DOI:
10.1039/C5CE01815C
(Paper)
CrystEngComm, 2016,
18, 130-142
A series of Zn(II) and Cd(II) coordination compounds based on 4-(4H-1,2,4-triazol-4-yl)benzoic acid: synthesis, structure and photoluminescence properties†
Received
11th September 2015
, Accepted 9th November 2015
First published on 10th November 2015
Abstract
Seven coordination compounds, namely [Zn(4-tba)2(H2O)2] (1), [Zn(4-tba)Cl]·CH3OH (2), [Zn(4-tba)2] (3), [Zn(4-tba)2]·DMA·3.5H2O (4), [Cd(4-tba)2]·DMF·3.6H2O (5), [Cd(4-tba)2] (6) and [Cd5Cl4(4-tba)6]·1.5DMF·4H2O (7) (4-Htba = 4-(4H-1,2,4-triazol-4-yl)benzoic acid, DMA = N,N-dimethylacetamide and DMF = N,N-dimethylformamide), have been synthesized under hydro/solvothermal conditions by using a triazolate–carboxylate bifunctional organic ligand, 4-Htba. Complexes 1 and 2 exhibit a mononuclear motif and a two-dimensional (2D) network with sql topology, respectively, whereas 3–6 display different interpenetrating structures. Compound 3 shows a 2-fold interpenetrating 2D net with sql topology, 4 and 5 display uninodal 4-connected 4-fold interpenetrating 3D nets with dia (66) topology belonging to class Ia and class IIIa, respectively, and compound 6 exhibits a 5-fold interpenetrating dia net. Complex 7 exhibits a non-interpenetrating 3D framework constructed from decanuclear cadmium–chloride chain units and 4-tba ligands with various coordination modes. All the complexes were characterized using single-crystal X-ray diffraction, powder X-ray diffraction (PXRD), IR spectroscopy and elemental analyses. In addition, the thermogravimetric analysis and solid-state photoluminescence results for 1–7 were also investigated.
1. Introduction
The rational design and construction of coordination compounds have received considerable attention over the past ten years because of their intriguing molecular topologies1 and wide range of potential applications such as in gas or liquid adsorption,2 separation,3 ion exchange,4 catalytic activity,5 magnetism6 and sensing.7 Generally, the construction of coordination compounds is dependent on a variety of factors such as the organic ligands,8 solvent,9 metal ion,10 ratio of reactants,11 and counterion.12 Of all these, appropriate organic ligands are some of the most important factors because their length, steric effects and flexibility will lead to diverse structures of coordination compounds. Aromatic polycarboxylate, polypyridine and deprotonated polyazaheterocycles are the most extensively exploited ligands for the construction of metal complexes.13 More recently, azolate–carboxylate bifunctional ligands have attracted considerable attention as another type of useful ligands for the construction of functional metal coordination polymers.14–16 Among them, the triazolate–carboxylate bifunctional organic ligand has received special attention owing to their multi-connectivity abilities and interesting luminescence, magnetic and gas adsorption properties.15,16
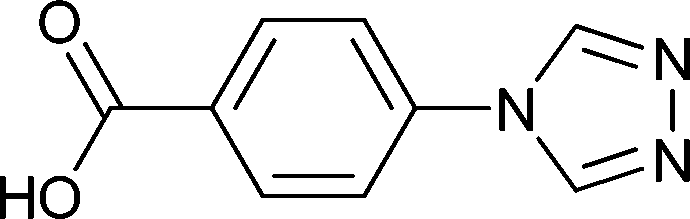
The bifunctional ligand 4-(4H-1,2,4-triazol-4-yl)benzoic acid (4-Htba, Scheme 1) with two nitrogen and two oxygen potential coordination sites has multiple coordination modes and can also construct diverse coordination polymers with various secondary building units (SBUs).16 For example, Murugesu and coworkers utilized 4-Htba as the organic bridging ligand and synthesized a MIL88 structure with a trinuclear cobalt core as the triangular SBU and cubic coordination polymers with an octanuclear Co(II) cubane core as the SBU.16b,c Cheng et al. reported a series of 4-tba-based Cu(II) MOFs containing various SBUs such as mononuclear, binuclear, trinuclear, tetranuclear and chloride-centered square-planar [Cu4Cl]7+ units.16d,e However, the most reported coordination polymers based on this ligand are usually restricted to magnetic metal ions, and their properties are mainly confined to the fields of magnetism and gas or liquid adsorption. The investigation of the 4-tba ligand as a building block for constructing d10 luminescent coordination polymers has been less developed so far.16a
 |
| | Scheme 1 Structure of 4-(4H-1,2,4-triazol-4-yl)benzoic acid (4-Htba). | |
Herein, a series of new Zn(II) and Cd(II) coordination compounds based on the 4-tba ligand, namely [Zn(4-tba)2(H2O)2] (1), [Zn(4-tba)Cl]·CH3OH (2), [Zn(4-tba)2] (3), [Zn(4-tba)2]·DMA·3.5H2O (4), [Cd(4-tba)2]·DMF·3.6H2O (5), [Cd(4-tba)2] (6) and [Cd5Cl4(4-tba)6]·1.5DMF·4H2O (7), were synthesized under hydro/solvothermal conditions and characterized via single-crystal X-ray diffraction. Moreover, the thermogravimetric analysis and solid-state photoluminescence results for 1–7 were also studied.
