
Truncated tetragonal bipyramidal anatase nanocrystals formed without use of capping agents from the supercritical drying of a TiO2 sol†
D. M.
Tobaldi
*a,
R. C.
Pullar
*a,
L.
Durães
b,
T.
Matias
b,
M. P.
Seabra
a and
J. A.
Labrincha
a
aDepartment of Materials and Ceramic Engineering/CICECO – Aveiro Institute of Materials, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: david.tobaldi@ua.pt; david@davidtobaldi.org; rpullar@ua.pt; Tel: +351 234 370 041
bCIEPQPF, Department of Chemical Engineering, Faculty of Sciences and Technology, University of Coimbra, Pólo II, Rua Sílvio Lima, 3030-790 Coimbra, Portugal
First published on 25th November 2015
Abstract
Titanium dioxide (TiO2) nanoparticles are extremely attractive materials for numerous applications, especially in the anatase form. We have made these shaped, <10 nm anatase nanoparticles (NPs) via the supercritical (SC) drying of a titania sol, made by a “green” aqueous sol–gel nanosynthesis route. The SC drying was carried out in alcohol at 255–260 °C, and no further heating or processing of the NPs was required. The true phase composition (crystalline and amorphous phases) and the microstructure of the NPs was thoroughly characterised by the advanced X-ray methods, such as Rietveld-reference intensity ratio (RIR) and the whole powder pattern modelling (WPPM) technique, and HR-TEM analysis. Furthermore, the NPs were also characterised by Raman, FT-IR and optical spectroscopy. These anatase NPs showed themselves to exhibit a truncated tetragonal bipyramidal shape, exposing the {101} (side) and {001} (top) faces. They had a euhedral crystal habit, with sharply defined and easily recognised faces, and were very homogeneous and monodisperse in both shape and size. The photocatalytic activity (PCA) of the samples was assessed in gas–solid phase by monitoring the degradation of nitrogen oxides (NOx), a major atmospheric pollutant. Results showed that the particular shape of these anatase NPs played a key role in their photocatalytic behaviour. In fact, these truncated tetragonal bipyramidal nanocrystals exhibited an enhanced photocatalytic activity, double that of spherical anatase NPs of a similar size reported previously by the authors. This was attributed to the exposure of mainly the {101} and, to a lesser extent, {001} crystal faces, which are more reactive under photocatalysis for redox reactions.
Introduction
The development of nanotechnologies is currently a key topic in Materials science, because nanomaterials possess unique properties, providing solutions to problems that cannot be dealt with using conventional technologies. Nanocrystalline titanium dioxide (TiO2) is of particular importance, and TiO2 nanoparticles (NPs) are extremely attractive materials because of their vast range of potential applications, from common products (sunscreens, cosmetics and paints) to advanced devices and a series of environmental and biomedical applications, e.g. photovoltaic cells, photocatalytic degradation of indoor and outdoor pollutants, water purification, antibacterial activity, biosensing and drug delivery.1 Interest in TiO2 as a semiconductor photocatalyst has grown steadily since the discovery in 1972 of the Honda–Fujishima effect,2 as shown by the large number of review articles published on this topic,3–12 making photocatalysis one of the best known applications of TiO2.The photocatalytic activity (PCA) of TiO2 based materials is due to the absorption on the photocatalyst surface of a photon, with energy equal-to or greater-than the photocatalyst's energy band gap (Eg).13–15 This absorption leads to charge separation: an electron (e−) in the valence band of the semiconductor is promoted to the conduction band, thus leaving a hole (h+) in the valence band. This photogenerated electon–hole pair (e−–h+) is able to act as a redox source, mineralising organic and/or inorganic matter adsorbed on the surface of the photocatalyst via the creation of free radical species.14,16,17 On the other hand, the main disadvantage of TiO2 as a photocatalyst is widely recognised to be the fast recombination rate of that photogenerated pair, though it is still greatly controversial where, when and how this phenomenon occurs.18 Amongst the large number of TiO2 polymorphs,19 anatase is widely recognised to be a better photocatalyst than rutile20 – although rutile, when nanostructured, is reckoned to be a visible-light active photocatalyst, as opposed to anatase, which absorbs only in the UV region.21 PCA is affected by several factors, such as TiO2 phase composition, NP size of the photocatalyst, level of crystallinity, and of great significance, surface properties.22–24
Regarding surface phenomenon,25 there has been a focus on designing TiO2 structures able to yield the maximum advantage through morphology.26 Spherical TiO2 NPs have usually been employed as photocatalysts, because of their high surface-to-volume ratio providing a huge number of surface active sites, and their ease of synthesis. On the other hand, this advantage is partially cancelled out by the disadvantage of the fast electron–hole recombination process, which is favoured by the small size of the NPs, and by their lack of geometric anisotropy.22,27 As these regular isotropic shapes have drawbacks, anisotropic TiO2 nanomaterials have been made with many different morphologies, i.e. nanorods,28–30 nanowires,31,32 nanobelts,33,34 nanosheets35,36 and nanotubes.37–39 The aim is to obtain better photocatalytic performance, by simultaneously providing a high surface-to-volume ratio and a lower recombination rate for the photogenerated pair.
Furthermore, as photocatalysis is a surface phenomenon, it is strictly associated with which crystal faces are exposed. Therefore, not only shape is a key factor, but also which of the anatase TiO2 faces are exposed: Ohno et al. recently suggested that the oxidation site is mainly on the {001} faces, whilst the reduction site is mainly on the {101} faces40 – thus proposing an anisotropic behaviour for the e− and h+ on the external anatase crystal structure. D'Arienzo et al., taking advantage of electronic spin resonance (ESR) upon UV excitation in anatase synthesised via a solvothermal synthesis method and making use of capping agents, showed that, in vacuum conditions, the {001} surfaces mainly provided oxidation sites for the photocatalytic reaction, whilst the {101} surfaces gave reductive sites. In an O2 atmosphere, they conversely showed that oxidant species were located mostly on {101} surfaces.41 Lu's group used hydrofluoric acid as a morphology controlling agent to engineer anatase showing a dominance of exposed {001} faces, which had superior PCA.42 On the other hand, Pan et al., also using HF as a controlling agent, stated that anatase {001} faces exhibited lower PCA than {101} faces.43 However, in both cases, it is not still clear whether that superior PCA is actually due to the presence of those highly exposed energy surfaces, or to the pivotal role of residual fluorine on the surface due to the preparation method,44,45 and hence it is still unclear which is the more photoreactive anatase face.43,46,47 Literature data about which of the exposed anatase faces provides better conditions for photocatalytic applications are, therefore, contradictory and confusing, as most involve the use, and probable residual incorporation, of fluoride ions.
