
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmmonia-modified Co(II) sites in zeolites: spin and electron density redistribution through the CoII–NO bond†
Adam
Stępniewski
a,
Mariusz
Radoń
b,
Kinga
Góra-Marek
b and
Ewa
Broclawik
*a
aJerzy Haber Institute of Catalysis PAS, Niezapominajek 8, 30-239 Krakow, Poland. E-mail: broclawi@chemia.uj.edu.pl
bFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Krakow, Poland
First published on 5th January 2016
Abstract
Electronic factors essential for the bonding of a non-innocent NO ligand to ammonia-modified Co2+ sites in cobalt-exchanged zeolites are examined for small cluster models using DFT and advanced correlated wave function calculations. The analysis of charge transfer processes between the NO ligand and the cobalt center involves two protocols: valence-bond expansion of the multiconfiguration CASSCF wave function (in terms of fragment-localized active orbitals) and spin-resolved natural orbitals for chemical valence (SR-NOCV). Applicability of SR-NOCV analysis to transition metal complexes involving non-innocent fragments is critically assessed and the approach based on the CASSCF wave function turns out to be much more robust and systematic for all studied models. It is shown that the character and direction of electron density redistribution through the Co–N–O bond, quantified by relative share of the CoII–NO0, CoIII–NO−, and CoI–NO+ resonance structures in the total wave function, fully rationalize the activation of the N–O bond upon NH3 co-ligation (evidenced by calculated and measured red-shift of the NO stretching frequency and commonly ascribed to enhanced backdonation). The huge red-shift of νN–O is attributed to an effective electron transfer between the ammonia-modified Co(II) centers and the NO antibonding π*-orbitals (related to the increased share of the CoIII–NO− form). Unexpectedly, the effect is stronger for the singlet complex with three NH3 ligands than for that with five NH3 ligands bound to the cobalt center. Our results also indicate that high-efficiency electron transfers between the Co(II) center and the NO ligand may be enabled for the selected spin state and disabled for the other spin state of the adduct. This illustrates how the cobalt center may serve to fine-tune the electronic communication between the NO ligand and its binding site.
1. Introduction
The interest in cobalt exchanged zeolites stems from their catalytic activity, e.g. with respect to the removal of nitric oxides by selective catalytic reduction (SCR) with ammonia or hydrocarbons, in particular the SCR with methane.1–5 The catalytic properties of a cobalt site depend on many factors: they may be modified by a zeolite framework type, Si/Al ratio, cobalt setting (position, coordination or oxidation status) and by coadsorption of ligands. Furthermore, an in-depth description of the ligand bonding in cobalt complexes (where the high-spin electronic state is the most common, but low-spin complexes are also known) has been a topical research target of unremitting interest for a long time.6–9 For zeolites, these factors are not easily accessible from experiment thus molecular modeling by quantum chemical methods is a desirable complementary technique to help clear all quandaries.10,11On the other hand, nitric oxide is a well-known redox-noninnocent ligand, which piles up difficulties in the description of the Co–NO bonding. The chameleon nature of NO adducts is well expressed by the Enemark–Feltham notation for its complexes with dn transition metal (TM), namely {TM–(NO)y}n+y (where y is the number of NO ligands and n + y denotes the number of electrons delocalized within the fragment in the braces). The notation stems from the known fact that a strict allocation of electrons to TM or NO species is intrinsically disputable and constitutes a big challenge for computational chemistry, calling for the involvement of high-level correlated methods.12,13
In our recent work14 we have already discussed the dependence of the activity of cobalt sites in zeolites towards NO on the coordination of additional electron donor ligands. The calculation results served to interpret the IR spectra measured for nitric oxide sorbed on cobalt sites in zeolites after controlled ammonia pretreatment. Both IR experiment and DFT modeling recorded a strong red-shift of the NO stretching frequency after ammonia adsorption on zeolite samples14,15 which complies with a commonly accepted notion that co-adsorption of electron-donating ligands should enhance the backdonation from the cobalt center to the NO ligand and thus increase the deNOx activity of cobalt sites.16 Also for the isolated pentaamminecobalt(II) complex a strong red-shift of the NO stretching frequency was calculated, in accord with the experimental IR data for the black isomer of nitrosylpentaamminecobalt(III) dichloride.17 Interestingly enough, our previous study indicated that the spin state of the Co(II)–NO adduct (triplet in the native site) may evolve upon bonding of consecutive NH3 ligands to either the singlet or the triplet, these two spin states having comparable energies but showing strikingly different activation abilities towards NO.14 This finding increases the prospective relevance of such systems since the controlled tuning of the spin state in atoms/molecules has already met profound interest in view of prospective spin-based devices.18 For example, it was reported that the surface-induced magnetic moment in (S = 1/2) CoII porphyrin could be switched-off by axial coordination of the NO ligand whereas the chemical switching-on of the spin in organometallic complexes could be imposed by non-spin-bearing (S = 0) external NH3 ligands.19 Therefore, an in-depth investigation of the character of the Co(II)–NO bond in the considered co-adducts with ammonia emerges as a consequent extension of the previous work.
In this work we present an advanced study on the nature of the Co–NO interaction and its dependence on the coordination sphere of the cobalt center. The electronic factors essential for the bonding between the zeolite-exchanged Co2+ cation and the NO ligand are examined by DFT (density functional theory) calculations for small cluster models of a zeolitic cobalt site, corroborated by CASSCF/CASPT2 (complete active space multireference SCF/perturbative treatment of dynamic correlation) and CCSD(T) (coupled clusters with explicit double and noniterative triple excitations) wave function methods. The use of the DFT cluster approach as the main working machinery has already been justified by comparing the calculated NO stretching frequencies (from vibrational analysis) with those measured by IR spectroscopy. Here, it is further validated by applying high-level correlated quantum chemical methods. Owing to the modest size of the basic model used here, it is practical to apply correlated wave function methodologies (CASSCF and CCSD(T)), serving to calibrate DFT methods as well as to recover the missing rigorous information on multiconfiguration character of the system. In addition, we employ the SR-NOCV (spin resolved natural orbitals for chemical valence) analysis20–22 to investigate the global flow of electron density along the bond between the NO ligand and the cobalt center in terms of independent electron and spin transfer channels. In our former work14 we showed that electron transfer channels between co-ligated ammonia molecules (closed-shell fragment) and the Co–NO core rationalized the significant red-shift of νNO through additional population of the πNO* orbitals by electron density transfer from donor ligands, mediated by the Co(II) center. Here, we attempt to discuss electron transfer channels between the Co(II) center and the NO ligand (both inherently open-shell fragments) in terms of the Chatt–Duncason model, based on two major components of the dative bond:23 backdonation to π*-antibonding orbitals on NO and donation to metal d orbitals. We have already tested a similar approach to explain spin and charge flow through the bond between the Fe(II) and NO fragments in two {Fe–NO}7 complexes: FeIIP(NH3)NO (P – porphin ligand) and [FeII(H2O)5(NO)]2+.22 However, these electron transfer channels to/from a redox active ligand were found to be heavily perturbed by a weak covalent coupling along the Fe–NO bond which obfuscated the interpretation of electron density transfers in {FeNO}7. In this view, we critically assess the limitations of the NOCV analysis applied to the complexes involving non-innocent ligands. Our study is in line with the interpretation of ligand redox-non-innocence for the {CoNO}9 and {NiNO}10 complexes by Tomson et al. in ref. 12 (based on the analysis of broken-symmetry UDFT solution in terms of unrestricted corresponding orbitals, UCOs24).
2. Methodology and models
2.1 DFT calculations and cluster models
DFT calculations were done for cluster models of the studied systems (to obtain the structures for stable electronic states and to obtain other properties) using the BP86 potential and the def2-TZVP basis set provided by the Turbomole 5.9 package,25 following the methodology used in ref. 14. Good performance of the BP86 exchange–correlation functional for structural properties and vibrational frequencies of transition metal complexes is well known;26 nonhybrid functionals like BP86 were also shown to reasonably reproduce CASSCF spin densities of {Fe–NO}7 and {Co–NO}8 systems.14,22 Relative spin-state energies were additionally calculated using other functionals (B3LYP, TPSSh, PBE0, and PBE) and compared with correlated wavefunction methods (see below). All calculations for open-shell species were spin-unrestricted (UDFT). Frequencies were obtained from the harmonic approximation while force constants of the N–O bond (kN–O) were computed numerically at the DFT:BP86/def2-TZVP level based on energies of the equilibrium structure and two distorted structures, where the terminal O atom was moved by ±0.005 Å out of the equilibrium geometry along the direction of the N–O bond.Working cluster models are constructed on the basis of the simplest fragment of the zeolite framework, i.e. a single aluminum tetrahedron [Al(OH)4]− (labeled T1) binding the Co2+ cation via two oxygens, thus they are positively charged. For nitrosyl complexes of Cu(I/II) sites in zeolites this simple approximation sufficed for the interpretation of IR characteristics of the NO bond in Cu(I/II)–NO adducts.27–30 However, Co(II) centers show preferentially fourfold coordination to basic oxygens (as also pointed by periodic modeling31–34) thus rough extension of this working model by including two additional water ligands to cobalt was proposed and found useful in our previous work.14 In addition, the charge of the Co2+ cation is not fully compensated by a single Al tetrahedron. This doubt may be partly dispelled in view of recently revived discussion on the catalytic properties of bivalent cations in high-silica zeolites, localized at the isolated aluminum–oxygen tetrahedron and truly compensated by the electrostatic interaction with distantly placed aluminum atoms in the framework.35,36 Formation of such sites has been proposed to explain the unusual catalytic properties of the high-silica zeolites modified by divalent transition metal ions,37 in particular towards H2 and CH4.38 Furthermore, we have tested the extended T12 model composed of two six-member hexagonal rings, each containing six T atoms (Si or Al) and representing the realistic fragment of a zeolite framework (vide infra).