2. Experimental
2.1. Materials and methods
The ligand 4-Htba was synthesized according to a literature method.15b Other reagents and solvents were commercially available and were used without further purification. IR spectra were obtained from KBr pellets on a Bruker EQUINOX 55 FTIR spectrometer in the 400–4000 cm−1 region. Elemental analyses (C, H, N) were performed with a Vario EL elemental analyzer. The phase purity and crystallinity of each product were examined by powder X-ray diffraction (PXRD) using a Rigaku D/M-2200T automated diffractometer (CuKα, 1.5418 Å). The observed and simulated powder XRD patterns of all compounds are displayed in Fig. S1–S7 in the ESI.† Thermogravimetric analysis (TGA) was performed using a Netzsch STA 449C instrument. Each sample was heated from room temperature to 700 °C with a heating rate of 10.0 °C min−1 under nitrogen atmosphere. Room-temperature photoluminescence (PL) spectra were recorded using a Hitachi F-4600 fluorescence spectrophotometer equipped with a xenon lamp. For zinc complexes 1–4, the slit widths of excitation and emission were fixed at 2.5 and 1.0 nm, respectively. For cadmium complexes 5–7, the slit widths of excitation and emission were fixed at 5.0 and 2.5 nm, respectively.
2.2. Preparation of complexes 1–7
2.2.1. Synthesis of [Zn(4-tba)2(H2O)2] (1).
A mixture of ZnCl2 (14 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol), H2O (4 mL) and MeOH (1 mL) was placed in a Teflon-lined stainless steel vessel (15 mL) and heated at 120 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. Colorless, needle-like crystals were collected by filtration, washed with water and dried in air to afford 13 mg (55% based on 4-Htba) of the product. Anal. calcd. for C18H16N6O6Zn: C, 45.25; H, 3.38; N, 17.59. Found: C, 45.23; H, 3.39; N, 17.62. IR (cm−1, KBr): 3317(br, m), 1719(m), 1606(s), 1536(s), 1469(w), 1423(s), 1306(w), 1248(m), 1093(m), 1007(m), 861(m), 784(m), 696(w), 523(m).
2.2.2. Synthesis of [Zn(4-tba)Cl]·CH3OH (2).
A mixture of ZnCl2 (14 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol) and MeOH (5 mL) was placed in a Teflon-lined stainless steel vessel (15 mL) and heated at 120 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. Colorless, needle-like crystals were collected by filtration, washed with MeOH and dried in air to afford 22 mg (70% based on 4-Htba) of the product. Anal. calcd. for C10H10ClN3O3Zn: C, 37.41; H, 3.14; N, 13.09. Found: C, 37.45; H, 3.12; N, 13.12. IR (cm−1, KBr): 3442(m, br), 3116(w), 1710(w), 1608(s), 1535(s), 1459(s), 1379(s), 1237(m), 1178(w), 1095(m), 1016(m), 866(w), 781(m), 696(w), 624(w), 527(w).
2.2.3. Synthesis of [Zn(4-tba)2] (3).
A mixture of ZnCl2 (14 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol) and DMF (5 mL) was placed in a Teflon-lined stainless steel vessel (15 mL) and heated at 120 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. Colorless, block crystals were collected by filtration, washed with MeOH and dried in air to afford 10 mg (45% based on 4-Htba) of 3. Anal. calcd. for C18H12N6O4Zn: C, 48.94; H, 2.74; N, 19.03. Found: C, 48.92; H, 2.76; N, 19.00. IR (cm−1, KBr): 3447(br, m), 3118(m), 1633(s), 1581(m), 1531(s), 1366(s), 1235(m), 1095(m), 1034(s), 1015(m), 842(m), 780(m), 696(w), 655(w), 506(w).
2.2.4. Synthesis of [Zn(4-tba)2]·DMA·3.5H2O (4).
A mixture of ZnCl2 (14 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol) and DMA (5 mL) was placed in a Teflon-lined stainless steel vessel (15 mL) and heated at 95 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. A few colorless, block crystals were collected by filtration, washed with ether and dried in air to afford 11 mg (38% based on 4-Htba) of 4. Anal. calcd. for C22H28N7O8.5Zn: C, 44.64; H, 4.77; N, 16.57. Found: C, 44.62; H, 4.79; N, 16.54. IR (cm−1, KBr): 3220(br, m), 3106(m), 1607(s), 1529(s), 1398(s), 1313(w), 1243(m), 1098(m), 1010(m), 865(m), 782(m), 705(w), 625(w), 566(m). The framework of 4 can also be obtained by another procedure as follows: the colorless filtrate resulting from the synthesis of 3 was allowed to stand at room temperature, and slowly evaporated in air for over two weeks to give colorless block crystals of the framework of 4 (yield: 40% based on 4-Htba). Elemental and thermal gravimetric analyses reveal that the molecular formula of the product synthesized through this method is [Zn(4-tba)2]·0.5DMF·2H2O (4′).
