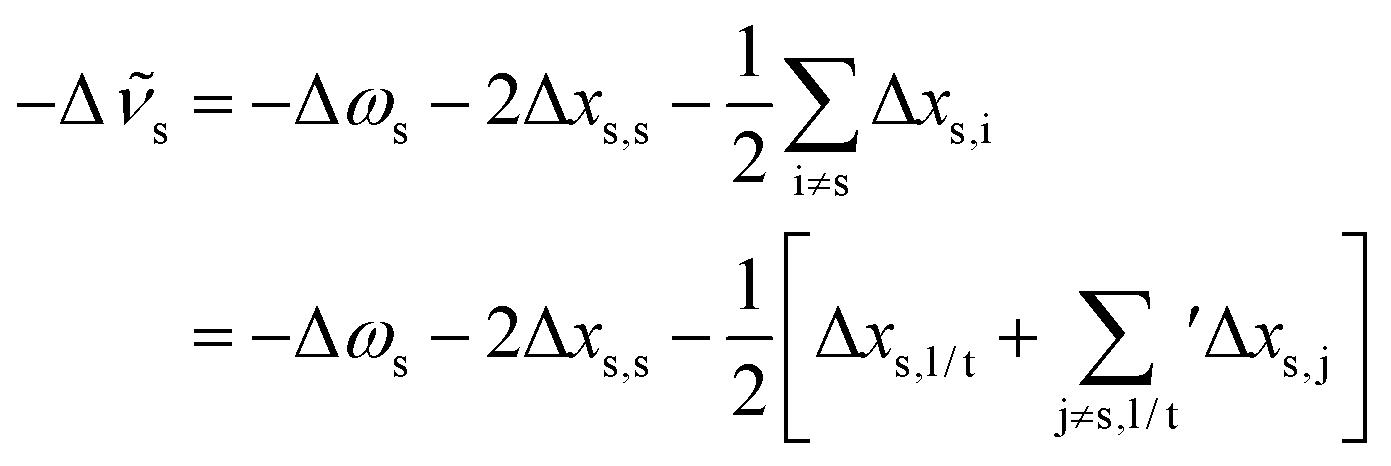 | (3) |
One fundamental error in the monomer calculations is that they do not honor the three-fold symmetry of the OH torsional motion and consequently neglect the resulting strong tunneling splittings into the A and E sub-states. Gas-phase studies by Hunt et al.25 and Rueda et al.26 have shown that the torsional barrier increases upon excitation of the OH stretching oscillator, thereby reducing the tunneling splittings in the stretching–torsion vs, vt = 1, 1 state relative to vs, vt = 0, 1. One should thus obtain two distinct values for xMs,t, depending on whether it is calculated from the A or E states. Using energy levels determined from the OH stretching fundamental25 and the first overtone26 transitions together with ground-state torsional references,27 we find average gas-phase values of xMs,t = +0.4(3) cm−1 for the A states and xMs,t = +13.5(3) cm−1 for the E states, with surprisingly little variation for the fundamental and overtone values. To compare these with the matrix environment, we make use of earlier monomer studies by Perchard et al.6,17 Their band assignments suggest a coupling constant of +12.8 cm−1 for the E ← A stretching–torsion combination transition, and since the perturbations from the Ne matrix host are moderate for the fundamentals,17 this compares quite well with the analogous gas-phase value of +10.9 cm−1.25,27 (For details on the involved transitions, see ESI.†) We are thus confident that the experimental dimer coupling constant determined from our matrix measurements is in similarly good agreement with the gas-phase situation.