In the case of models for ammonia-modified sites, initial displacement of the two water ligands (mimicking less tightly coordinated O-donors in the zeolite framework) by two ammonia molecules is presumed. This may be rationalized by the energetics of the corresponding complexes14 as well as by experimental reaction enthalpies and DFT:X3LYP formation energies available for complexes of the form [Co(H2O)6−n(NH3)n]2+, which point to a stabilizing effect of each water-to-ammonia exchange: the average exchange enthalpy of 2.8 kcal mol−1 per single substitution was found by experiment while DFT calculations yielded an increase in complex stability by 6 ± 1 kcal mol−1 per additional NH3 ligand.39,40 On the other hand, the group of Wichterlova directly addressed the state and coordination of Co2+ ions in zeolites to lattice oxygens after binding additional ligands:41,42 the authors postulated from the analysis of the shift of skeletal vibrations that upon adsorption of ‘strong ligands’ like NH3 the bonding of the cation became gradually loosened, until a complete detachment of Co2+ from the framework oxygen atoms occurs. Therefore, we felt entitled to assume that already after binding two ammonia ligands the number of coordinating oxygens is reduced to 2 and binding of the next ammonia molecules gradually weakens the Co–O bonds (these trends are reasonably reproduced by the present small cluster results, see Table 1), until the full release of the five-ammonia adduct. We must stress here again that the modest size of the models is indispensable to enable correlated wave function calculations to corroborate UDFT computations for non-innocent, redox-active systems.13,22 It also facilitates the analysis of the wave function and the emerging electron transfer channels in chemical terms.
| (a)T | (a*)S | (a*)T | (b)S | (b)T | (c)S | (c)T | |
|---|---|---|---|---|---|---|---|
| Angle (deg) | |||||||
| Co–N–O | 180.0 | 180 | 150 | 122 | 148 | 122.5 (119.0) | 150.5 |
| Bond length (Å) | |||||||
| Co–NO | 1.69 | 1.63 | 1.71 | 1.79 | 1.70 | 1.84 (1.87) | 1.72 |
| N–O | 1.14 | 1.16 | 1.16 | 1.19 | 1.16 | 1.17 (1.15) | 1.15 |
| Co–NH3 | — | 1.98; 1.98 | 2.10; 2.10 | 2.00; 2.00; | 2.19; 2.19; | 2.03eq(1.98) | 2.24eq |
| 2.00 | 2.15 | 2.40ax(2.22) | 2.18ax | ||||
| Co–OT1 | 1.97; 1.97 | 2.01; 2.01 | 1.98; 2.02 | 2.20; 1.94 | 2.12; 2.03 | — | — |
| Co–OH2O | 2.19; 2.19 | — | — | — | — | — | — |
In consequence, we designate the following small-cluster models for a detailed analysis of electron density and wave function properties (a): [(T1)Co(H2O)2NO]+, (a*): [(T1)Co(NH3)2NO]+ and (b): [(T1)Co(NH3)3NO]+ (with T1 = [Al(OH)4]−), completed by the complex (c): [Co(NH3)5NO]2+. Here (a) denotes models with two additional ligands, (b) those with three ligands, and (c) those with five additional ligands to the cobalt–NO center. Equilibrium structures of the models (in two low-lying spin states) obtained from DFT are shown in Fig. 1.
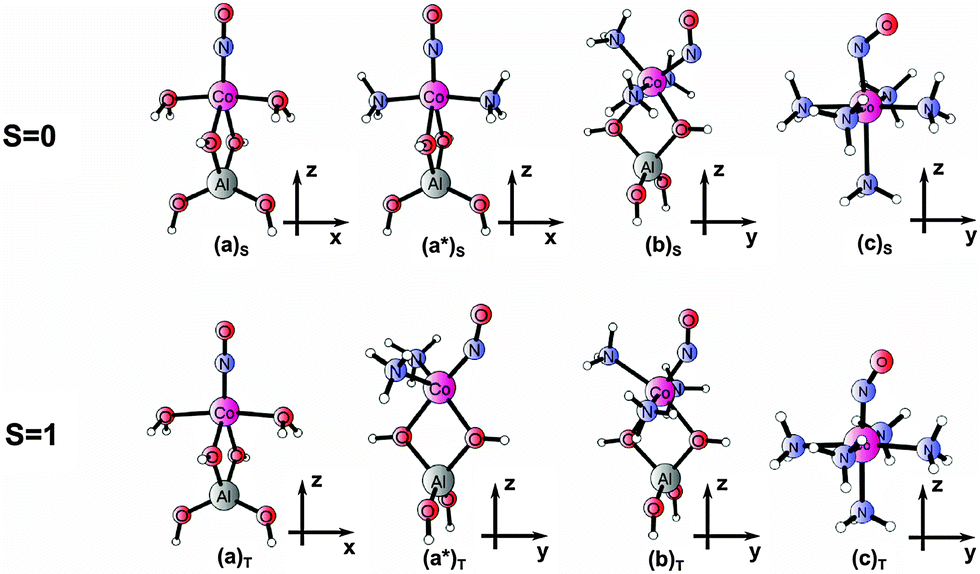 | ||
| Fig. 1 Optimized geometries for models (a), (a*), (b), and (c) in singlet and triplet states (shorthand notation for models following ref. 14). | ||
The extended T12 cluster model is additionally investigated to verify model (a) for the parent cobalt site. It is composed of two double six-membered rings (D6R), with next T atoms replaced by hydrogens to saturate peripheral bonds and represents the fragment of chabazite framework. The initial (T12)2Al structure (neutral) has been taken from preliminary periodic DFT:PBE minimization for Co-exchanged chabazite with Si/Al = 10, containing two Al substitutions (details of periodic minimization are described and the relevant fragment of the periodic structure is shown in Fig. S1 in the ESI†).43 To test the validity of small, +1 charged cluster models (and in accord with recent suggestions35–38) the cluster model with one of the aluminum atoms re-substituted by silicon and bearing +1 charge (labeled as (T12)1Al) is also analyzed. The comparison between the T12 models with two or one aluminum atom in one hexagonal ring may also give some clues on the dependence of the site properties on the Al distribution and location of the charge. The DFT optimized structures of T12 models (for the triplet ground state, postulated also in the literature10,11,32–34) are shown in Fig. S2 in the ESI.†
Following our previous study,14 model (a) serves to mimic the parent Co(II) site in zeolite while models (b) and (c) are set to reproduce the properties of experimentally suggested forms under intermediate (b) or complete (c) ammonia-saturation conditions. Model (a*) has been included in the set as an interjacent species in the course of saturating the zeolite catalyst with ammonia, assumed as a transient step in modeling nitrosyl adducts for ammonia pre-treated cobalt sites in zeolites. Labeling of the models and optimized geometries follows that from ref. 14, except for (a*) which was not considered previously.
The subsequent full optimization of the electronic and geometric parameters of all small models resulted in two energetically close-lying (singlet and triplet) states, labeled by subscripts (![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) )S and ()T, respectively. Both (a) models have a linear arrangement of the Co–N–O motif for two spin states and are symmetrized to the C2v point group. The model (a)T is taken hereafter as the reference system since the triplet state has been argued as the ground state for the parent cobalt site with bound NO10,11,31–34 (in addition, the two spin states of (a) differ significantly either in geometry or in vibrational properties). The structures of T1-based models (a) cannot be fully analogous to the relevant part of (T12)1Al (compare Fig. 1 and Fig. S2, ESI†) due to either symmetry constraints (indispensable to avoid spurious hydrogen bonding in small clusters) or fixed positions of hydrogen atoms, leading to C1 symmetry for large clusters. Nevertheless, the comparison of geometry parameters for (a)T and (T12)1Al models (cf. Table S1 in ESI†) indicates that the small model fairly reproduces well bond distances of extended models. Although the Co–N–O bent is not closely reproduced, this may be ascribed to the lack of symmetry in the bigger model rather than to actual changes in the character of the Co–N–O unit. We believe that it should not significantly influence other properties since charge and spin distributions as well as NO stretching frequencies for the two models are very much alike (Table S1, ESI†). The model (T12)2Al may serve as a crude probe of the difference between Co(II) sites in high silica versus low silica zeolites. The distances between Co and framework oxygens in extended models (more realistically mimicking part of the chabazite structure, Table S1 in ESI†) indicate that the cobalt binds similarly to two Al tetrahedra in (T12)2Al while for a single Al substitution one Co–OSi bond is substantially elongated (by 0.57 Å, compared to that in (T12)2Al) and the two bonds to the oxygens of an Al tetrahedron become shorter. Let us note, however, that when going from two to one aluminum atom in the vicinity of cobalt, neither the Co–N–O angle nor the Co–N and N–O bonds differ significantly. Mulliken populations only slightly depend on the position of two aluminum atoms (either in one hexagonal ring or well-spaced by the zeolite framework), while the spin density and total charge distributions on NO and Co are in line with those for the small cluster (a). Thus it may be assumed that the description of cobalt coordination by two water ligands and two oxygens of AlO− tetrahedra mimics partial neutralization of the extraframework Co2+ cation fairly well in comparison with T12 clusters. Small as well as enlarged models of the parent cobalt site give a blue-shift of the NO stretching frequencies.