2.2.5. Synthesis of [Cd(4-tba)2]·DMF·3.6H2O (5).
A mixture of Cd(NO3)2·4H2O (31 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol) and DMF (5 mL) was placed in a Teflon-lined stainless steel vessel (15 mL) and heated at 95 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. Colorless, block crystals were collected by filtration, washed with ether and dried in air to afford 19 mg (62% based on 4-Htba) of the product. Anal. calcd. for C21H26.2CdN7O8.6: C, 40.25; H, 4.21; N, 15.65. Found: C, 40.20; H, 4.25; N, 15.68. IR (cm−1, KBr): 3289(br, m), 3099(m), 1632(m), 1607(s), 1538(s), 1409(s), 1305(w), 1248(m), 1092(m), 1009(w), 860(m), 783(m), 696(w), 519(m).
2.2.6. Synthesis of [Cd(4-tba)2] (6).
A mixture of Cd(NO3)2·4H2O (31 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol), 4,4′-bipyridine (16 mg, 0.1 mmol), DMF (4 mL) and MeOH (1 mL) was placed in a Teflon-lined stainless steel vessel (15 mL) and heated at 120 °C for 3 days and then cooled to room temperature at a rate of 5 °C h−1. Colorless, block crystals were collected by filtration, washed with EtOH and dried in air to afford 8 mg (32% based on 4-Htba) of the product. Anal. calcd. for C18H12CdN6O4: C, 44.23; H, 2.47; N, 17.20. Found: C, 44.20; H, 2.51; N, 17.22. IR (cm−1, KBr): 3384(br, m), 3105(m), 1607(s), 1541(s), 1408(s), 1313(m), 1248(m), 1092(m), 1034(w), 856(m), 784(m), 696(w), 625(w), 510(m).
2.2.7. Synthesis of [Cd5Cl4(4-tba)6]·1.5DMF·4H2O (7).
A mixture of CdCl2·2.5H2O (23 mg, 0.1 mmol), 4-Htba (19 mg, 0.1 mmol), DMF (3 mL) and CH3CN (1 mL) was heated at 95 °C for 3 days under autogenous pressure. The mixture was slowly cooled to room temperature at a rate of 5 °C h−1. Colorless, block crystals were collected by filtration, washed with EtOH and dried in air to afford 5 mg (15% based on 4-Htba) of the product. Anal. calcd. for C58.5H54.5Cd5Cl4N19.5O17.5: C, 34.88; H, 2.73; N, 13.56. Found: C, 34.95; H, 2.80; N, 13.61. IR (cm−1, KBr): 3544(br, m), 3120(m), 1631(m), 1603(s), 1561(s), 1538(s), 1388(s), 1360(m), 1260(s), 1092(m), 862(m), 783(m), 691(w), 642(w), 510(m).
2.3. X-ray crystallography
The diffraction intensities of the seven compounds were collected on a Bruker Apex CCD diffractometer or a Rigaku R-AXIS SPIDER diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Absorption corrections were applied by using the multi-scan program SADABS17 or the program ABSCOR.18 The structures were solved by direct methods and refined using a full-matrix least-squares technique with the SHELXTL program package.19 Anisotropic thermal parameters were applied to all non-hydrogen atoms. All the hydrogen atoms were generated geometrically expect for the hydrogen atoms of the disordered water molecules in 7. Due to the presence of large cavities in the structure and heavily disordered solvent molecules in the cavities, the crystals of 4 and 5 scattered weakly and only low-angle data could be detected. An attempt to locate and refine the solvent molecules failed. Thus, the SQUEEZE routine of PLATON was applied for 4 and 5 to remove the contributions to the scattering from the solvent molecules.20 The reported refinement is of the guest-free structure using the *.hkp file produced by the SQUEEZE routine. Crystal data and structure refinement are summarized in Table S1.† The void volume (excluding guest solvent molecules) in the crystal cell was calculated using the program PLATON.20 Crystal data as well as the details of data collection and refinements for the complexes are summarized in Table 1.