In this work, we synthesised anatase through supercritical (SC) drying of titanium(IV) hydroxide sols, prepared following a “green chemistry” aqueous procedure previously reported, and without the use of surfactant or shape controller.21,48 Therefore, our anatase NPs definitely contain no fluoride ions, and changes in PCA can be attributed purely to physical/crystallographic/shape effects – SC drying represents a great advantage for engineering porous solids when compared to evaporative methods, since it avoids the agglomeration and shrinkage of the particles normally observed when capillary pressures are developed. Euhedral nanocrystalline anatase is usually obtained using hydrothermal/solvothermal conditions,49–51 and/or through shape controlling agents – commonly hydrofluoric acid (an environmentally unfriendly and extremely corrosive chemical)42–45 – or surfactants such as diethanolamine52,53 or oleic acid and oleylamine.41 However, the hydrothermal technique requires a complex autoclave with a well-controlled temperature gradient along the vessel height. Wang et al. recently synthesised TiO2 mesoporous nanosheets with dominant {001} faces under SC conditions, but still using fluorine ions as shape control agents, and obtaining large NPs with average lengths of 120–150 nm and 20 nm in thickness.54
The nanostructured anatase obtained in the present work (<10 nm) exhibited a euhedral truncated tetragonal bipyramidal crystal habit, exposing the {101} and {001} faces. Its micro-/nanostructure (shape of the crystalline domains, their size distribution, and also edge and screw dislocation densities) was thoroughly characterised via an advanced X-ray method (whole powder pattern modelling, WPPM), and high resolution transmission electron microscope (HR-TEM) analysis. Furthermore, as a functional property of the prepared anatase, photocatalysis (i.e. PCA), was assessed against the removal of nitrous oxides (NOx = NO + NO2), that are known to be amongst the major air pollutants produced by the combustion of fuels in air from automobiles and power stations, and strongly contribute to both outdoor (formation of acid rain, photochemical smog) and indoor (sick building syndrome) pollution.55,56
Experimental
Sample preparation
Aqueous titanium(IV) hydroxide sols were made via the controlled hydrolysis of titanium(IV) isopropoxide (Ti-i-pr, Ti(OCH(CH3)2)4) with distilled water diluted in isopropyl alcohol (IPA), following a low-cost environmental friendly “green nanosynthesis” protocol, as previously reported in detail by these authors.21,48 In brief, one part of Ti-i-pr (Aldrich, 97%) was added to four parts of isopropyl alcohol to make a 20 vol% Ti-i-pr solution. That Ti-i-pr solution was hydrolysed by the controlled addition of an excess of water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:Ti-i-pr), employing a 20 vol% solution of water in IPA. Concentrated HNO3 (Aldrich, 65%) was simultaneously employed as the acid necessary to peptise the sol: this was also added to the water–IPA solution, in a molar ratio of Ti4+:acid of 2.5:1. The water–IPA–acid solution was added dropwise to the Ti-i-pr solution at room temperature, whilst being mechanically stirred. The precipitated mixture was evaporated to a white jelly-like mass on a rotary evaporator, removing the IPA. Distilled water was added to restore the mixture to the original volume, and the gelatinous mass was then redispersed to form a titania sol, which can be dried to a gel consisting of <10 nm TiO2 NPs.
:1 water:Ti-i-pr), employing a 20 vol% solution of water in IPA. Concentrated HNO3 (Aldrich, 65%) was simultaneously employed as the acid necessary to peptise the sol: this was also added to the water–IPA solution, in a molar ratio of Ti4+:acid of 2.5:1. The water–IPA–acid solution was added dropwise to the Ti-i-pr solution at room temperature, whilst being mechanically stirred. The precipitated mixture was evaporated to a white jelly-like mass on a rotary evaporator, removing the IPA. Distilled water was added to restore the mixture to the original volume, and the gelatinous mass was then redispersed to form a titania sol, which can be dried to a gel consisting of <10 nm TiO2 NPs.
For SC drying, we ideally need an alcoholic solvent. To achieve this, the prepared aqueous Ti sol was dried to glassy gel on the rotary evaporator at the same temperature as that used in the original synthesis (60 °C), and then redispersed in alcohol, either IPA or ethanol (EtOH) – purity of the alcohols used was >99.5% – to give a sol which was much less stable than the aqueous sol, resulting in a very fine precipitation of titania NPs over time upon standing. This alcoholic dispersion of the titania sol nanoparticles was then dried by high-temperature SC drying, by placing it in an autoclave, with an equivalent amount of alcohol (IPA or EtOH) being added. The autoclave was then sealed and pressurised with nitrogen gas up to 7 bar. At this point, the system was heated to 80 °C h−1, leading to a gradual increase in pressure under isochoric conditions, which avoids the crossing of the liquid–vapour equilibrium line and, thus, avoiding the development of capillary pressure in the particles' interstices, and agglomeration. Once the SC region of the alcohol was achieved (T ≈ Tc + 20 °C; P ≈ Pc + 20 bar; Tc, Pc – critical temperature and pressure, respectively), the pressure in the autoclave was slowly decreased down to atmospheric pressure by release of the SC alcohol, under constant temperature. When IPA was used as the SC fluid, the temperature and pressure used were 255 °C and 70 bar, whereas in the case of EtOH, these were 260 °C and 80 bar. After, the sample was cooled to ambient pressure and the dried particles were removed. These samples are referred to as Ti-IPA and Ti-EtOH, respectively.