)S and ()T, respectively. Both (a) models have a linear arrangement of the Co–N–O motif for two spin states and are symmetrized to the C2v point group. The model (a)T is taken hereafter as the reference system since the triplet state has been argued as the ground state for the parent cobalt site with bound NO10,11,31–34 (in addition, the two spin states of (a) differ significantly either in geometry or in vibrational properties). The structures of T1-based models (a) cannot be fully analogous to the relevant part of (T12)1Al (compare Fig. 1 and Fig. S2, ESI†) due to either symmetry constraints (indispensable to avoid spurious hydrogen bonding in small clusters) or fixed positions of hydrogen atoms, leading to C1 symmetry for large clusters. Nevertheless, the comparison of geometry parameters for (a)T and (T12)1Al models (cf. Table S1 in ESI†) indicates that the small model fairly reproduces well bond distances of extended models. Although the Co–N–O bent is not closely reproduced, this may be ascribed to the lack of symmetry in the bigger model rather than to actual changes in the character of the Co–N–O unit. We believe that it should not significantly influence other properties since charge and spin distributions as well as NO stretching frequencies for the two models are very much alike (Table S1, ESI†). The model (T12)2Al may serve as a crude probe of the difference between Co(II) sites in high silica versus low silica zeolites. The distances between Co and framework oxygens in extended models (more realistically mimicking part of the chabazite structure, Table S1 in ESI†) indicate that the cobalt binds similarly to two Al tetrahedra in (T12)2Al while for a single Al substitution one Co–OSi bond is substantially elongated (by 0.57 Å, compared to that in (T12)2Al) and the two bonds to the oxygens of an Al tetrahedron become shorter. Let us note, however, that when going from two to one aluminum atom in the vicinity of cobalt, neither the Co–N–O angle nor the Co–N and N–O bonds differ significantly. Mulliken populations only slightly depend on the position of two aluminum atoms (either in one hexagonal ring or well-spaced by the zeolite framework), while the spin density and total charge distributions on NO and Co are in line with those for the small cluster (a). Thus it may be assumed that the description of cobalt coordination by two water ligands and two oxygens of AlO− tetrahedra mimics partial neutralization of the extraframework Co2+ cation fairly well in comparison with T12 clusters. Small as well as enlarged models of the parent cobalt site give a blue-shift of the NO stretching frequencies.
Ammonia ligands are introduced in other models to construct NH3 co-ligated, zeolite-bound adducts (a*) and (b); the pentaamminecobalt(II)–NO complex (c) represents the model for a fully ammonia-saturated adduct, interacting only non-covalently with the zeolite framework.14,15 The displacement of two water molecules by two NH3 ligands in (a*) does not change the fivefold coordination (distorted trigonal bipyramid) of the Co center. When going from (a) to (a*), linear geometry of the Co–N–O motif is preserved only in the low-spin state whereas in the high-spin state the Co–N–O unit is bent and the symmetry is lowered to Cs.
Models (b) and (c) comprise a six-coordinated Co center and correspond to distorted octahedral geometry (with the bent NO ligand in the axial position). They differ by the nature of ligands coordinated to the cobalt center (apart from axial NO): in model (b) there are three ammonia ligands (each donating a lone pair) and two framework oxygens; in complex (c) all five ligands correspond to NH3. For the complexes with three ammonia ligands (singlet (b)S as well as triplet (b)T adducts), the bent Co–NO motif and the location of ammonia ligands result in the Cs symmetry. All Co–N bonds (both for NH3 and NO) are longer by 0.08–0.13 Å for the triplet state (b)T than those for the singlet state (see Table 1), which is a typical behavior (caused by occupation of an antibonding metal–ligand orbital in the triplet state). Let us also recall that the adsorption of consecutive ammonia ligands noticeably loosens the bonding of cobalt to the [Al(OH)4]− cluster; at variance, the Co–NH3 bonds do not change significantly upon binding consecutive ligands. The pentaammine complex has the bent Co–N–O motif (and thus the Cs symmetry) for both spin states; for the singlet (c)S structure, the axial Co–NH3 bond is significantly longer, which nicely mimics the crystal structure,44 while for the triplet (c)T it is somewhat shorter than the equatorial bonds. It is worth pointing that geometries of small models with two or three ammonia ligands (in the singlet spin state) correspond well to the relevant fragments of larger structures obtained from independent periodic DFT calculations (shown in Fig. S3 in ESI†).
Table 1 shows the structural parameters for models of the triplet (a)T (native Co–NO site) and ammonia-modified adducts (a*), (b) and (c) in singlet or triplet spin states (other properties will be consecutively discussed in next sections). Only in the case of (c) the computed structural parameters may be compared to the crystal structure of the [Co(NH3)5NO]2+ unit in nitrosylpentaamminecobalt(II) dichloride.44 Here, the experimental geometry is much closer to the one calculated for the singlet than that for the triplet state, which is a strong argument in favor of the singlet ground state for (c). Therefore, only model (c)S will be taken under further scrutiny concerning the complete ammonia saturation.
2.2 Correlated wave function methods
The active space was constructed in accord with the standard rules for transition metal systems.48 All molecular orbitals arising from 3dCo orbitals, the πNO*, πNO and σNO (lone pair) orbitals, and selected double-shell orbitals (dCo′) were made active. The choice of active space for each model is described in detail in the ESI† (Section II and Table S4, ESI†). Contour plots of the active orbitals can be found in Fig. S4–S10, ESI.†
Valence-bond (VB)-like expansion of the CASSCF wave function in terms of resonance structures was performed analogously as in ref. 13, i.e., by expressing the multiconfigurational CAS wave function in terms of (partly) localized active orbitals. By doing this transformation it is possible to read the wave function in terms of resonance structures with a definite number of electrons assigned to the πNO and πNO* orbitals (NO fragment), and the dCo orbitals (Co fragment). Natural or canonical orbitals, obtained by standard from CASSCF calculations (i.e., prior to the localization), are not suitable for such an interpretation because they are considerably delocalized to both fragments (due to great covalency of the metal–nitrosyl bond). Compared with our previous work in ref. 13, there are several improvements in the present methodology, with the aim of recovering appropriate shapes of the localized active orbitals, in particular to better separate the dCo′ (double-shell) and πNO* contributions. The details are described in the ESI† (Section II).
2.3 SR-NOCV analysis
Our previous studies on the interaction of NO with transition metal sites showed that the interpretation of charge flow channels (resulting from NOCV analysis) required spin resolution due to non-innocence of the open-shell NO ligand.14,22,29,30 The SR-NOCV method decomposes the differential density (arising from the bond formation between specified non-interacting fragments, constituting the complex) into one-particle contributions (named, in brief, NOCVs), separately for the α and β spins. The NOCV orbitals are intrinsically paired,54 which enables the extraction of independent electron transfer channels, to help in understanding the diversity of charge transfers through the TM–NO bond in transition metal complexes.To extract electron transfer channels, single point DFT:BP86/def2-TZVP calculations were performed for each complex and for a corresponding promolecule built of two non-interacting fragments: the NO molecule (fragment 1) and the remainder of the complex (fragment 2), promoted from their equilibrium geometry and the electronic ground state to the geometry and the electronic state in the final compound. Special care must be taken to appropriately select the electron configurations on the open-shell fragments to make them consistent with the electronic configuration of the final complex (to avoid non-physical effects like spurious orbital rotations). An analysis of spin density and natural spin orbitals (NSOs) for open-shell adducts helps to select the appropriate fragments' orbital occupancies: this procedure qualitatively corresponds to the ‘valence bond reading’ of broken-symmetry DFT results. Our former experience14,22 revealed that a number of electron pairs, (partly) separated in space but coupled to the singlet (with α electron on one fragment and β electron on the other one), show up which pointed to the importance of static correlation. Therefore CASSCF calculations were invoked, serving not only to assist the choice of a promolecule for SR-NOCV analysis, but also to independently estimate the shares of ionic versus radical structures in the nitrosyl adducts by means of the valence-bond analysis of CASSCF wavefunction13 (see above) and thus to support DFT in performing a charge transfer analysis.
3. Results and discussion
3.1 Electronic structure and spin-state energetics of {Co–NO}8 complexes
As already pointed out, assigning an unambiguous electron configuration to the complex (and, for the sake of NOCV analysis, also to its fragments) is not always a straightforward task. For the studied {Co–NO}8 complexes, already establishing the ground spin state constitutes a non-trivial issue. Table 2 lists relative adiabatic energies of the singlet (S = 0) and triplet (S = 1) spin states for models (a), (a*), (b) and (c), computed with various DFT methods and with two wave function theory methods: CASPT2 and CCSD(T). Unfortunately, no experimental data on spin-state energetics are available for these {CoNO}8 species, except for complex (c), for which the known crystal structure of pentaamminenitrosylcobalt(II) dichloride44 points to the singlet ground state (cf. Section 2.1).| Method | Relative energy (kcal mol−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| (a)S | (a)T | (a*)S | (a*)T | (b)S | (b)T | (c)S | (c)T | |
| a PBE0 alone points to the other triplet (with slightly different configuration) as the ground state for model (a). | ||||||||
| BP86 | 1.0 | 0 | 0 | 10.0 | 0 | 5.2 | 0 | 2.4 |
| PBE | 1.1 | 0 | 0 | 10.3 | 0 | 5.0 | 0 | 2.4 |
| B3LYP | 11.6 | 0 | 4.2 | 0 | 3.3 | 0 | 0.9 | 0 |
| PBE0 | 14.0 | 0.5a | 8.6 | 0 | 7.0 | 0 | 4.7 | 0 |
| TPSSh | 7.6 | 0 | 0 | 4.2 | 0 | 1.9 | 1.0 | 0 |
| CASPT2 | 25.9 | 0 | 3.8 | 0 | 8.4 | 0 | 9.8 | 0 |
| CCSD(T) | 3.6 | 0 | 0 | 5.4 | 0 | 10.5 | 0 | 13.4 |
Already a first glance at Table 2 reveals that the DFT relative energies are highly variable with the choice of the exchange–correlation functional. This is consistent with typical trends observed for transition metal complexes.55 Hybrid functionals (here: PBE0, B3LYP, and TPSSh) point to a greater stability of the triplet with respect to the singlet state than non-hybrid functionals (here: BP86 and PBE). Interestingly, with the exception of model (a), even the ordering of the spin states may be reversed by the changing functional (cf.Table 2). The high-level CASPT2 and CCSD(T) calculations were performed with the hope of clarifying these doubts, but unfortunately even these correlated methods lead to a contradictory prediction of the ground state for models (a*), (b), and (c).