Table 1 Crystal data for 1–7
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Empirical formula |
C18H16N6O6Zn |
C10H10ClN3O3Zn |
C18H12N6O4Zn |
C22H28N7O8.5Zn |
C21H26.2CdN7O8.6 |
C18H12CdN6O4 |
C58.5H54.5Cd5Cl4N19.5O17.5 |
| Formula weight |
477.74 |
321.03 |
441.71 |
591.89 |
626.69 |
488.74 |
2014.55 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Triclinic |
| Space group |
C2/c |
P21/c |
C2 |
Pbcn
|
Pnc2 |
Fdd2 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
a (Å) |
13.519(2) |
9.097(3) |
13.664(2) |
20.0621(18) |
8.3848(17) |
16.680(3) |
12.946(3) |
|
b (Å) |
9.8960(17) |
17.622(5) |
7.8616(13 |
15.5930(14) |
14.808(3) |
32.764(7) |
17.321(3) |
|
c (Å) |
14.609(2) |
7.464(2) |
8.2311(14) |
17.2349(15) |
11.494(2) |
6.5537(13) |
22.153(4) |
|
α (°) |
90 |
90 |
90 |
90 |
90 |
90 |
70.13(3) |
|
β (°) |
112.259(3) |
92.337(6) |
91.400(3) |
90 |
90 |
90 |
76.08(3) |
|
γ (°) |
90 |
90 |
90 |
90 |
90 |
90 |
73.31(3) |
|
V (Å3) |
1808.9(5) |
1195.6(6) |
883.9(3) |
5391.6(8) |
1427.1(5) |
3581.8(12) |
4418.3(15) |
|
Z
|
4 |
4 |
2 |
8 |
2 |
8 |
2 |
|
D
c (g cm−3) |
1.754 |
1.784 |
1.660 |
1.458 |
1.458 |
1.813 |
1.508 |
|
μ (mm−1) |
1.413 |
2.280 |
1.430 |
0.971 |
0.791 |
1.260 |
1.370 |
|
R
int
|
0.0201 |
0.0392 |
0.0296 |
0.0503 |
0.0295 |
0.0299 |
0.0598 |
| GOF |
1.123 |
1.126 |
1.012 |
1.012 |
1.074 |
1.199 |
1.020 |
|
R
1 [I > 2σ(I)] |
0.0432 |
0.0505 |
0.0390 |
0.0441 |
0.0284 |
0.0176 |
0.0545 |
| wR2 [all data] |
0.1210 |
0.1322 |
0.0755 |
0.1143 |
0.0779 |
0.0522 |
0.1700 |
3. Results and discussion
3.1. Crystal structure of [Zn(4-tba)2(H2O)2] (1)
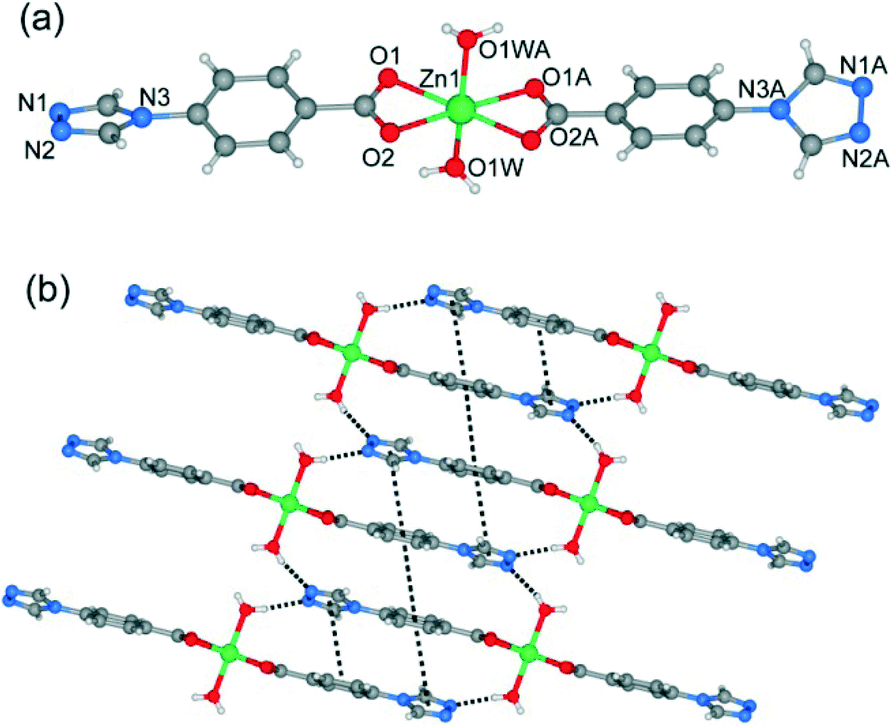