Sample characterisation
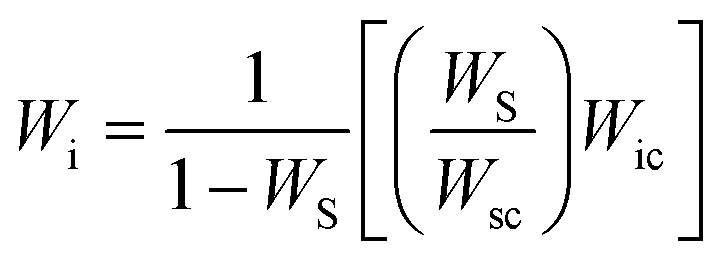 | (1) |
| Wa = 1 − ∑iWi | (2) |
XRPD data for the microstructural analysis were collected using the same instrument and the same set up as per the QPA, but, with the purpose of handling data with a higher signal-to-noise ratio, these were collected in the 20–125° 2θ range, with virtual step scan of 0.1050° 2θ and virtual time per step of 500 s. Microstructural features were analysed via the whole powder pattern modelling (WPPM) method,62 through the PM2K software.63 This novel yet state-of-the-art procedure is based on physical models describing the microstructure by refining model parameters via a non-linear least squares routine, so as to fit the experimental peaks, without any use of arbitrary analytical functions (i.e. Gaussian, Lorentzian, or Voigtian). As the diffraction peak profile is the result of a convolution of instrumental and sample-related physical effects, with this procedure we are able to truly extract microstructural information (crystalline domain shape and size distribution, dislocation density) from a diffraction pattern,64,65 as well as additional sources of line broadening, such as domain size and/or lattice strain,66 that are not properly considered by line profile analysis (LPA) methods, like the Scherrer formula, or the Williamson–Hall method.67,68
The instrumental contribution was obtained by modelling 14 hkl reflections from the NIST SRM 660b standard (LaB6), according to the Caglioti et al. relationship.69 Afterward, anatase SG I41/amd, was included in the WPPM modelling, and the following parameters were refined: background (modelled using a 4th-order of the shifted Chebyshev polynomial function), peak intensities, specimen displacement, and lattice parameters. Dislocations were supposed to be the principal source of anisotropy, thus being the major defect present in anatase; the presence of both edge and screw dislocations (with densities of ρe and ρs, respectively) have been supposed to be in the 〈10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 〉{101} slip system, with the Burgers vector being equal to (α20 + c20)1/2 – with a0 and c0 being the lattice parameters.70,71 Furthermore, crystalline domains were assumed to be octahedral in shape, and their edges distributed according to a lognormal size distribution – this was an adaptation of the actual shape of the anatase NPs, which was found to actually be a truncated tetragonal bipyramid, as proved by HR-TEM analysis (see below). As a matter of fact, the shape of naturally occurring anatase crystals is a (often truncated) tetragonal bipyramid, also according to the Wulff construction.72
〉{101} slip system, with the Burgers vector being equal to (α20 + c20)1/2 – with a0 and c0 being the lattice parameters.70,71 Furthermore, crystalline domains were assumed to be octahedral in shape, and their edges distributed according to a lognormal size distribution – this was an adaptation of the actual shape of the anatase NPs, which was found to actually be a truncated tetragonal bipyramid, as proved by HR-TEM analysis (see below). As a matter of fact, the shape of naturally occurring anatase crystals is a (often truncated) tetragonal bipyramid, also according to the Wulff construction.72
XRPD data were also used to model the (101) and (004) anatase reflections with a pseudo-Voigt (pV) analytical profile function, using ProFit software (v. 1.0c, Philips Electronics, NL). These parameters were refined: background parameters, peak position and full width at half maximum (FWHM), the maximum intensity and the shape parameter (η) for the pV function. Intensity of these two reflections was then extracted, and used to obtain the (I(004)/I(101)) ratio.
HR-TEM analysis and selected area electron diffraction (SAED) patterns were assessed using a JEOL 2200FS microscope with a field emission gun, operated at 200 kV, and equipped with a Gatan Ultrascan 4000 SCCD camera. Samples were prepared by dispersing the NPs in isopropanol, and evaporating the suspension drops on carbon-coated copper grids. Fast Fourier transform (FFT) patterns were analysed through the JEMS software package.73
The specific surface area (SSA) was evaluated by the Brunauer–Emmett–Teller (BET) method (Micromeritics Gemini 2380, US) using N2 as the adsorbate gas, on samples degassed at 120 °C. Pore volume (desorption cumulative pore volume, DCPV) and pore size distribution were estimated via the Barrett–Joyner–Halenda (BJH) method.
One is the differential reflectance method – applied mainly to well crystalline semiconductor materials. This method supposes that, plotting the first derivative of reflectance (dR/dλ) versus the wavelength (λ), the maximum value of such a plot corresponds to the optical Eg of the semiconductor material.74,75 The resulting curves were fitted adopting a Gaussian function (Origin ProLab, version 8.5.0), and the maximum values were found from the fitting itself.
The other method we used is the Tauc plot.76 This latter method assumes that the optical Eg of a semiconductor material can be calculated from this equation:
| (αhv) = A(hv − Eg)γ | (3) |
 | (4) |
Furthermore, for anatase NPs, the type of band-to-band transition (i.e. whether directly allowed or indirectly allowed) is still controversial – most authors assume it to be indirectly allowed, although it has been shown that a direct transition can appear when in a colloidal nanomaterial form.77,79 Thus, in this work, we considered both the direct and indirect Eg models.
Raman spectra were acquired in the 50–700 cm−1 wavenumber range, with 2 cm−1 resolution, on a RFS 100/S (Bruker, DE) equipped with a Nd:YAG laser (1064 nm) as the excitation source.
FT-IR (in attenuated total reflectance (ATR) mode) analyses aimed at detecting the occurrence of OH groups and/or water adsorbed on the photocatalyst surface. This was evaluated on a Bruker Tensor 27 (DE) spectrometer. The measurements were carried out over the wavenumber range of 4000–350 cm−1.
 | (5) |
Results and discussion
QPA and microstructural analyses: XRPD and HR-TEM
XRPD patterns of the prepared samples are shown in Fig. 1; a graphical output of a Rietveld refinement is reported in Fig. S2,† while QPA results employing the Rietveld-RIR method are described in Table 1. As is seen from Fig. 1, all the prepared samples crystallised in the TiO2 anatase polymorph. This is not surprising, as at the nanoscale, anatase, having the lowest surface free energy amongst TiO2 polymorphs, is the most thermodynamically stable phase.83 Furthermore, in Table 1 it can be seen that some amorphous material still remained in the samples after the SC drying of the sols, namely 9.3 and 5.8 w% for Ti-IPA and Ti-EtOH, respectively. The presence of amorphous phase was also confirmed by HR-TEM analysis (see below), and it might be a legacy of the un-crystallised titania present in the starting sols. | ||