Both CCSD(T) and CASPT2 calculations were used recently to obtain accurate spin-state energetics of transition metal complexes (see, for instance, ref. 13, 51, 56, 57, and 58). It is difficult to judge a priori which of these two high-level methods is more accurate for transition metal complexes in general. We note, however, that here CCSD(T) is able to correctly reproduce the experimentally suggested low-spin state for model (c), which is not the case for the present CASPT2 calculations.59 Moreover, CCSD(T) is capable of predicting the experimental ground state for hexaammine complexes of both Co(II) (high-spin) and Co(III) (low-spin);60 see the results in Table S4, ESI.† Interestingly, CCSD(T) correctly recovers the experimental ground state for model (c) even though this species, like the other {CoNO}8 complexes studied here, features a noticeable multi-reference character. The latter is reflected in the elevated values of, so-called, multi-reference diagnostics61,62 (T1, D1; see Table S5, ESI†) and presumably is related to the left–right correlation in the Co–NO bond. There are many other cases reported in the literature where CCSD(T) provides reliable relative energies despite the pronounced multi-reference character.62 For instance, the CCSD(T) estimate of the Co–NO bond energy in CoP(NO) (P – porphin) is only 2 kcal mol−1 above the experiment, despite the very high value (0.43) of the D1 diagnostics.63 Moreover, when relative energies of spin-states are in focus, one should expect a partial cancellation of the left–right correlation effects in the Co–NO bonding for the two spin states whose energies are compared. Hence, the differential correlation effect between the two spin states may be still reasonably accounted for by single-reference CCSD(T) treatment, despite the moderate multi-reference character of both states. In contrast, the previous experience with CASPT2 calculations of spin-state energetics13,56 indicates that this method may have a tendency to overstabilize the high-spin state (at least when used with the standard choice of the active space). A comparison of the CASPT2 results with the experimental data for model (c) seems to confirm this tendency for the present case too.59
As already mentioned above, due to very limited amount of the relevant experimental data and contradictory results of the two highest-level methods, one should be extremely careful in reaching conclusions about the ground state of the studied models. This being said, the present CCSD(T) calculations (believed by us to provide reliable results here – see previous paragraph) point to the triplet ground state for model (a), and to the singlet one for models (a*), (b), and (c). Designation of the triplet GS for (a) is consistent with all DFT calculations, where even non-hybrid functionals BP86 and PBE (known for their tendency to favor the low-spin states) point to the high-spin triplet ground state. The triplet GS for (a) is also supported by periodic DFT calculations for cobalt sites in zeolites.31,34 In addition, neither geometric features nor vibrational properties differ significantly between the two spins for the parent model, thus further scrutiny concerns mostly the triplet state, (a)T.
In contrast, the ground state assignment is less certain for models (a*) and (b). Moreover, the two spin states for these models have disparate properties: they significantly differ with respect to the equilibrium structure (in particular the Co–N–O geometry, cf.Fig. 1) and we shall see below that their ability to red-shift the NO stretching frequency with respect to a free NO molecule (the signature of ligand activation upon adsorption) is dramatically spin-dependent. In the case of the Co(II) center co-ligated by three ammonia molecules (represented here by model (b)), the appearance of both singlet and triplet adducts has been already postulated in our former work (ref. 14) upon re-interpretation of the registered IR spectra. The complex with two co-ligands, (a*) serves merely to mechanistically mimic the progress of sample saturation by ammonia where the high-spin to low-spin transformation might occur at an arbitrary point of the assumed scenario. Finally, under real conditions (at finite temperature and variable coordination environment) the equilibrium between the triplet and singlet intermediate adducts is hardly predictable. Therefore both spin states are scrutinized below for models (a*) and (b).
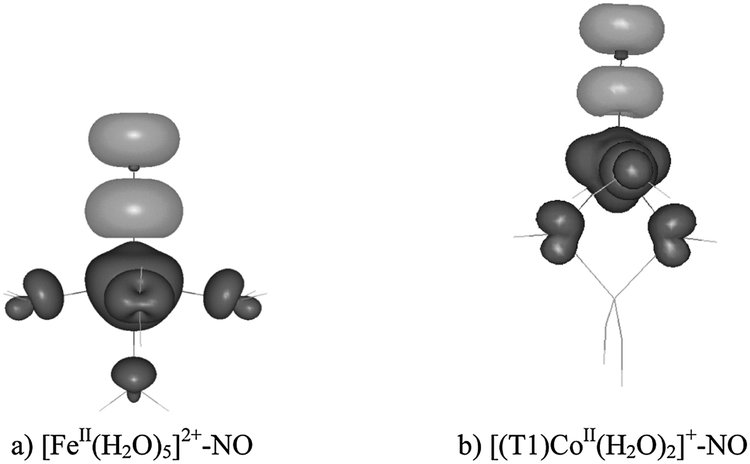 | ||
| Fig. 2 BP86 spin densities for the complexes with FeII–NO (a) and CoII–NO (b) core; black – positive spin density, grey – negative spin density (contour value 0.003 a.u.). | ||
For the iron adduct, we have previously assigned (based on natural spin orbitals for the complex) the quartet promolecule as the NO0 (S = 1/2) fragment antiferromagnetically coupled to the [Fe2+(H2O)5] (S = 2) fragment.22 By analogy, here we propose the promolecule for the aluminanitrosylcobalt(II) adduct to be composed of the NO0 (S = 1/2) fragment, antiferromagnetically coupled to the high-spin [(T1)Co2+] (S = 3/2) one.
An in-depth analysis of the origin of DFT spin density polarization in [(T1)CoII(H2O)2]+–NO is given here as the case study, to illustrate the procedure followed in other cases. An inspection of natural spin orbitals and their occupancies (Fig. 3) reveals that two NSOs with unitary positive integer occupations show a clear 3d character and correspond to two unpaired (α-spin) electrons localized on the cobalt. In addition, there are two pairs of coupled NSOs (with eigenvalues of ±0.30 or ±0.26) which may be ascribed to two pairs of weakly spin-coupled electrons: one (β-spin) mostly localized on NO and another one (α-spin) on Co. These electron pairs are effectively delocalized in the xz and yz planes, and may be interpreted as a signature of two weak, partly decoupled covalent π-bonds emerging between the fragments. A σ-bonding through the NO lone pair should not play a major role here, despite the linear Co–N–O geometry, because the respective antibonding orbital (i.e., one of the dCo orbitals pointing towards the NO ligand) is occupied.
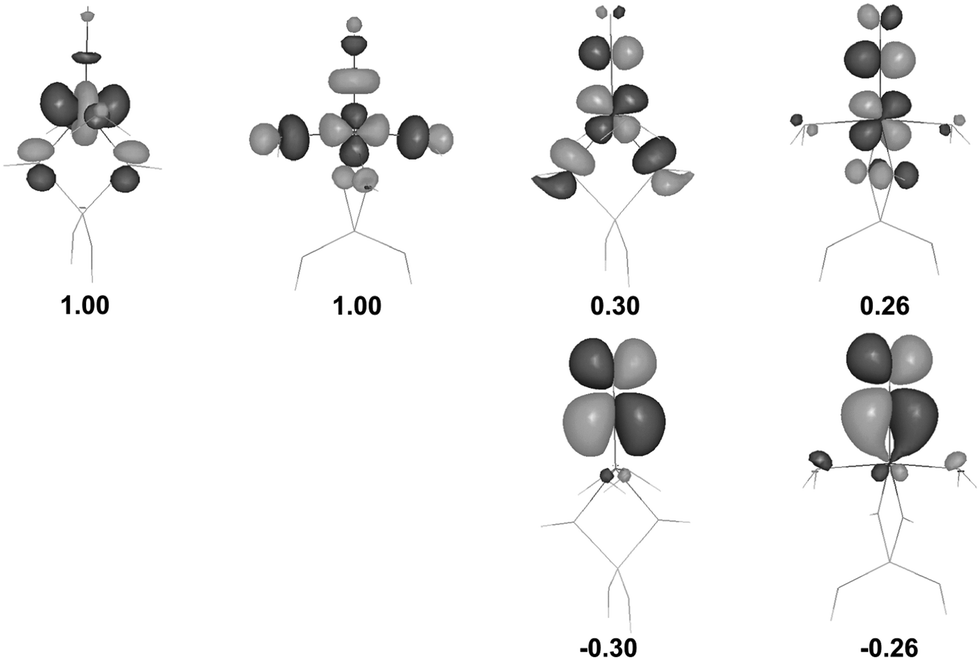 | ||
| Fig. 3 UDFT natural spin orbitals (NSOs) for the complex (a)T; two pairs with ±λ eigenvalues correspond to weakly coupled α and β electrons (contour value ±0.04 a.u.). | ||
CASSCF results of model (a)T illustrate a notably multiconfiguration character of this system, where the leading configuration covers roughly 67% of the wave function (the full diagram of CASSCF orbitals is shown in Fig. S4, ESI†). The leading configuration contains one doubly occupied Co dxy orbital, two occupied, nearly equivalent π-orbitals (weakly-bonding with respect to the Co–NO bond, composed of (dxz,dyz) and (πx*,πy*)), and singly occupied dz2–y2 and dx2 orbitals. Moreover, VB-like representation of the total CASSCF wavefunction (i.e., its decomposition into VB configurations constructed from the localized active orbitals, see Section 2.2.1 and Section II in ESI†) yielded three dominant contributions (each of them denotes a spin-symmetrized combination of the Slater determinants):
| Φ1 = |dxy2dxz2dz2−x2↑dy2↑dyz↑πy*↓πx*0|(41%) |
| Φ2 = |dxy2dyz2dz2−x2↑dy2↑dxz↑πx*↓πy*0|(20%) |
| Φ3 = |dxy2dxz2dz2−x2↑dy2↑dyz2πx*0πy*0|(12%) |
The first two configurations describe antiferromagnetically coupled electron pairs (delocalized in the xz and yz planes, respectively) which may be assigned to the CoII–NO0 resonance structure, and the third one corresponds to the CoI–NO+ resonance structure. For a quantitative analysis of the participating resonance structures for this and other models, refer to Section 3.2.1. As the expansion of the CASSCF wave function in terms of the fragment-localized orbitals contains at least two contributions of comparable weights (Φ1 and Φ2), the assignment of electrons and spins to the orbitals of open-shell fragments in the promolecule (necessary to perform SR-NOCV analysis) is rather arbitrary.