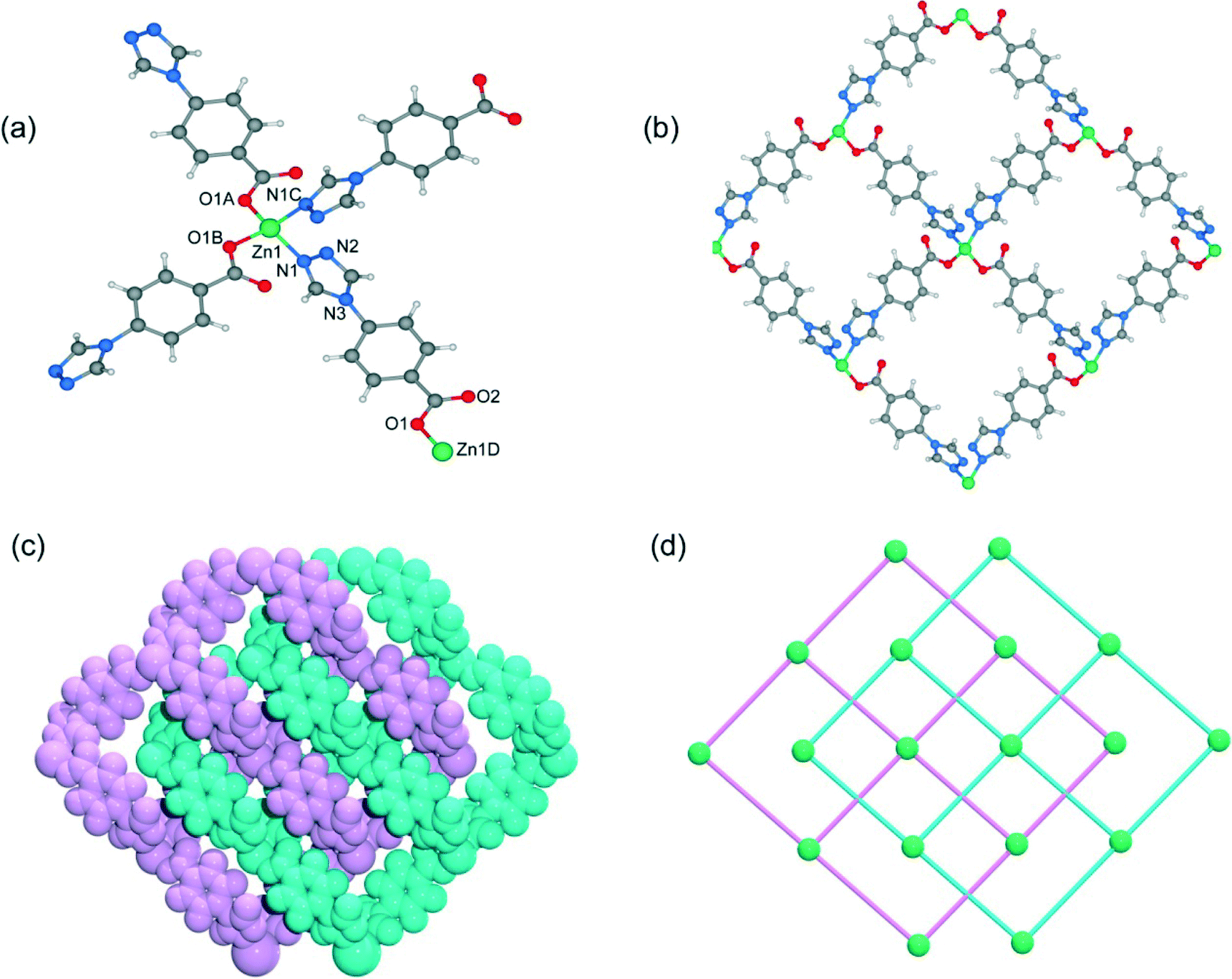
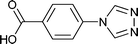
Single-crystal X-ray analysis reveals that complex 1 shows a mononuclear 0D structure, and is isomorphous to previously reported complexes constructed from first-transition metal ions (such as Mn2+, Co2+, Ni2+, Cu2+ and Cd2+) and 4-tba anions.16a,21 As shown in Fig. 1a, the asymmetric unit contains a half molecule of [Zn(4-tba)2(H2O)2], and a crystallographic mirror plane passes through the Zn1 site. Zn1 adopts an octahedral coordination geometry, being coordinated by four oxygen atoms from two different 4-tba ligands and two coordinated water molecules. In complex 1, the deprotonated 4-tba ligand coordinates with ZnII ions in an O,O-chelating mode, and the nitrogen atoms from the 4-tba ligand are not coordinated to the metal atoms (Fig. 2a). However, the uncoordinated nitrogen atoms from 4-tba anions (N1 and N2) are involved in two intermolecular O–H⋯N hydrogen bonds (O⋯N = 2.746(17)–2.790(17) Å, H⋯N = 2.04–2.06 Å, O–H⋯N = 155.8–164.5°) with two neighbouring coordinated water molecules to generate a 2D supramolecular structure (Fig. 1b). The corresponding hydrogen bonding parameters are summarized in Table S1.† Moreover, the π–π interactions between 4-tba further stabilize the 2D supramolecular layer (the closest interplanar and centroid–centroid separations are 3.34–3.78 and 3.87–4.13 Å, respectively). These 2D layers are further assembled by weak C–H⋯O interactions (C⋯O = 3.295(15)–3.329(4) Å, H⋯O = 2.40–2.49 Å, C–H⋯O = 145.3–174.3°) between adjacent layers into an infinite 3D supramolecular structure (Fig. S8†).