| Fig. 1 XRD patterns of synthesised samples. The vertical black bars represent anatase reflections, from ICDD powder diffraction card # 21-1272. | ||
| Sample | No. of variables | Agreement factors | Phase composition (wt%) | |||
|---|---|---|---|---|---|---|
| R(F2) (%) | R wp (%) | χ 2 | Anatase | Amorphous | ||
| Ti-IPA | 14 | 3.25 | 4.70 | 2.17 | 90.7 ± 1.0 | 9.3 ± 1.0 |
| Ti-EtOH | 14 | 4.19 | 5.22 | 2.48 | 94.2 ± 1.0 | 5.8 ± 1.0 |
WPPM results are shown in Table 2 and Fig. 2. A graphical output of WPPM modelling is shown in Fig. S3.† As per the crystalline domain dimensions, from Table 2, it can be seen that the average dimension of anatase in Ti-EtOH is 9.7 nm. On the contrary, anatase in Ti-IPA has a slightly smaller size of 8.6 nm, most likely due to the lower temperature and pressure used in the SC drying. Also, both specimens exhibit a narrow size distribution (the mode of the lognormal size distribution being 7.0, and 8.2 nm, for anatase Ti-IPA, and Ti-EtOH, respectively), with a little dispersion around the tails, cf.Table 2 and Fig. 2. Regarding the dislocation density, the number of edge dislocations was greater than that of screw dislocations (within experimental error) in both samples studied. These values are comparable to those attained by Ghosh et al. for an anatase synthesised by flame combustion chemical vapour condensation and annealed at 200 °C.84 On the other hand, dislocation densities in these anatase NPs were shown to be higher (by approximately one order of magnitude) compared to those of anatase NPs synthesised by the authors from the same starting sol, but thermally treated in air at 450 and 600 °C.70 Literature data showed that the lowering in dislocation density is due to their polygonisation, as a consequence of crystal growth during the thermal treatment.84
| Agreement factors | Unit cell parameters (nm) | Edge of the octahedron (nm) | Dislocation density (×1016 m−2) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | R wp (%) | R exp (%) | χ 2 | a = b | c | Average | Mode | ρ e | ρ s |
| Ti-IPA | 5.65 | 2.09 | 2.71 | 0.3787(1) | 0.9499(1) | 8.6(3) | 7.0(2) | 2.3(9) | 1.0(6) |
| Ti-EtOH | 5.58 | 2.13 | 2.62 | 0.3787(1) | 0.9502(1) | 9.7(4) | 8.2(3) | 3.7(7) | 1.0(4) |
 | ||
| Fig. 2 Size distribution, as obtained from the WPPM modelling of synthesised samples. | ||
Descriptive HR-TEM micrographs of anatase Ti-IPA, taken as a representative sample, are depicted in Fig. 3a–e. It is seen that the large majority of the anatase NPs have a euhedral crystal habit (that is, with clearly defined edges and faces), and are nearly homogeneous in shape and in size, although some other crystals with less defined shapes can also be seen in the HR-TEM images. HR-TEM analysis confirmed both the QPA and WPPM results. As a matter of fact, a halo of amorphous phase around some anatase NPs is clearly visible in Fig. 3c and d. Moreover, the anatase NPs are actually exhibiting a narrow size distribution as suggested by WPPM, and the length of the edge of the octahedron they were approximated to is consistent with the WPPM results (see Fig. 3e, where the edge of the (101) anatase crystal plane is approximately 7.8 nm). Going into more detail, with the aim of showing the shape and the exposed crystal faces of anatase in Ti-IPA, higher magnification HR-TEM micrographs are depicted in Fig. 4a–d. In Fig. 4a and b are shown well crystallised anatase NPs, exhibiting the euhedral truncated tetragonal bipyramidal habit, thus exposing the {101} and {001} faces (cf.Fig. 4a). Another anatase NP is shown at even greater magnification in Fig. 4c, in which the anatase (101) and (002) crystallographic planes, having d-spacing of around 0.35 and 0.47 nm, respectively, are clearly seen (i.e. lattice fringes along the [010] direction). In the inset is depicted the corresponding fast Fourier transform (FFT) pattern, in which the angle between the (01) and (004) crystal planes of anatase is around 68.2°, thus perfectly matching the theoretical values for the angles between the {101} and {001} faces (i.e. 68.3°).43,51,85 This proves that synthesised anatase NPs mainly show exposed {101} and {001} faces. The higher amount (compared to a standard anatase) of exposed {001} faces is also well-confirmed by XRD analysis: the ratio between the intensity of the (004) and (101) anatase reflections (I(004)/I(101)) in the ICDD powder diffraction card #21-1272 is 0.20, whilst in Ti-IPA and Ti-EtOH is 0.24 and 0.23, respectively.
 | ||
| Fig. 3 HR-TEM micrographs of anatase Ti-IPA (a, b). In Fig. 3c and d is visible a halo of amorphous phase around anatase nanoparticles. In Fig. 3e is shown an anatase NP whose edge is 7.8 nm in length. | ||
 | ||
| Fig. 4 HR-TEM micrographs of anatase Ti-IPA. In Fig. 4a are highlighted the {001} and {101} facets of anatase; the truncated bipyramid crystal form is also reported, showing the (001) and (101) crystallographic planes. Fig. 4b shows euhedral anatase NPs. In Fig. 4c the d(101) and d(002) crystallographic planes are shown; in the inset is reported the resultant FFT pattern of that NP, zone axis [100]. Fig. 4d shows the view along the [001] direction, also reporting the d(101) and d(011) crystallographic planes; the inset reports the corresponding FFT pattern. | ||
Fig. 4d is taken along the [001] direction of another anatase NP: the (101) and (011) crystallographic planes, having d-spacing of ∼0.35 and ∼0.36 nm, are marked. In the inset, the FFT pattern shows the angle between these two crystallographic planes to be approximately 82.3°, once again in line with theoretical data.43,51,85 Furthermore, the percentage of exposed {101} and {001} faces was calculated and averaged over a hundred anatase NPs, according to the model defined by Jaroniec and colleagues:86,87
 | (6) |
In (6), θ is the theoretical value for the angle between the {101} and {001} faces of anatase (i.e. 68.3°), and A/B is the aspect ratio, that is the ratio between the side of the truncation face B, and the side of the bipyramid A, cf.Fig. 4a. Obtained results are in line with those expected for euhedral anatase having a truncated tetragonal bipyramidal crystal habit: A/B = 0.3, and %S{001}exp = 5%46,88 – those values are obviously only intended to be relative, as a TEM image only shows a projection of the real object.
Proposed formation mechanism
At the nanoscale, anatase is the stable TiO2 polymorph because of its lower surface free energy.83 Therefore, as stated previously, it is expected to find anatase as the TiO2 nanocrystalline polymorph. Also, we know that:• When the prepared aqueous Ti sol was dried to glassy gel on the rotary evaporator at 60 °C (prior to being redispersed in alcohol and then subjected to SC drying), it was composed of mostly amorphous/poorly crystalline anatase (cf. Fig. S4†).
• We made no use of shape-controlling agents, such as HF: thus, we did not modify the surface energy of the anatase crystallographic planes.