The present DFT calculations for the (a*)S (singlet) model result in the geometry very much alike that of the parent (a) system. An inspection of CASSCF natural orbitals (Fig. S5 in ESI†) confirms that upon NH3 ligation the total spin is dumped by pairing of the two cobalt-centered electrons, leaving the remainder of electron configuration nearly unchanged. The leading configuration (covering 77% of the CASSCF wave function) has doubly occupied dxy and dy2 orbitals, two doubly occupied, weakly-bonding π orbitals, and an empty dz2−x2 orbital. Therefore, the character of the Co–NO bond in this complex is similar to that in the native [(T1)Co(H2O)2NO]+ adduct: two weak π-bonds are formed by two partially decoupled electron pairs (with three electrons of dπ origin and one from πNO*); the only difference is somewhat stronger donor character of the Co(II) center due to direct donation from the ammonia lone pairs.
Unexpectedly, the triplet state of the same adduct, (a*)T, shows markedly different properties from the singlet. The triplet has a slightly bent Co–N–O unit oriented in the yz plane, and the local z axis of the complex (fixed by the Co–N vector) no longer coincides with the original z axis. To simplify the notation, appropriate combinations of cobalt d orbitals (labeled with respect to the local z axis) will be further considered along with π||* and π*⊥ orbitals on NO. Accordingly, the inspection of the electronic structure reveals a significant change in the bonding pattern: UDFT natural spin orbitals as well as CASSCF orbitals point to the formation of one covalent bond by the coupling of d||↑ and π*||NO↓ electrons, accompanied by a donor contribution involving the d⊥ electron pair and the empty π⊥NO orbital (UDFT natural spin orbitals are shown in Fig. S11 (ESI†), CASSCF orbitals may be found in Fig. S7 in ESI†). However, the dominant configuration covers only 68% of the CASSCF wave function while decomposition of the latter into VB-like structures (in terms of fragment-localized orbitals) yields a few configurations of comparable weights. In regard to the SR-NOCV analysis we thus anticipate an analogous problem as for model (a)T: the independent charge flow channels may be dimmed by spurious orbital rotations due to the uncertainty in selecting the unique occupations of the fragments' orbitals in a respective promolecule.
The singlet adduct (b)S ([(T1)Co(NH3)3]+–NO, S = 0) has a significantly bent Co–N–O unit oriented in the yz plane and the original Co d orbitals are again rotated in the local coordinate system. The electron configuration of fragments (based on relevant CASSCF orbitals of dCo and π*NO provenience, see Fig. S9 in ESI†) points to a covalent σ-bond formed by the coupling of electrons in a (d||↑ π*||↓) pair and a strong donor π-bond, due to the donation of an electron pair from cobalt d⊥ to the empty π⊥* on NO.
For the singlet (c)S model corresponding to the Co(II) complex with the axial NO and five NH3 ligands (showing nearly octahedral coordination apart from the bent NO unit), the conceivable bonding scheme is very much alike that of the singlet adduct, (b)S. CASSCF molecular orbitals clearly point to a strong σ-bond formed by covalent coupling of an electron pair (d||↑, π*||↓), accompanied by a typical donor π-bond, formed by the donation of electron pair from d⊥2 to π*⊥0 (cf. Fig. S10 in ESI†).
3.2 Electron density redistribution through the Co(II)–NO bond
| Model: ligands to Co(II), spin state | Contribution to CAS wave functiona | ΔνN–Ob (cm−1) | ΔkN–Ob (mdyn Å−1) | |||
|---|---|---|---|---|---|---|
| NO0 (%) | NO+ (%) | NO− (%) | Other | |||
| a From VB-like expansion of the CASSCF wave function in terms of localized active orbitals; see Section 2.3 for details. b With respect to free N–O: νN–O = 1884 cm−1, kN–O = 15.62 mdyn Å−1 (DFT:BP86). | ||||||
| (a)T (H2O)2, T | 73.3 | 18.2 | 8.2 | NO2− 0.2% | +74 | +0.45 |
| NO2+ 0.2% | ||||||
| (a*)S (NH3)2, S | 68.1 | 16.0 | 15.4 | NO2− 0.3% | +6 | −0.99 |
| NO2+ 0.2% | ||||||
| (a*)T (NH3)2, T | 73.4 | 12.4 | 13.8 | NO2− 0.3% | −58 | −1.16 |
| NO2+ 0.1% | ||||||
| (b)S (NH3)3, S | 68.0 | 6.1 | 25.1 | NO2− 0.7% | −226 | −3.42 |
| (b)T (NH3)3, T | 72.2 | 10.9 | 16.4 | NO2− 0.4% | −80 | −1.49 |
| NO2+ 0.1% | ||||||
| (c)S (NH3)5, S | 75.4 | 7.5 | 16.6 | NO2− 0.4% | −166 | −2.47 |
It is clearly visible that the relative weights of the major resonance structures (CoII–NO0, CoIII–NO− or CoI–NO+) align very well with the vN–O shift and the change of the force constant, ΔkN–O. (Intuitively, an increasing share of the NO−/NO+ structure should weaken/strengthen the N–O bond, due to increasing/decreasing population of antibonding πNO*, which is indeed observed.) This definite interdependence nicely illustrates the role played by the redistribution of electron density between the Co(II) site and the NO ligand for the strengthening/weakening of the NO bond.
In addition, our results may also serve to upgrade the understanding of the character of the Co–NO bonding. In the preceding sections we have extensively discussed the electronic structures of studied complexes; here we partially recall the reasoning to illustrate chemically relevant issues. In the native, (a)T ([(T1)Co(H2O)2]+–NO) complex, there is a sharp predominance of the CoII–NO0 and CoI–NO+ resonance structures (with the latter raised up to 18% and the share of the CoIII–NO− structure of only 8%), which results in a significant strengthening of the N–O bond (i.e., the increase of the NO force constant by 0.45 mdyn Å−1 compared to free NO and the computed blue-shift of νNO). For all other complexes the share of the CoIII–NO− resonance structure gradually increases and peaks (at 25%) for the one with three ammonia ligands in the singlet state (showing an outstanding weakening of the N–O bond). Interestingly enough, NO stretching frequencies calculated for this complex resulted in the novel, unforeseen interpretation of the IR spectra taken for nitric oxide adsorbed onto ammonia pre-saturated zeolites:14 surprisingly huge red-shift of νN–O was ascribed by us to the opening of an effective electron transfer channel between lone pairs of ammonia and the NO antibonding π*-orbital, yet more efficient for the complex with three NH3 than that for five ammonia ligands bound to the cobalt center. The present results impart to the interpretation of the NO bond weakening as being due to the occupancy of the antibonding π*-orbital on NO by an electron pair (cf. 25% of NO− structure), only slightly opposed by the NO → Co donation (cf. 6% of the CoI–NO+ resonance structure).
The complex (a*) with two ammonia ligands is discussed here to clarify the influence of water-to-ammonia exchange on the bonding scheme. For (a)T (no ammonia ligands) the electronic configuration and linear geometry imply no σ-bond and two weak π-bonds with partially decoupled α and β spins, suggesting only scant backdonation (see Section 3.1.1). The exchange of water by ammonia ligands accompanied by the pairing of cobalt-centered electrons in the (a*)S adduct results in a comparable share of the NO+ structure in the two adducts. Noticeable lowering of the NO force constant for the (a*)S adduct (compared to (a)T) may be ascribed to an increased population of the πNO* orbitals (cf. 15% of the CoIII–NO− structure) due to strong donor properties and the electron density-pushing effect of NH3 ligands. At variance, the triplet (a*)T adduct (lying close in energy) shows already a bent structure, some red-shift of the NO frequency and a further lowering of the force constant, mainly due to the diminished NO → Co donation.
The triplet adduct with three ammonia ligands, (b)T, reveals similar properties as the triplet (a*)T one, but as well, the share of the CoI–NO+ structure is further decreased as the π* backdonation becomes strengthened by reinforced donor properties of the cobalt center due to the increased number of donor ligands. Therefore, the decrease of the N–O force constant for (b)T is larger by 33% than that for (a*)T. As already pointed out in preceding sections, a strikingly remarkable weakening of the N–O bond is observed upon spin change (i.e. for the singlet (b)S adduct). Here, we put stress on the electronic origin of this behavior. The share of the CoI–NO+ resonance structure is reduced in (b)S by 44% and that of the CoIII–NO− resonance structure is increased by 53%, which results in more than twice larger reduction of the force constant with respect to free NO, compared with (b)T. This effect might be ascribed to the formation of two strong bonds: covalent σ-bond and donor π-bond. Let us also recall the suggestion from our former work14 that in the case of the Co(II) center hosting three ammonia ligands, the high-efficiency direct electron transfer between co-ligated ammonia and NO is enabled only in the singlet state of (b)S, whereas it is inactive for the triplet adduct (b)T.