Still, the ambiguities caused by the tunneling splittings prompt for some sort of localization approach in order to be directly comparable to quantum-chemical calculations. In a simple state-specific picture, we regard the A/E triplet of each vs, vt ensemble as the eigenvalues of a 3 × 3 matrix with localized, three-fold degenerate “single-well” energy levels on the diagonal, and uniform inter-level couplings among the potential wells as the off-diagonal elements. From the eigenvalues of these matrices, the coupling elements amount to a third of the observable A–E splittings, and the localized energy levels represent the center of gravity of the A and E levels. We concede that this is a rather coarse approach to the problem, neglecting all interactions between the sub-levels of different torsional states. On the other hand, the moderate difference in the A and E xs,t values calculated above suggests that its errors will likely be exceeded by those of the VPT2 calculations. An energy level scheme of the delocalized (experimental) and localized vs, vt states for vs = 0 to 2 is given in the ESI.† Using both the OH fundamental and overtone transitions, we find xMs,t values of +9.3 and +8.8 cm−1, respectively. Seeing the high anharmonicity content of the torsional motion, it is surprising to find the vs = 1 and 2 results being in such good agreement; however, we cannot rule out error compensation with the localization approach as the cause for this without further analysis.
In addition to the stretching–torsion coupling, we find a localized OH-stretching fundamental wavenumber of 3683.4 cm−1 and a diagonal anharmonicity constant of xMs,s = −85.7 cm−1. Together with the 3574.5 cm−1 donor OH stretching wavenumber observed for the dimer, the overall approximate single-well dimerization shift relevant to eqn (3) is about 109 cm−1.
Having established a monomer reference comparable to our calculations, we can now set out to fill in the quantities in eqn (3). The best estimate for the harmonic dimerization shift is 121 cm−1,11 for which we assume a ±5 cm−1 uncertainty. The experimental gas-phase diagonal anharmonicity contribution in the dimer, −99.2 cm−1, together with the localized gas-phase monomer reference from above, yields −2Δxs,s = 27 cm−1.7 Meeting the overall 109 cm−1 dimerization red shift then requires an off-diagonal correction of 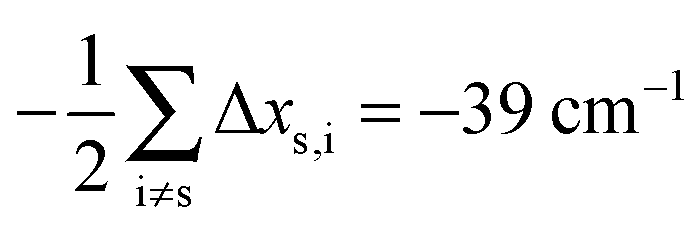 , containing an experimental torsional/librational contribution of
, containing an experimental torsional/librational contribution of 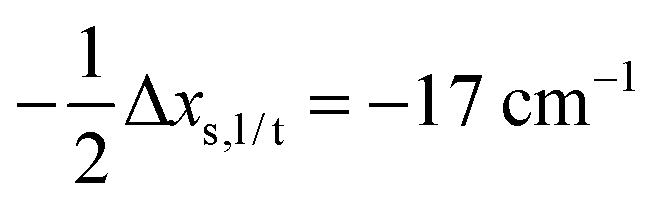 (see Table 2). From an experimental perspective, the 10 remaining off-diagonal differences and 18 unique dimer coupling terms together must thus contribute −22 cm−1, but considering the cumulative errors, this residual (one half the difference of the primed sums) is only determined within about ±10 cm−1. Again, the VPT2 calculations – particularly B3LYP – have some difficulty in predicting the quantities listed in Table 2, which we further detail in the ESI.† Given the availability of experimental values for the most important off-diagonal contributions, however, we are free of the burden to rely on these predictions alone, and can further confirm that at least a qualitative agreement between theory and experiment can be reached.
(see Table 2). From an experimental perspective, the 10 remaining off-diagonal differences and 18 unique dimer coupling terms together must thus contribute −22 cm−1, but considering the cumulative errors, this residual (one half the difference of the primed sums) is only determined within about ±10 cm−1. Again, the VPT2 calculations – particularly B3LYP – have some difficulty in predicting the quantities listed in Table 2, which we further detail in the ESI.† Given the availability of experimental values for the most important off-diagonal contributions, however, we are free of the burden to rely on these predictions alone, and can further confirm that at least a qualitative agreement between theory and experiment can be reached.