• A recent investigation by Iversen's group, via the pair distribution function (PDF) method applied to total X-ray scattering data, revealed that – in a TiO2 prepared via hydrothermal hydrolysis of Ti-i-pr – the amorphous solid precursor structure can be modelled by titanium hydroxide clusters consisting of TiO6/TiO5 units, with an arrangement related to that of bulk crystalline anatase.89
Hence, we can argue that here the solid precursor displayed the anatase structure and that, as a result of the high pressure and supersaturation, as well as the relatively high temperature, crystal growth was allowed within the supercritical fluid.90,91 This led to the nucleation and growth of anatase crystals, similar to that observed in hydrothermal conditions.49 Furthermore, the reported surface energy values of {101} and {001} faces are 0.44 J m−2 and 0.90 J m−2, respectively.25 Therefore, the crystal growth of anatase NPs proceeded by lowering the surface energy through minimising the area of the high-energy {001} faces, as reduction in surface energy is the primary driving force for particle growth. Thus, by analogy to what was proposed by Penn and Banfield, in the first stage of crystal growth we had the formation of distinctly faceted crystalline domains dominated by {101} surfaces; this was followed by a shrinkage (but not a disappearance) of the higher-energy {001} faces.49 As a result, euhedral anatase nanocrystals showing a truncated tetragonal bipyramidal habit were formed. These NPs also displayed little agglomeration, and no clear sign of oriented attachment, as is frequently seen, on the contrary, with hydrothermal conditions,49 because with SC drying the capillary pressure in inter-particles interstices is avoided. This is shown by their sorption isotherms (Fig. S5†), which are type IV, typical of a mesoporous material. Furthermore, from the t-plot (inset of Fig. S5†), we detected the presence of no micropores (all pores were <5 nm) – this result being close to that shown by D'Arienzo et al. on similarly shaped anatase NPs.41
Spectroscopic analyses
DRS data are reported in Fig. 5 and Table 3. All the spectra consist of one single absorption band, at around 380 nm. This band is due to the metal–ligand charge transfer (MLCT) in TiO2.92 The apparent optical Eg values obtained for both samples, using the dR/dλ method, are around 376 nm (3.30 eV) – values consistent with that expected for anatase (∼388 nm; ∼3.2 eV).20 Furthermore, the values obtained from the Tauc plot (Table 3, third and fourth columns) using the indirect Eg model were (3.36 ± 0.04) eV and (3.37 ± 0.06) eV for Ti-IPA and Ti-EtOH, respectively. This agrees well with the dR/dλ method, and other literature data.20,76,93 On the contrary, adopting the direct Eg model, we obtained a larger optical Eg of 3.60 eV (344 nm) for both the specimens, which contrasts with most of the available literature data. Therefore, the indirect model would seem more applicable. | ||
| Fig. 5 a) DRS spectra of synthesised anatase; in the inset is reported a plot of the first derivative of diffuse reflectance as a function of wavelength λ (dR/dλ) against the photon energy. The vertical dashed black line represents the optical Eg of anatase. b) Kubelka–Munk elaboration versus energy according to the direct Eg model (γ = 1/2). The dashed black line represents the x-axis intercept of the line that is tangent to the inflection point of the curve (i.e. apparent optical direct Eg calculated using the Tauc procedure). c) Kubelka–Munk elaboration versus energy according to the indirect Eg model (γ = 2). The dashed black line represents the x-axis intercept of the line that is tangent to the inflection point of the curve (i.e. apparent optical indirect Eg calculated using the Tauc procedure). | ||
| Sample | Optical Eg (eV) | Raman Eg mode (cm−1) | |||||
|---|---|---|---|---|---|---|---|
| Tauc plot | |||||||
| dR/dλ | Direct allowed, γ = 1/2 | Indirect allowed, γ = 2 | SSABET (m2 g−1) | Pore volume (DCPV, cm3 g−1) | FWHM | Position | |
| Ti-IPA | 3.30 ± 0.01 | 3.60 ± 0.05 | 3.36 ± 0.04 | 105.3 ± 2.1 | 0.49 | 13.8 ± 0.4 | 144.8 ± 0.1 |
| Ti-EtOH | 3.30 ± 0.01 | 3.60 ± 0.05 | 3.37 ± 0.06 | 108.6 ± 1.5 | 0.55 | 14.1 ± 0.4 | 145.1 ± 0.1 |
Raman spectra (Fig. S6† and Table 3, seventh and eighth columns) essentially confirmed the XRPD data: only the Raman modes of anatase appears. Furthermore, the Raman Eg mode of anatase, located at approximately 145 cm−1, being (qualitatively) broad in all the samples, is red-shifted compared to the anatase standard value (142.5 cm−1),94,95 thus confirming the reduced dimensions of the anatase crystalline domain size.96
FT-IR spectra are shown in Fig. S7.† The two samples display the same characteristics: the absorption bands in the 400–600 cm−1 range are attributed to Ti–O–Ti stretching modes,71 while the broad band centred at approximately 3250 cm−1 is due to hydroxyl groups. The peak at around 1620 cm−1 has been attributed to O–H bending vibrations.97 The band centred at around 3250 cm−1 is slightly stronger in Ti-EtOH, suggesting that this sample might have more hydroxyl groups adsorbed on its surface.
Photocatalytic activity