The pentaamminenitrosylcobalt(II) complex, (c)S, is very similar to (b)S with respect to the character of the occupied CASSCF molecular orbitals (compare Fig. S9 and S10 in ESI†) as well as to the bending of the Co–N–O unit. However, it is intriguing that the backdonation in (c)S is less efficient than in (b)S, despite a larger number of donor NH3 ligands.14 This trend is evidenced not only by a less pronounced red-shift and a smaller decrease of the N–O force constant, but also by a smaller share of CoIII–NO− and a larger share of CoI–NO+ resonance structures for (c)S compared with (b)S. This substantially lower efficiency of the backdonation in the case of the pentaammine complex might be ascribed to the still stronger donor character of the oxygens mimicking the zeolite framework (two O atoms of the O–Al–O moiety) than that of nitrogen atoms from ammonia ligands. However, this should be taken as a tentative suggestion rather than a strong conclusion since our simplified model obviously does not allow us to describe the basicity of the zeolite framework accurately. Studies on realistic models of the zeolite framework to address this suggestion more comprehensibly are currently underway in our group.
However, one should be aware of limitations inherent to this approach stemming from the arbitrariness in selecting fragments' occupations in the promolecule (already pointed above). The previous paper14 was focused on the direct influence of ammonia co-ligation on donor properties of the adduct. In that, an alternative partitioning (with ammonia ligands taken as the first fragment) was applied to extract the information on direct charge transfer from ammonia co-ligands to NO thus the analysis of the electronic structure of the promolecule was much simplified due to the null spin on the first fragment. Herein, both fragments are open-shell species and apart from the spin state of the entire complex, care must be taken with respect to not only populating one of the two π* spin orbitals on the NO fragment by one electron, but also the spin state of the metal fragment and populating the singled-out spin orbitals of the dCo provenience. Analyses of both the DFT natural spin orbitals and the CASSCF wave function (expressed in fragment-localized orbitals) greatly help in determining physically reasonable fragment occupations in the promolecule for SR-NOCV analysis (cf. Section 2.3). Nevertheless, the selection of a single determinant to represent electron configuration on open-shell fragments frequently must be arbitrary.
Due to these intrinsic methodological reasons, one may expect that SR-NOCV analysis is capable to yield complete information on the charge density and specified independent electron density transfer channels only when comparing systems with alike multiconfigurational character and same type of spin-couplings in the promolecule. Therefore, a quantitative discussion of NOCV results should be limited to such cases when similar errors may be expected (i.e., for the (b)S and (c)S complexes with comparable geometries and electronic structure, but with various activation abilities).
The relevant SR-NOCV channels (presented in Fig. 4a and b) are plotted assuming red contours for the depletion of electron density and blue contours for the increased electron density, and may be interpreted as corresponding to the effective electron density flow from the red to blue region of space. Conventional labels depicting donation or backdonation (with σ- or π-indices describing local symmetry) are qualitatively assigned after a visual inspection of the contours. The corresponding eigenvalue moduli are given for each channel to quantify the redistributed electron density (i.e., channel efficiency in the total charge transfer between the fragments, triggered by the bond formation). The last row in Table 4 lists the measures for backdonation (from dCo to πNO*, estimated from NOCV eigenvalues), presumably ascribed to the ligation of donor ammonia ligands to cobalt; later they will be discussed in conjunction with calculated and experimental relative shifts of NO frequency (with respect to the shift registered for NO bound to the native Co(II) site), and electron and spin densities on NO.
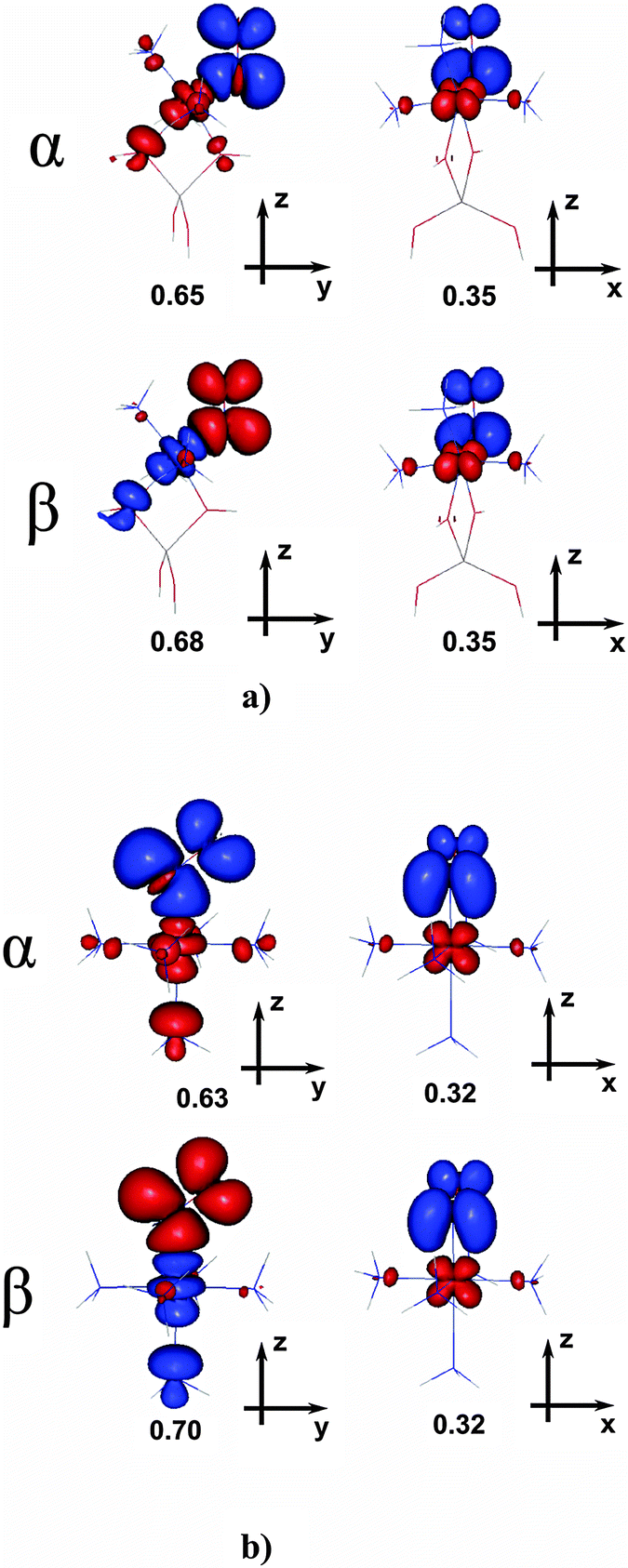 | ||
| Fig. 4 Dominant electron transfer channels for singlet complexes: (a) [CoAl(OH)4(NH3)3]+–NO (bS) and (b) [Co(NH3)5]2+–NO (cS); red – depletion, blue – accumulation of electron density (contour value ±0.001 a.u.). | ||
| Property | [T1Co(H2O)2]+–NO | [T1Co(NH3)3]+–NO | [Co(NH3)5]2+–NO | |
|---|---|---|---|---|
| (a)T | (b)S | (b)T | (c)S | |
| a Reference values ΔvNO are +74 cm−1 (calc.) and −7 cm−1 (exp.). b From Mulliken populations. c In spin minority. d Only qualitative estimates may be provided (see text). | ||||
| ΔΔvcalcNO (cm−1) | 0a | −300 | −154 | −240 |
| ΔΔvNOexpav (cm−1) | 0a | −247 | −102 | −200 |
| Q NO | +0.22 | −0.03 | +0.10 | +0.07 |
|
ρ
SNOb |
0.43c | 0 | 0.13c | 0 |
| ΔρNOCVNO | <0d | 0.67 | >0d | 0.57 |
For both relevant singlet adducts ((b)S and (c)S) a major activation of the NO bond has been evidenced by the huge red-shift of the NO stretching frequency (IR experiment and DFT calculations) as well as by the decrease of the NO force constant and a significant elongation of the N–O bond (DFT, cf.Tables 1 and 3). In view of the bonding scheme discussed in Sections 3.1.3 and 3.2.1, the analogous electron configurations of the respective promolecules (composed of fragments “prepared” to bind in the singlet (b)S and (c)S complexes) may be assumed. Thus we believe that the differences in density transfer channels discussed below are truly due to the variation of donor properties of Co(II) centers in the two adducts. Indeed, the overall shapes of electron density transfer channels (shown in Fig. 4a and b, respectively) are very similar for both adducts. The first pair of channels (in the α- and β-spin manifolds, left panels in Fig. 4) represent an electron density flow towards the bonding region due to the formation of a σ-bond. Two unpaired electrons (one with α-spin, originating from Co; another with β-spin, originating from NO) couple to form a covalent σ-bond. However, in the case of the pentamminenitrosyl adduct (c)S the outflow of β-spin density from NO along this channel (−0.70e) is less exactly balanced by the inflow of α-spin density (+0.63e) than in the case of the (b)S adduct (i.e., −0.68e outflow and +0.65e inflow, respectively). The second pair of channels (right panels in Fig. 4) clearly represent a cumulative (α + β) π*-backdonation from the cobalt d orbital of appropriate symmetry to the NO π⊥* orbital. Again, the cumulative electron density transfer into the empty NO π*-orbital is slightly larger for (b)S than for (c)S. In consequence, the net increase of electron population on NO antibonding orbitals is predicted (from the sum of eigenvalues, taking the sign depending on the flow direction) to be roughly 0.67e for (b)S, while for (c)S it falls to only 0.57e. Inspection of Table 4 indicates a satisfactory agreement between the frequency red-shift and the reinforced backdonation. This result is also in line with the high-efficiency of direct electron transfer channels between co-ligated ammonia molecules and NO, found in our former work for (b)S and (c)S complexes.14
At variance, let us briefly analyze and compare electron and spin transfers for triplet states of (a) and (b) adducts (Fig. S13 in the ESI†), of which the first one deactivates (shortening of the NO bond and blue-shift of the NO stretching frequency), while the second one shows a minute activation of the NO ligand compared to (bS) (red shift by −80 cm−1vs. −226 cm−1). In the case of (a)T, however, the electron configuration of the promolecule has been chosen based on the leading VB configuration covering merely 41% of total wave functions (see Section 3.1.1). Since πx*- and πy*-orbitals are nearly equivalent in (a)T, but cannot be equivalent in the promolecule, one may expect that spurious features would appear in the SR-NOCV electron density flow channels, serving to recover cylindrical symmetry of the spin density as well as to appropriately symmetrize the antiferromagnetic coupling of electrons.22 A similar bonding situation is predicted for (b)T, where two π-bonds are formed: a mixture of a dative one (donation of an electron pair from the occupied d orbital to the empty π* orbital) and a weak covalent bond (coupling of d↑ and π*↓ electrons). However, the assignment of promolecular configuration is arbitrary (acceptable configurations have comparable shares of only 20–30% in the total CASSCF wave function). Hence, it is clear that SR-NOCV analysis cannot offer complete information on the electron density transfer in any of these cases.