 |
| | Fig. 1 (a) The coordination environment of the zinc atoms in 1. Only one position of disordered water molecules is shown for clarity. Symmetry codes: A: −x, y, 1/2 − z. (b) 2D supramolecular network of 1 along the b-axis constructed by hydrogen bonds. | |
 |
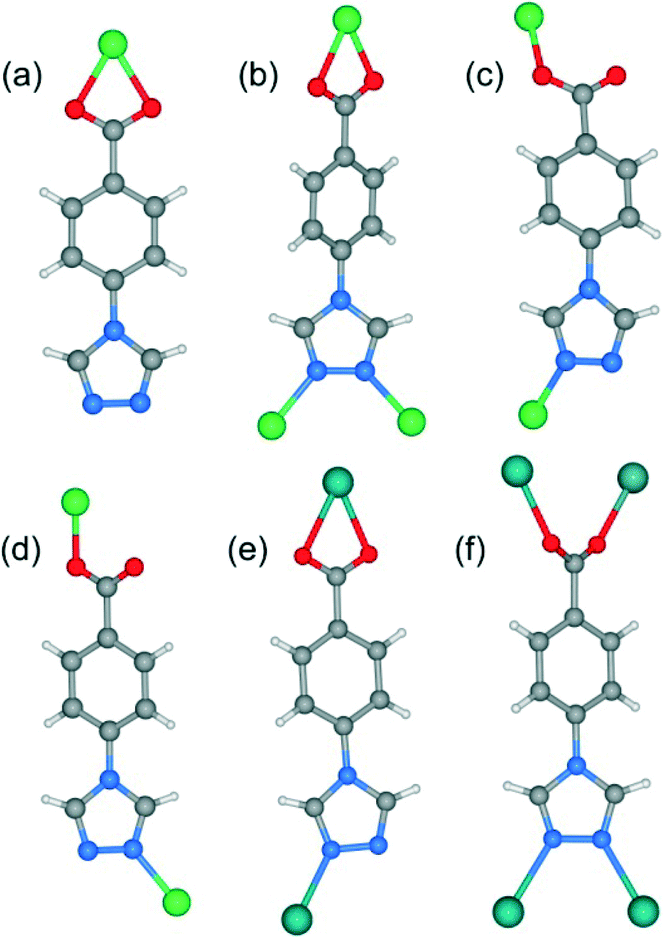
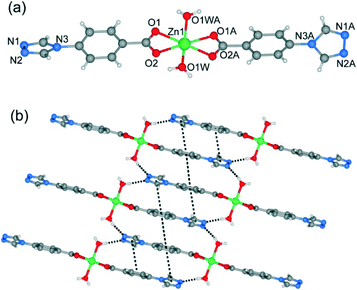
| | Fig. 2 (a–f) The coordination environment of 4-tba ligands in complexes 1–7. | |
3.2. Crystal structure of [Zn(4-tba)Cl]·CH3OH (2)
When MeOH was utilized as the reaction solvent in place of water under similar synthetic conditions to those for 1, a new complex 2 with a different structure was isolated. Compound 2 crystallizes in the monoclinic space group P21/c and exhibits a two-dimensional (2D) layer. As depicted in Fig. S9,† each Zn1 atom is five-coordinated to two N atoms from two 4-tba ligands, two oxygen atoms from one chelating carboxylate group and one chloride ion to form a highly distorted square-pyramidal coordination geometry (trigonality index τ = 0.39).22 The chloride ion only acts as an auxiliary ligand in a monodentate coordination mode, while each 4-tba ligand adopts a μ3-bridging mode to connect three zinc(II) ions through two N atoms from the triazolyl group and two O atoms from one carboxylate group with a μ1-η1:η1 chelating mode (Fig. 2b). The Zn ions are linked together by 4-tba in μ3-η1:η1:η1:η1 mode to generate a 2D network with a 3-connected 4·82 topology when Zn and 4-tba are regarded as two kinds of 3-connected nodes (Fig. S10†). If the dinuclear [Zn2(triazole)2] subunits are further simplified as 4-connecting nodes and the 4-tba ligands as linkers, compound 2 has a simple 44-sql network (Fig. 3b). The 2D layers are further linked together via intermolecular weak C–H⋯O hydrogen bonds (C⋯O = 2.965(6)–3.496(7) Å, H⋯O = 2.25–2.57 Å, C–H⋯O = 112.8–171.5°) to generate a 3D structure (Fig. S11†). Furthermore, the π–π stacking interactions between the adjacent benzene rings (the closest interplanar and centroid–centroid separations are 3.36 and 3.78 Å, respectively) may further stabilize the structure (Fig. S12†).