The output of PCA, against NOx abatement, is depicted in Fig. 6. Ti-IPA and Ti-EtOH anatase were shown to have comparable PCA, after the first photocatalytic run, degrading approximately 37% of the initial NOx concentration (0.2 ppm) after only 20 minutes irradiation. This similar activity is certainly due to the comparable characteristics of the two samples, irrespective of small post-synthetic variables from the SC drying procedure. As a matter of fact, both samples consist of the same TiO2 polymorph (anatase), and have comparable size, shape, optical Eg, SSA and hydroxyl groups attached to the surface (cf.Tables 1–3). Furthermore, all the samples exhibited great stability, the PCA being virtually identical after three consecutive repetitions of the test, using the same specimen and protocols as in the first run. The reaction path for NOx conversion is mediated by OH˙ radicals, as through the photocatalytic reaction O2˙− and OH˙ radicals are formed, and these react with the pollutant gas to generate HNO3.98,99 This repeatability means that the HNO3 produced on the surface of the catalyst had no effect on the stability of the photocatalysts, with no inhibition of reaction, nor dissolution of the photocatalyst. Thus, the SC drying of such a sol not only gave the anatase a euhedral crystal habit (i.e. truncated tetragonal bipyramid, exposing {101} and {001} faces), but also a superior PCA. To have a like-with-like comparison, the PCA of a commercial nano-anatase K7000 was also tested. From Fig. 6, we can clearly see that both Ti-IPA and Ti-EtOH possess a higher PCA than the commercial K7000 (∼37% for Ti-IPA and Ti-EtOHversus ∼30% for K7000), despite the fact that K7000 has smaller crystalline domains, and the SSA is more than double that of our samples.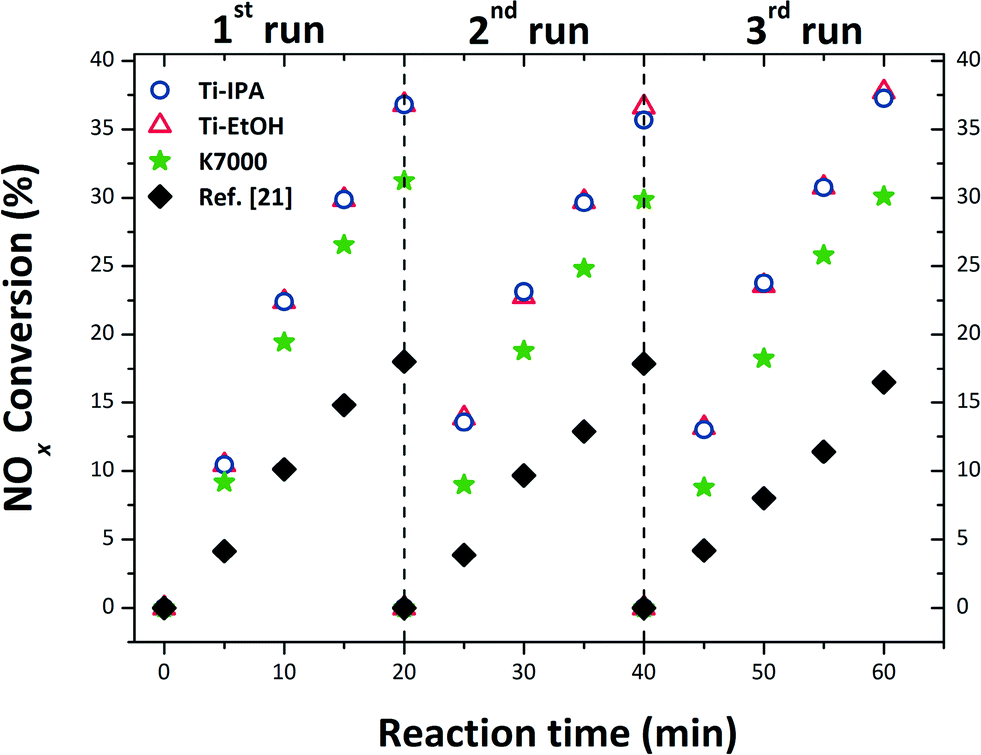 | ||
| Fig. 6 PCA result of the prepared samples, and of the commercial anatase K7000, in triplicate, using the solar lamp. | ||
Indeed, TiO2 prepared by these authors from the same starting sol, but thermally treated in air at 450 °C, led to similarly-sized sub-spherical NPs, with a PCA for NOx abatement of only 18% after 20 min irradiation time, measured using the very same protocol as in this manuscript, cf.Fig. 6. This is also supported by the findings of Li et al.,100 who compared the PCA of nitrogen doped TiO2 synthesised under SC conditions (in EtOH solution), and the same TiO2 calcined at 300 °C: they found that the SC TiO2 had superior PCA, and this was attributed to a well crystalline anatase having higher SSA, compared to that calcined.
Thus, the particular anisotropic euhedral shape of these anatase NPs surely plays a key role in their superior photocatalytic behaviour. From literature data, we actually know that exposed {101} faces of anatase NPs yield not only an enhanced reactivity with molecular O2, thus enabling the generation of superoxide radicals, but also less recombination of the photogenerated pair compared to spherical anatase NPs.33 Moreover, Murakami et al. demonstrated that anatase NPs (prepared by hydrothermal treatment of peroxo-titanic acid solution with polyvinyl alcohol as a shape-control agent) had a truncated tetragonal bipyramidal shape, and had more reduction sites available on their {101} faces.101 Furthermore, it has been recently shown that strongly reductive electrons are generated on the {101} faces, due to their higher conduction band minimum.43 This readily points out the reason for superior PCA: having more reduction sites available (and also less recombination of the e−–h+, as in reference 33), the photocatalytic reaction path is shifted to the generation of more superoxide radicals, according to these reactions:
| TiO2 + hν → eCB− + hVB+ | (7) |
| eCB− + O2 → O•−2 | (8) |
| NO + O•−2 → NO3− | (9) |
| NO−3 + H+ → HNO3 | (10) |
| TiO2 + hν → eCB− + hVB+ | (7) |
| hVB+ + H2O(ads) → OH• + H+ | (11) |
| hVB+ + OH− → OH• | (12) |
| NO + OH• → NO2 + H+ | (13) |
| NO2 + OH• → NO3− + H+ → HNO3 | (14) |
Conclusions
Anatase NPs were made from the supercritical drying of a titanium(IV) hydroxide sol, made via a green aqueous sol–gel route, and with no use of any surfactant or capping/morphology-controlling agent – in particular, without the use of HF. A quantitative phase analysis of the microstructure (through advanced X-ray techniques such as WPPM and HR-TEM analysis), the optical properties, and the PCA of the NPs were thoroughly evaluated. Synthesised anatase NPs showed themselves to exhibit very similar properties (mineralogical, morphological, optical and functional), irrespective of the alcohol used in the SC drying (i.e. IPA or EtOH). Moreover, these anatase NPs had euhedral crystal habit, being nearly homogeneous in both shape and size, and they exhibited a clear truncated tetragonal bipyramidal shape, exposing {101} and {001} faces.It was proposed that the particular shape of these anatase NPs (truncated tetragonal bipyramid) played a key role in their superior photocatalytic behaviour. Having mainly exposed {101} faces, and many fewer {001} faces, the photocatalytic NOx conversion is promptly mediated and enhanced by the generation of reactive oxygen species. This means that strongly reductive electrons can be generated on the {101} side faces, giving superoxide (O2˙−), and hydroxyl radicals (OH˙) at the {001} top faces. This is because the reduction sites are preferentially located at the {101} faces, whilst those for oxidation are accommodated on the {001} faces.
Acknowledgements
D. M. Tobaldi is grateful to the ECO-SEE project (funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no 609234. Note: the views expressed are purely those of the authors and may not in any circumstances be regarded as stating an official position of the European Commission). R. C. Pullar acknowledges the support of FCT grant SFRH/BPD/97115/2013. This work was developed in the scope of the project CICECO – Aveiro Institute of Materials (ref. FCT UID/CTM/50011/2013), financed by national funds through the FCT/MEC and when applicable co-financed by FEDER under the PT2020 Partnership Agreement. M. Ferro and RNME – University of Aveiro, FCT Project REDE/1509/RME/2005 – are also acknowledged for HR-TEM analysis.Notes and references
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 839–840 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- T. Fröschl, U. Hörmann, P. Kubiak, G. Kučerová, M. Pfanzelt, C. K. Weiss, R. J. Behm, N. Hüsing, U. Kaiser, K. Landfester and M. Wohlfahrt-Mehrens, Chem. Soc. Rev., 2012, 41, 5313 RSC.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC.
- X. Chen and A. Selloni, Chem. Rev., 2014, 114, 9281–9282 CrossRef CAS PubMed.
- A. Fujishima, K. Hashimoto and T. Watanabe, TiO2 photocatalysis: fundamentals and applications, BKC, Tokyo, Japan, 1999 Search PubMed.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- K. Sunada, Y. Kikuchi, K. Hashimoto and A. Fujishima, Environ. Sci. Technol., 1998, 32, 726–728 CrossRef CAS.