Here, even if the overall shapes of charge flow channels for models (a)T and (b)T (Fig. S13 in the ESI†) are seemingly equivalent, some corresponding channels either only partly or not at all participate in the inter-fragment electron redistribution (serving merely to cover for incomplete representation of the molecular electronic structure by the arbitrarily selected promolecular configuration, or for intra-fragment polarization). Moreover, while the second pair of channels in Fig. S13 (ESI†) (panels a and b, respectively) could be roughly attributed to the formation of a dative π-bond for both complexes, the first pair represents an in-plane bond only for the (b)T adduct. For (a)T the first channel in the α-manifold has a large intra-fragment (NO) reorganization character and hence its eigenvalue (0.31e) can hardly be treated as a measure of backdonation. Thus one can only speculate that the net unbalanced donation of the β-density from NO to the Co center arises for the (a)T complex. For the (b)T adduct, the backdonation of the Co d electrons towards NO along the second channel seems to be partly cancelled by the donation along the first channel; yet, some residual net backdonation might be anticipated.
Overall, a quantitative interpretation of the NOCV results turns out ambiguous for cases as complicated as the present (a)T and (b)T complexes. Fortunately, all trends in NO activation/deactivation are more robustly rationalized by a pronounced admixture of the CoI–NO+ resonance structure in the case of (a)T and a growing admixture of the CoIII–NO− resonance structure in the case of (b)T, as was revealed above from the VB-like expansion of the CASSCF wave function.
4. Summary and conclusions
We should recall here that interpreting the electronic structure of complexes with {Mn+–NO}n+1 core in terms of either pure ionic ({M(n+1)+–NO−} and {M(n−1)+–NO+}) or pure radical ({Mn+–NO0}) structures seems highly oversimplified. For NO complexes with Fe(II) (ref. 22) as well as with Co(II) centers (this work), the electronic structure should be described as an appropriate mixture (quantum-mechanical superposition) of resonance structures. Moreover, we have shown that populating or depopulating the π*-antibonding orbitals on NO (another factor frequently used to rationalize the activation of the N–O bond) may be accomplished in such systems through several independent electron density transfer channels of various provenience and direction, active either cumulatively or selectively for spin majority and spin minority manifolds. Hence, a simple correlation with donor properties of the center, estimated from the SR-NOCV analysis, does not always hold good here as it did in the cases with simpler electronic structures (ref. 29 and 30). It must be also reminded that the results regarding all experimentally relevant adducts are based on small models, not capable to fully mimic any actual zeolite. Therefore the calculation results are related to experimental IR data averaged over several zeolite types (Co-MOR, Co-FER, Co-ZSM5) while our conclusions concern general donor properties of cobalt sites in a zeolite framework.Tables 3 and 4 evidence clearly that, in accord with the expectations, pre-adsorption of electron-donor ammonia ligands modifies the donor properties of the cobalt site and shifts the stretching frequency registered for the co-adsorbed NO to lower values. However, the extent of NO activation significantly depends not only on the number of donor ligands, but also on the spin state of the complex, an effect which could not be intuitively anticipated. In the case of three ammonia co-ligands, the triplet state (of distinct provenience but with the same total spin as that of the parent, unmodified adduct) reveals only a minute activation compared with the singlet state. The latter shows a paramount red-shift of the NO stretching frequency, even bigger than that of the pentaammine complex in its ground singlet state. This effect was experimentally confirmed after reinterpretation of relevant IR spectra (suggested by our DFT modeling). The weakening of the N–O bond (well reproduced also by the calculated force constant, Table 3) is in line with its elongation, but neither conventional correlation with the negative charge accumulated on the NO molecule (after binding of additional electron-donor ammonia ligands) holds strictly nor the bending angle is a sufficient descriptor to fully explain the range of the red-shift of the NO stretch (cf. also Table 1). Mulliken spin populations (a measure of the radical character of the NO ligand) do not explain the electronic origin of the bond weakening either (note that they are null by definition for all singlet species with a closed-shell structure). At variance, the character and direction of the electron density redistribution (within the Co–N–O unit) nicely rationalize the observed modification of the NO bond. According to Table 4, already the estimated efficiency of relevant electron density transfer channels explains why the singlet adduct [CoAl(OH)4(NH3)3]+–NO shows more pronounced weakening of the NO bond than the [Co(NH3)5]2+–NO complex. Nevertheless, the protocol based on the CASSCF wave function (represented by the valence-bond type resolution of the multiconfigurational wavefunction) turns out more robust in a systematic analysis of electron density redistribution along the Co–NO bond in all presently studied complexes.
Finally, this work reinforces our former suggestion that the cobalt center (e.g. the Co2+ cation exchanged in a zeolite framework) may be a tunable electron and spin transmitter between the adsorption site and the NO adsorbate. Detailed results of the adduct with three ammonia ligands (some of them shown already in ref. 14, but considerably extended in this work) indicate that the high-efficiency electron transfers between the Co(II) center and the NO ligand are enabled only in the singlet state, whereas they are disabled for the triplet state of the adduct. This illustrates clearly how the cobalt center (depending on its electronic status, in particular the spin state) may either block or enhance the favorable (spin) electron density transfer towards the NO ligand.
Acknowledgements
We kindly acknowledge financing provided by Grants No. 2013/11/N/ST4/00984, 2015/17/B/ST5/00023 from the National Science Centre, Poland; Marian Smoluchowski Krakow Research Consortium (Leading National Research Centre, KNOW) and the departmental funds of Jagiellonian University (project no. K/DSC/002853). Part of this work was supported by the COST Action CM1305 (ECOSTBio), including STSM-CM1305-151114-051084 and STSM-CM1305-150215-051085. We thank Tomas Bucko and Lubomir Benco for their contribution to calculations in VASP. Periodic calculations were performed in the Computing Centre of the Slovak Academy of Sciences using the supercomputing infrastructure (project ITMS 26230120002 and 26210120002, Slovak infrastructure for high-performance computing) supported by the Research & Development Operational Programme funded by the ERDF. The use of facilities at Center of Computational Material Science of Vienna University is also kindly acknowledged. Other calculations were supported in part by PL-Grid Infrastructure.References
- D. Pietrogiacomi, M. C. Campa and V. Indovina, Catal. Today, 2010, 155, 192–198 CrossRef CAS.
- M. C. Campa, S. De Rossi, G. Ferraris and V. Indovina, Appl. Catal., B, 1996, 8, 315–331 CrossRef CAS.
- Y. Traa, B. Burger and J. Weitkamp, Microporous Mesoporous Mater., 1999, 30, 3–41 CrossRef CAS.
- M. Iwamoto, Stud. Surf. Sci. Catal., 2000, 130A, 23 CrossRef CAS.
- A. E. Palomares, C. Franch and A. Corma, Catal. Today, 2011, 176, 239–241 CrossRef CAS.
- K. Góra-Marek, B. Gil and J. Datka, Appl. Catal., A, 2009, 353, 117–122 CrossRef.
- K. Góra-Marek, H. Mrowiec and S. Walas, J. Mol. Struct., 2009, 923, 67–71 CrossRef.
- A. J. Timmons and M. D. Symes, Chem. Soc. Rev., 2015, 44, 6708–6722 RSC.
- P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1017 CrossRef CAS PubMed.
- I. Georgieva, L. Benco, D. Tunega, N. Trendafilova, J. Hafner and H. Lischka, J. Chem. Phys., 2009, 131, 054101 CrossRef PubMed.
- S. Sklenak, P. C. Andrikopoulos, S. R. Whittleton, H. Jirglova, P. Sazama, L. Benco, T. Bucko, J. Hafner and Z. Sobalik, J. Phys. Chem. C, 2013, 117, 3958–3968 CAS.