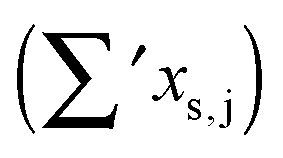 as per eqn (2). The last line gives experimental values. All data are in cm−1
as per eqn (2). The last line gives experimental values. All data are in cm−1
| Monomer | Dimer | Dimer–monomer | ||||
|---|---|---|---|---|---|---|
| x s,t |
|
x s,l |
|
|
|
|
| B2PLYP-D3/VTZ | +4 | −28 | +58 | +15 | −27 | −22 |
| B3LYP-D3/VTZ | +3 | −26 | +58 | +25 | −28 | −26 |
| MP2/VTZ | +9 | −30 | +59 | +9 | −25 | −20 |
| Exp. | +9 | +43 | −17 | −22 | ||
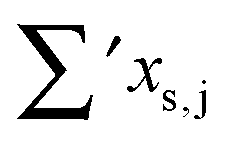
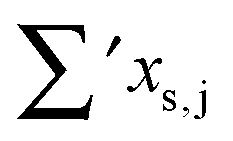
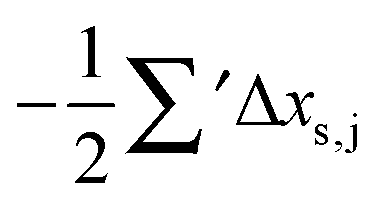
5 Summary
FTIR absorption spectra of the methanol dimer embedded in neon matrices have been analyzed with respect to the combination band of the donor OH stretching and libration motions, and the overtone band of the latter. A co-investigation of the stretching fundamental is necessary to correlate the complicated band patterns and extract the stretching–libration coupling constant. Together with libration fundamental band positions from previous investigations, we find a stretching–libration coupling constant of xs,l = +43(5) cm−1 for the methanol dimer. Anharmonic VPT2 calculations mostly overestimate these values by some 35(5)%, while the experimentally determined diagonal anharmonicity constant of the librational motion (xl,l = −71(5) cm−1) is underestimated in magnitude by a similar amount. With the new experimental finding, we are able to update previous analyses of the observable dimerization shift in the donor OH stretching band.Using a state-specific deperturbation approach to approximate tunneling-independent monomer reference values, we explain the observable 109 cm−1 gas-phase OH stretching red-shift of the methanol dimer as being comprised of about 121(5) cm−1 (theoretical) harmonic and 27(2) cm−1 (experimental) diagonal anharmonic contributions, counteracted in part by a single −17(5) cm−1 (experimental) stretching–libration contribution. This leaves a −22(10) cm−1 gap to be explained by the remaining off-diagonal couplings, which VPT2 helps to do in a qualitative way. Our data support the prediction that the stretching–libration coupling is the most important contribution to the off-diagonal anharmonic shift, which is necessary to compensate for the red-shifting diagonal weakening of the OH stretching oscillator and explain the observable dimerization red-shift. A thorough variational treatment in an accurate stretching–libration potential would be desirable to gain further insight into these important dynamics.
As a bottom line, we have now characterized the two major and tendentially canceling anharmonic contributions to the overall dimerization red-shift of the methanol dimer in a direct spectroscopic manner, and the next challenge in this regard may be the influence of the torsional motion of the free OH group in the acceptor molecule.
Acknowledgements
Funding from the German Research Foundation (Su 121/4) is greatly appreciated. RWL acknowledges financial support from the Danish Council for Independent Research's Sapere Aude Progamme (Grant Ref.: 12-125248).References
- F. Huisken, A. Kulcke, C. Laush and J. M. Lisy, J. Chem. Phys., 1991, 95, 3924–3929 CrossRef.
- T. Häber, U. Schmitt and M. A. Suhm, Phys. Chem. Chem. Phys., 1999, 1, 5573–5582 RSC.