- J.-M. Herrmann, C. Duchamp, M. Karkmaz, B. T. Hoai, H. Lachheb, E. Puzenat and C. Guillard, J. Hazard. Mater., 2007, 146, 624–629 CrossRef CAS PubMed.
- B. Ohtani, Catalysts, 2013, 3, 942–953 CrossRef CAS.
- H. Zhang and J. F. Banfield, Chem. Rev., 2014, 114, 9613–9644 CrossRef CAS PubMed.
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2014, 4, 4043 Search PubMed.
- D. M. Tobaldi, R. C. Pullar, R. Binions, A. Belen Jorge, P. F. McMillan, M. Saeli, M. P. Seabra and J. A. Labrincha, Catal. Sci. Technol., 2014, 4, 2134 CAS.
- Z. Zhang, C.-C. Wang, R. Zakaria and J. Y. Ying, J. Phys. Chem. B, 1998, 102, 10871–10878 CrossRef CAS.
- G. Balasubramanian, D. D. Dionysiou, M. T. Suidan, I. Baudin and J.-M. Laîné, Appl. Catal., B, 2004, 47, 73–84 CrossRef CAS.
- A. G. Agrios and P. Pichat, J. Photochem. Photobiol., A, 2006, 180, 130–135 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- S. Mann, Angew. Chem., Int. Ed., 2000, 39, 3392–3406 CrossRef CAS.
- H. Kominami, S. Murakami, J. Kato, Y. Kera and B. Ohtani, J. Phys. Chem. B, 2002, 106, 10501–10507 CrossRef CAS.
- L. Manna, E. C. Scher, L.-S. Li and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 7136–7145 CrossRef CAS PubMed.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- J. Joo, S. G. Kwon, T. Yu, M. Cho, J. Lee, J. Yoon and T. Hyeon, J. Phys. Chem. B, 2005, 109, 15297–15302 CrossRef CAS PubMed.
- X. Peng and A. Chen, Adv. Funct. Mater., 2006, 16, 1355–1362 CrossRef CAS.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- N. Wu, J. Wang, D. N. Tafen, H. Wang, J.-G. Zheng, J. P. Lewis, X. Liu, S. S. Leonard and A. Manivannan, J. Am. Chem. Soc., 2010, 132, 6679–6685 CrossRef CAS PubMed.
- W. Zhou, L. Gai, P. Hu, J. Cui, X. Liu, D. Wang, G. Li, H. Jiang, D. Liu, H. Liu and J. Wang, CrystEngComm, 2011, 13, 6643–6649 RSC.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2004, 126, 5851–5858 CrossRef CAS PubMed.
- J. Yu, J. Fan and K. Lv, Nanoscale, 2010, 2, 2144 RSC.
- Z. Liu, X. Zhang, S. Nishimoto, T. Murakami and A. Fujishima, Environ. Sci. Technol., 2008, 42, 8547–8551 CrossRef CAS PubMed.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- K. Huo, H. Wang, X. Zhang, Y. Cao and P. K. Chu, ChemPlusChem, 2012, 77, 323–329 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167–1170 RSC.
- M. D'Arienzo, J. Carbajo, A. Bahamonde, M. Crippa, S. Polizzi, R. Scotti, L. Wahba and F. Morazzoni, J. Am. Chem. Soc., 2011, 133, 17652–17661 CrossRef PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- J. Pan, G. Liu, G. Q. M. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- Q. Xiang, K. Lv and J. Yu, Appl. Catal., B, 2010, 96, 557–564 CrossRef CAS.
- F. Dufour, S. Pigeot-Remy, O. Durupthy, S. Cassaignon, V. Ruaux, S. Torelli, L. Mariey, F. Maugé and C. Chanéac, Appl. Catal., B, 2015, 174–175, 350–360 CrossRef CAS.
- A. Selloni, Nat. Mater., 2008, 7, 613–615 CrossRef CAS PubMed.
- S. Selçuk and A. Selloni, J. Phys. Chem. C, 2013, 117, 6358–6362 Search PubMed.
- D. M. Tobaldi, R. C. Pullar, A. F. Gualtieri, M. P. Seabra and J. A. Labrincha, Chem. Eng. J., 2013, 214, 364–375 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549–1557 CrossRef CAS.
- B. Horvat, A. Rečnik and G. Dražić, J. Cryst. Growth, 2012, 347, 19–24 CrossRef CAS.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Max Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS PubMed.
- N. Roy, Y. Sohn and D. Pradhan, ACS Nano, 2013, 7, 2532–2540 CrossRef CAS PubMed.
- N. Roy, Y. Park, Y. Sohn, K. T. Leung and D. Pradhan, ACS Appl. Mater. Interfaces, 2014, 6, 16498–16507 CAS.
- J. Wang, P. Zhang, X. Li, J. Zhu and H. Li, Appl. Catal., B, 2013, 134–135, 198–204 CrossRef CAS.
- A. P. Jones, Atmos. Environ., 1999, 33, 4535–4564 CrossRef CAS.
- Air Quality in Urban Environments, ed. R. M. Harrison and R. E. Hester, Royal Society of Chemistry, Cambridge, 2009 Search PubMed.
- D. M. Tobaldi, R. C. Pullar, A. F. Gualtieri, M. P. Seabra and J. A. Labrincha, Acta Mater., 2013, 61, 5571–5585 CrossRef CAS.
- D. M. Tobaldi, C. Piccirillo, R. C. Pullar, A. F. Gualtieri, M. P. Seabra, P. M. L. Castro and J. A. Labrincha, J. Phys. Chem. C, 2014, 118, 4751–4766 CAS.
- D. M. Tobaldi, R. C. Pullar, A. F. Gualtieri, A. Belen Jorge, R. Binions, P. F. McMillan, M. P. Seabra and J. A. Labrincha, CrystEngComm, 2015, 17, 1813–1825 RSC.
- A. C. Larson and R. B. von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR, 2004 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- P. Scardi and M. Leoni, Acta Crystallogr., Sect. A: Found. Adv., 2002, 58, 190–200 CrossRef CAS.
- M. Leoni, T. Confente and P. Scardi, Z. Für Krist. Suppl., 2006, 23, 249–254 CrossRef.
- P. Scardi and M. Leoni, in Diffraction Analysis of the Microstructure of Materials, ed. E. J. Mittemeijer and P. Scardi, Berlin, Springer-Verlag., 2004, pp. 51–92 Search PubMed.
- P. Scardi and M. Leoni, J. Appl. Crystallogr., 2006, 39, 24–31 CrossRef CAS.
- P. Scardi and M. Leoni, Acta Mater., 2005, 53, 5229–5239 CrossRef CAS.