- N. C. Tomson, M. R. Crimmin, T. Petrenko, L. E. Rosebrugh, S. Sproules, W. C. Boyd, R. G. Bergman, S. DeBeer, F. D. Toste and K. Wieghardt, J. Am. Chem. Soc., 2011, 133, 18785–18801 CrossRef CAS PubMed.
- M. Radoń, E. Brocławik and K. Pierloot, J. Phys. Chem. B, 2010, 114, 1518–1528 CrossRef PubMed.
- K. Góra-Marek, A. Stępniewski, M. Radon and E. Broclawik, Phys. Chem. Chem. Phys., 2014, 16, 24089–24098 RSC.
- K. Góra-Marek and J. Datka, Catal. Today, 2011, 169, 181–185 CrossRef.
- F. Bin, C. Song, G. Lv, J. Song, X. F. Cao, H. T. Pang and K. P. Wang, J. Phys. Chem. C, 2012, 116, 26262–26274 CAS.
- E. E. Mercer, W. A. McAllister and J. R. Durig, Inorg. Chem., 1967, 6, 1816–1821 CrossRef CAS.
- C. Wäckerlin, D. Chylarecka, A. Kleibert, K. Müller, C. Iacovita, F. Nolting, T. A. Jung and N. Ballav, Nat. Commun., 2010, 1, 61 Search PubMed.
- (a) C. Wäckerlin, K. Tarafder, D. Siewert, J. Girovsky, T. Hahlen, C. Iacovita, A. Kleibert, F. Nolting, T. A. Jung, P. M. Oppeneer and N. Ballav, Chem. Sci., 2012, 3, 3154–3160 RSC; (b) C. Wäckerlin, K. Tarafder, J. Girovsky, J. Nowakowski, T. Hählen, A. Shchyrba, D. Siewert, A. Kleibert, F. Nolting, P. M. Oppeneer, T. A. Jung and N. Ballav, Angew. Chem., Int. Ed., 2013, 52, 4568–4571 CrossRef PubMed.
- M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347–355 CrossRef CAS PubMed.
- M. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- E. Broclawik, A. Stępniewski and M. Radoń, J. Inorg. Biochem., 2014, 136, 147–153 CrossRef CAS PubMed.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2942 RSC.
- B. Kirchner, F. Wennmohs, S. Ye and F. Neese, Curr. Opin. Chem. Biol., 2007, 11, 134 CrossRef CAS PubMed.
- R. Ahlrichs, H. Horn, A. Schaefer, O. Treutler, M. Haeser, M. Baer, S. Boecker, P. Deglmann and F. Furche, Turbomole v5.9, Quantum Chemistry Group, Universitaet Karlsruhe, Germany, 2006 Search PubMed.
- F. Neese, JBIC, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS PubMed.
- A. Pulido and P. Nachtigall, Phys. Chem. Chem. Phys., 2009, 11, 1447–1458 RSC.
- M. Radoń and E. Broclawik, J. Phys. Chem. A, 2011, 115, 11761–11774 CrossRef PubMed.
- P. Kozyra, M. Radoń, J. Datka and E. Brocławik, Struct. Chem., 2012, 23, 1349–1356 CrossRef CAS.
- M. Radoń, P. Kozyra, A. Stępniewski, J. Datka and E. Broclawik, Can. J. Chem., 2013, 91, 538–543 CrossRef.
- S. A. McMillan, L. I. Broadbelt and R. Q. Snurr, J. Catal., 2003, 219, 117 CrossRef CAS.
- F. Göltl and J. Hafner, J. Chem. Phys., 2012, 136, 64501 CrossRef PubMed.
- F. Göltl and J. Hafner, J. Chem. Phys., 2012, 136, 64502 CrossRef PubMed.
- F. Göltl and J. Hafner, J. Chem. Phys., 2012, 136, 64503 CrossRef PubMed.
- V. Kazansky and A. Serykh, Microporous Mesoporous Mater., 2004, 70, 151–154 CrossRef CAS.
- L. Benco, T. Bucko, J. Hafner and H. Toulhoat, J. Phys. Chem. B, 2005, 109, 20361–20369 CrossRef CAS PubMed.
- L. Benco, J. Catal., 2013, 298, 122–129 CrossRef CAS.
- (a) V. B. Kazansky, Kinet. Catal., 2014, 55, 492–501 CrossRef CAS; (b) V. B. Kazansky, Kinet. Catal., 2014, 55, 737–747 CrossRef CAS.
- P. R. Varadwaj and H. M. Marques, Phys. Chem. Chem. Phys., 2010, 12, 2126–2138 RSC.
- P. R. Varadwaj, A. Varadwaj and B.-Y. Jin, Phys. Chem. Chem. Phys., 2015, 17, 805 RSC.
- Z. Sobalik, J. Dedecek, I. Ikonnikov and B. Wichterlova, Microporous Mesoporous Mater., 1998, 21, 525–532 CrossRef CAS.
- J. Dedecek, Z. Sobalik and B. Wichterlova, Catal. Rev.: Sci. Eng., 2012, 54, 135–223 CAS.
- Models extracted from preliminary results of periodic VASP simulations (temporary access provided to one of the authors (AS) during STSM stay at Vienna University and Bratislava Comenius University).
- C. S. Pratt, B. A. Coyle and J. A. Ibers, J. Chem. Soc. A, 1971, 2146 RSC.
- (a) G. Karlström, R. Lindh, P.-Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrády and L. Seijo, Comput. Mater. Sci., 2003, 28, 222–239 CrossRef; (b) F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P.-Å. Malmqvist, P. Neogrády, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS PubMed.
- M. Reiher and A. Wolf, J. Chem. Phys., 2004, 121, 10945–10956 CrossRef CAS PubMed.
- (a) B. O. Roos, K. Andersson, M. P. Fulscher, P.-A. Malmqvist, L. Serrano-Andres, K. Pierloot and M. Merchan, Multiconfigurational Perturbation Theory: Applications in Electronic Spectroscopy, in Advances in Chemical Physics: New Methods in Computational Quantum Mechanics, ed. I. Prigogine and S. A. Rice, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1996, vol. 93 DOI:10.1002/9780470141526.ch5; (b) K. Pierloot, Mol. Phys., 2003, 101, 2083–2094 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz and others MOLPRO, version 2012.1, a package of ab initio programs, see http://www.molpro.net.
- (a) C. Hättig, W. Klopper, A. Köhn and D. P. Tew, Chem. Rev., 2012, 112, 4–74 CrossRef PubMed; (b) L. Kong, F. A. Bischoff and E. F. Valeev, Chem. Rev., 2012, 112, 75–107 CrossRef CAS PubMed.
- M. Radoń, J. Chem. Theory Comput., 2014, 10, 2306–2321 CrossRef PubMed.
- (a) T. F. Hughes, J. N. Harvey and R. A. Friesner, Phys. Chem. Chem. Phys., 2012, 14, 7724–7738 RSC; (b) A. S. Petit, R. C. R. Pennifold and J. N. Harvey, Inorg. Chem., 2014, 53, 6473–6481 CrossRef CAS PubMed.
- (a) T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed; (b) G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- M. Radoń, Theor. Chem. Acc., 2008, 120, 337–339 CrossRef.
- (a) J. N. Harvey, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 203–226 RSC; (b) C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC; (c) M. Swart, Int. J. Quantum Chem., 2013, 113, 2–7 CrossRef CAS.
- (a) S. Vancoillie, H. Zhao, M. Radoń and K. Pierloot, J. Chem. Theory Comput., 2010, 6, 576–582 CrossRef CAS PubMed; (b) S. Vancoillie, H. Zhao, V. T. Tran, M. F. A. Hendrickx and K. Pierloot, J. Chem. Theory Comput., 2011, 7, 3961–3977 CrossRef CAS PubMed.
- (a) L. M. Lawson Daku, F. Aquilante, T. W. Robinson and A. Hauser, J. Chem. Theory Comput., 2012, 8, 4216–4231 CrossRef CAS PubMed; (b) A. Vargas, I. Krivokapic, A. Hauser and L. M. Lawson Daku, Phys. Chem. Chem. Phys., 2013, 15, 3752–3763 RSC.
- (a) M. Kepenekian, V. Robert, B. Le Guennic and C. de Graaf, J. Comput. Chem., 2009, 30, 2327–2333 CAS; (b) M. Pápai, G. Vankø, C. de Graaf and T. Rozgonyi, J. Chem. Theory Comput., 2013, 9, 509–519 CrossRef PubMed.
- The present CASPT2 calculations seem to overstabilize the triplet state, not being able to reproduce the experimental singlet ground state for (c). It is possible that enlargement of the active space, such as making active the complete double-shell and/or the outer-core orbitals, may improve the spin-state energetics. However, for the sake of performing VB-like analysis below, which is the highlight of this work, further enlargement of the active space was not attempted.
- C. K. Jørgensen, Absorption Spectra and Chemical Bonding in Complexes, Pergamon Press, Oxford, England, 1962 Search PubMed.
- W. Jiang, N. J. DeYonker and A. K. Wilson, J. Chem. Theory Comput., 2011, 8, 460–468 CrossRef PubMed.
- J. N. Harvey, JBIC, J. Biol. Inorg. Chem., 2011, 16, 831–839 CrossRef CAS PubMed.
- M. Radoń, Inorg. Chem., 2015, 54, 5634–5645 CrossRef PubMed.
- A. M. Schmiedekamp, M. D. Ryan and R. J. Deeth, Inorg. Chem., 2002, 22, 5733–5743 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of periodic and extended cluster models, CCSD(T) and CASSCF calculations, VB-like analysis of CASSCF wave function, and additional computational results. See DOI: 10.1039/c5cp07452e |
| This journal is © the Owner Societies 2016 |