- R. A. Provencal, J. B. Paul, K. Roth, C. Chapo, R. N. Casaes, R. J. Saykally, G. S. Tschumper and H. F. Schaefer, J. Chem. Phys., 1999, 110, 4258–4267 CrossRef.
- J. P. Perchard and Z. Mielke, Chem. Phys., 2001, 264, 221–234 CrossRef.
- R. Wugt Larsen, P. Zielke and M. A. Suhm, J. Chem. Phys., 2007, 126, 194307 CrossRef PubMed.
- J. P. Perchard, F. Romain and Y. Bouteiller, Chem. Phys., 2008, 343, 35–46 CrossRef.
- F. Kollipost, K. Papendorf, Y.-F. Lee, Y.-P. Lee and M. A. Suhm, Phys. Chem. Chem. Phys., 2014, 16, 15948–15956 RSC.
- F. Kollipost, J. Andersen, D. W. Mahler, J. Heimdal, M. Heger, M. A. Suhm and R. Wugt Larsen, J. Chem. Phys., 2014, 141, 174314 CrossRef PubMed.
- M. Heger, R. A. Mata and M. A. Suhm, Chem. Sci., 2015, 6, 3738–3745 RSC.
- V. Barone, J. Chem. Phys., 2005, 122, 14108 CrossRef PubMed.
- M. Heger, M. A. Suhm and R. A. Mata, J. Chem. Phys., 2014, 141, 101105 CrossRef PubMed.
- V. Barone, M. Biczysko, J. Bloino, M. Borkowska-Panek, I. Carnimeo and P. Panek, Int. J. Quantum Chem., 2012, 112, 2185–2200 CrossRef CAS.
- R. Burcl, N. C. Handy and S. Carter, Spectrochim. Acta, Part A, 2003, 59, 1881 CrossRef.
- A. D. Boese and J. M. L. Martin, J. Phys. Chem. A, 2004, 108, 3085–3096 CrossRef CAS.
- A. Miani, E. Cané, P. Palmieri, A. Trombetti and N. C. Handy, J. Chem. Phys., 2007, 112, 248 CrossRef.
- J. F. Gaw, A. Willetts, W. H. Green and N. C. Handy, in Advances in Molecular Vibrations and Collision Dynamics, ed. J. Bowman, JAI Press Inc., Greenwich, CT, 1991, vol. 1B, pp. 169–185 Search PubMed.
- J. P. Perchard, Chem. Phys., 2007, 332, 86–94 CrossRef CAS.
- J. Ceponkus, P. Uvdal and B. Nelander, J. Chem. Phys., 2008, 129, 194306 CrossRef CAS PubMed.
- J. Andersen, J. Heimdal and R. Wugt Larsen, Phys. Chem. Chem. Phys., 2015, 17, 23761–23769 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- Y.-P. Lee, Y.-J. Wu, R. M. Lees, L.-H. Xu and J. T. Hougen, Science, 2006, 311, 365–368 CrossRef CAS PubMed.
- J. Ceponkus, A. Engdahl, P. Uvdal and B. Nelander, Chem. Phys. Lett., 2013, 581, 1–9 CrossRef CAS.
- R. H. Hunt, W. N. Shelton, F. A. Flaherty and W. B. Cook, J. Mol. Spectrosc., 1998, 192, 277–293 CrossRef CAS PubMed.
- D. Rueda, O. V. Boyarkin, T. R. Rizzo, I. Mukhopadhyay and D. S. Perry, J. Chem. Phys., 2002, 116, 91–100 CrossRef CAS.
- G. Moruzzi, F. Strumia, J. C. S. Moraes, R. M. Lees, I. Mukhopadhyay, J. W. Johns, B. P. Winnewisser and M. Winnewisser, J. Mol. Spectrosc., 1992, 153, 511–577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07387a |
| This journal is © the Owner Societies 2016 |