- H. P. Klug and L. E. Alexander, X-ray diffraction procedures for polycrystalline and amorphous materials, Wiley, New York, 2nd Edition., 1974 Search PubMed.
- G. K. Williamson and W. H. Hall, Acta Metall., 1953, 1, 22–31 CrossRef CAS.
- G. Caglioti, A. Paoletti and F. P. Ricci, Nucl. Instrum. Methods, 1960, 9, 195–198 CrossRef CAS.
- D. M. Tobaldi, R. C. Pullar, M. Leoni, M. P. Seabra and J. A. Labrincha, Appl. Surf. Sci., 2013, 287, 276–281 CrossRef CAS.
- M. Karmaoui, D. M. Tobaldi, A. Sever Skapin, R. C. Pullar, M. P. Seabra, J. A. Labrincha and V. Amaral, RSC Adv., 2014, 46762–46770 CAS.
- A. S. Barnard and L. A. Curtiss, Nano Lett., 2005, 5, 1261–1266 CrossRef CAS PubMed.
- P. Stadelmann, JEMS-SAAS, 2014 Search PubMed.
- S. Komornicki, M. Radecka and P. Sobaś, Mater. Res. Bull., 2004, 39, 2007–2017 CrossRef CAS.
- D. M. Tobaldi, L. Gao, A. F. Gualtieri, A. Sever Škapin, A. Tucci and C. Giacobbe, J. Am. Ceram. Soc., 2012, 95, 1709–1716 CrossRef CAS.
- M. T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, M. M. Müller, H.-J. Kleebe, K. Rachut, J. Ziegler, A. Klein and W. Jaegermann, J. Phys. Chem. C, 2013, 117, 22098–22110 CAS.
- K. A. Michalow, D. Logvinovich, A. Weidenkaff, M. Amberg, G. Fortunato, A. Heel, T. Graule and M. Rekas, Catal. Today, 2009, 144, 7–12 CrossRef CAS.
- A. S. Marfunin, Physics of Minerals and Inorganic Materials: An Introduction, Springer-Verlag, 1979 Search PubMed.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646–16654 CrossRef CAS.
- D. M. Tobaldi, R. A. S. Ferreira, R. C. Pullar, M. P. Seabra, L. D. Carlos and J. A. Labrincha, J. Mater. Chem. C, 2015, 3, 4970–4986 RSC.
- F. L. Toma, G. Bertrand, D. Klein and C. Coddet, Environ. Chem. Lett., 2004, 2, 117–121 CrossRef CAS.
- http://kronostio2.com/en/component/jdownloads/finish/99/288 [Accessed November 2015].
- H. Zhang and J. F. Banfield, J. Mater. Chem., 1998, 8, 2073–2076 RSC.
- T. B. Ghosh, S. Dhabal and A. K. Datta, J. Appl. Phys., 2003, 94, 4577 CrossRef CAS.
- Y. Dai, C. M. Cobley, J. Zeng, Y. Sun and Y. Xia, Nano Lett., 2009, 9, 2455–2459 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Commun., 2011, 47, 4532 RSC.
- J. Yu, G. Dai, Q. Xiang and M. Jaroniec, J. Mater. Chem., 2011, 21, 1049–1057 RSC.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- J.-L. Mi, K. M. Ø. Jensen, C. Tyrsted, M. Bremholm and B. B. Iversen, CrystEngComm, 2015, 17, 6868–6877 RSC.
- V. G. Courtecuisse, J. F. Bocquet, K. Chhor and C. Pommier, J. Supercrit. Fluids, 1996, 9, 222–226 CrossRef CAS.
- H. Li, X. Zhang, Y. Huo and J. Zhu, Environ. Sci. Technol., 2007, 41, 4410–4414 CrossRef CAS PubMed.
- Spectroscopic methods in mineralogy and geology, ed. F. C. Hawthorne and Mineralogical Society of America, Mineralogical Society of America, Washington, D.C., 1988 Search PubMed.
- S.-D. Mo and W. Y. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13023–13032 CrossRef CAS.
- S. Balaji, Y. Djaoued and J. Robichaud, J. Raman Spectrosc., 2006, 37, 1416–1422 CrossRef CAS.
- S. Ahmad, B. Jousseaume, T. Toupance, O. Babot, G. Campet, C. Labrugère, J. Brötz and U. Kunz, Dalton Trans., 2012, 41, 292–299 RSC.
- A. Li Bassi, D. Cattaneo, V. Russo, C. E. Bottani, E. Barborini, T. Mazza, P. Piseri, P. Milani, F. O. Ernst, K. Wegner and S. E. Pratsinis, J. Appl. Phys., 2005, 98, 074305 CrossRef.
- D. M. Tobaldi, A. Tucci, A. S. Škapin and L. Esposito, J. Eur. Ceram. Soc., 2010, 30, 2481–2490 CrossRef CAS.
- Y. Ohko, Y. Nakamura, A. Fukuda, S. Matsuzawa and K. Takeuchi, J. Phys. Chem. C, 2008, 112, 10502–10508 CAS.
- J. Lasek, Y.-H. Yu and J. C. S. Wu, J. Photochem. Photobiol., C, 2013, 14, 29–52 CrossRef CAS.
- H. Li, J. Li and Y. Huo, J. Phys. Chem. B, 2006, 110, 1559–1565 CrossRef CAS PubMed.
- N. Murakami, Y. Kurihara, T. Tsubota and T. Ohno, J. Phys. Chem. C, 2009, 113, 3062–3069 CAS.
- M. Lazzeri and A. Selloni, Phys. Rev. Lett., 2001, 87 Search PubMed.
- X.-Q. Gong, A. Selloni and A. Vittadini, J. Phys. Chem. B, 2006, 110, 2804–2811 CrossRef CAS PubMed.
- H. Cheng and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 092101 CrossRef.
- C. Sun, A. Selloni, A. Du and S. C. Smith, J. Phys. Chem. C, 2011, 115, 17092–17096 CAS.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS PubMed.
- X. Yao, X. Liu, T. Liu, K. Wang and L. Lu, CrystEngComm, 2013, 15, 10246–10254 RSC.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946 RSC.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197–7204 CrossRef CAS PubMed.
- M. M. Maitani, K. Tanaka, D. Mochizuki and Y. Wada, J. Phys. Chem. Lett., 2011, 2, 2655–2659 CrossRef CAS.
- J. Z. Bloh, A. Folli and D. E. Macphee, RSC Adv., 2014, 4, 45726–45734 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Emission spectrum of the solar lamp used in the PCA tests; Rietveld and WPPM graphical outputs; XRD pattern of the dried gel prior to SC drying; DRS, Raman and FT-IR spectra of synthesised anatase. See DOI: 10.1039/c5ce02112j |
| This journal is © The Royal Society of Chemistry 2016 |