 |
| | Fig. 3 (a) The two-dimensional network of 2. (b) The 44-sql net of 2. | |
3.3. Crystal structure of [Zn(4-tba)2] (3)
When DMF was utilized as the reaction solvent in place of water or MeOH under similar synthetic conditions to those for 1 and 2, complex 3 was obtained. Compound 3 crystallizes in the chiral space group C2, as a result of the unsymmetrical nature of the organic ligand. There are one half of a Zn2+ ion and one tba ligand in the asymmetric unit. As shown in Fig. 4a, the zinc ion (Zn1) is located on the crystallographic C2 axis and adopts a tetrahedral coordination geometry, being coordinated by two oxygen atoms from two symmetry-related 4-tba ligands and two nitrogen atoms from two other 4-tba ligands. The 4-tba ligand acts as an unsymmetrical, bidentate, linear linker (Fig. 2c), and each Zn(II) atom is connected to four adjacent Zn(II) atoms through four linear 4-tba linkers resulting in a 44-sql 2D network. Each 2D layer has an effective square window with dimensions of ca. 5.0 Å2 (Fig. 4b). Moreover, each 2D layer has enough “empty” space to allow another 2D layer to interpenetrate and form a two-fold parallel interwoven net (Fig. 4c and d), making 3 a nearly dense structure with the total solvent-accessible volume only comprising 2.1% (18.7 Å3) of the crystal volume.20 Furthermore, π–π stacking interactions can also be observed in 3 between the benzene rings from adjacent two-dimensional layers (Fig. S13,† the closest interplanar and centroid–centroid separations are 3.51 and 3.66 Å, respectively).
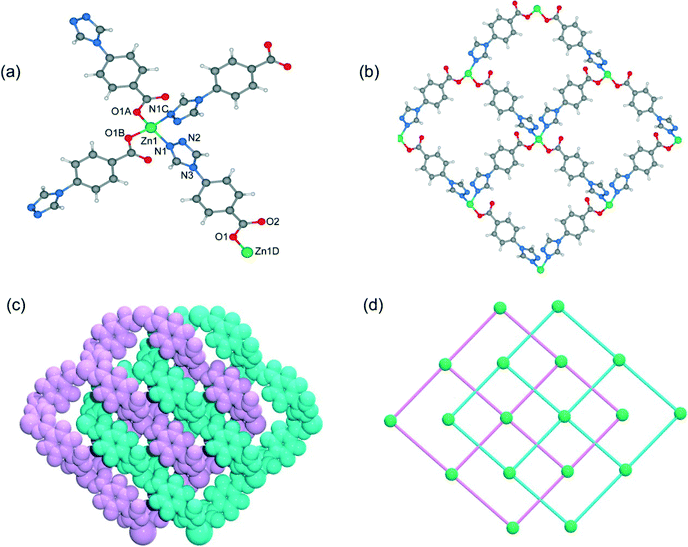 |
| | Fig. 4 (a) The coordination environment of the zinc atoms and 4-tba ligands in 3. Symmetry codes: A: x, y − 1, z + 1; B: −x + 2, y − 1, −z − 2; C: −x + 2, y, −z − 1. (b) A single 2D layer structure of 3. (c) Space-filling diagram of the 2-fold interpenetrating framework of 3. (d) Topological representation of the 2-fold interpenetration in 3. | |
3.4. Crystal structure of [Zn(4-tba)2]·DMA·3.5H2O (4)
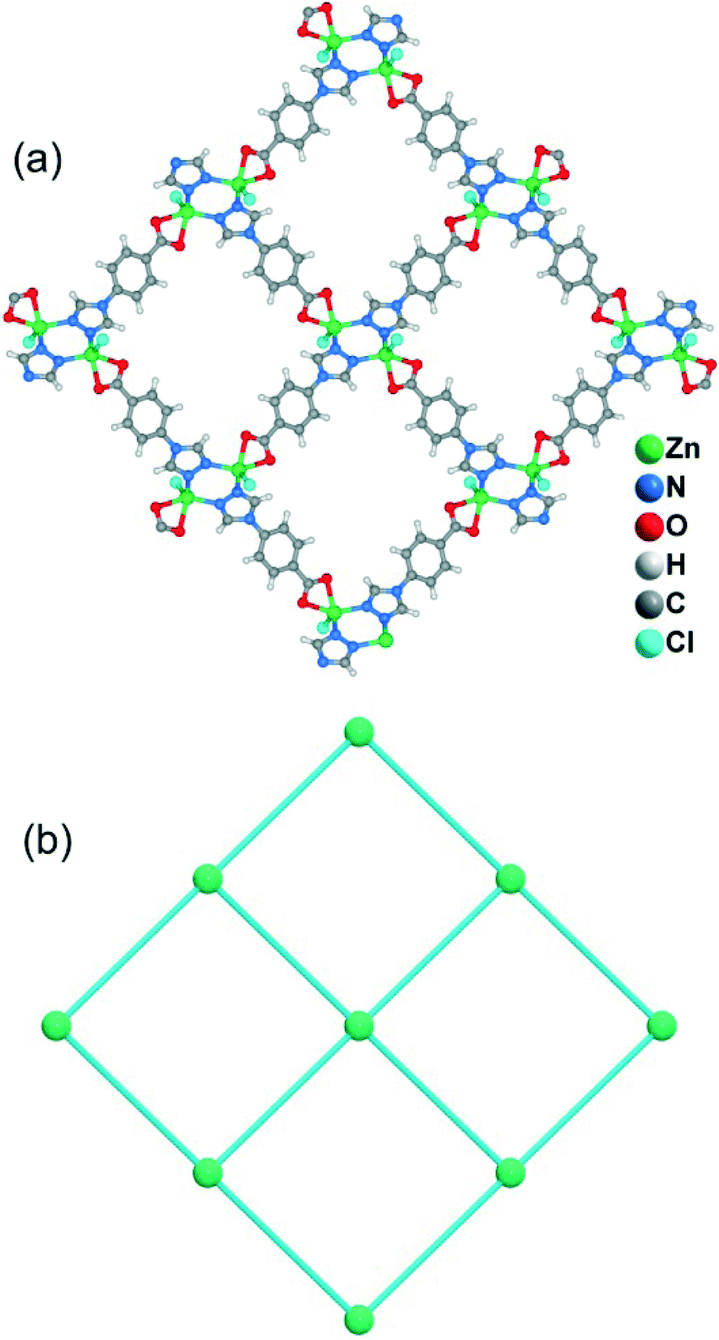
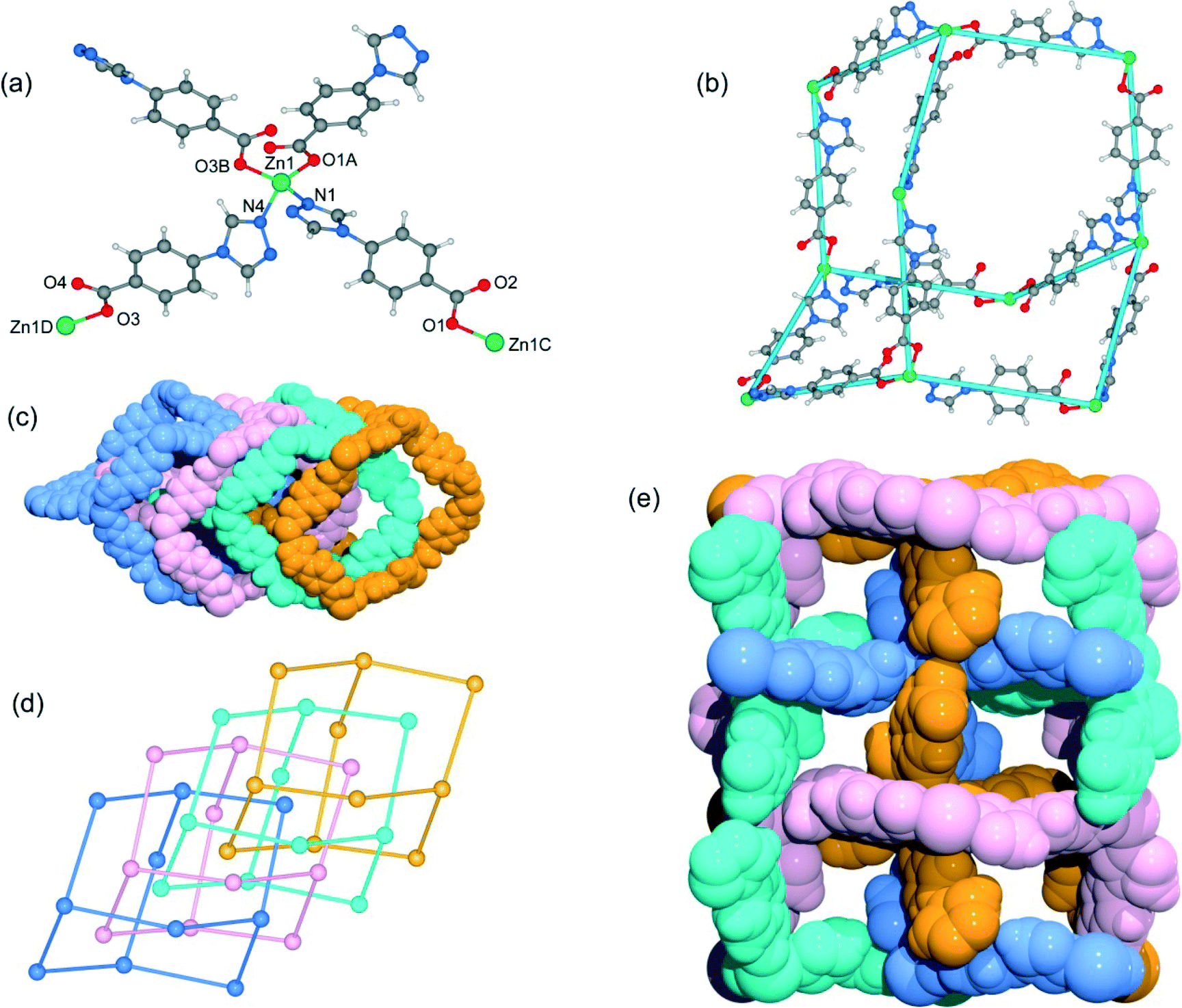
Although the framework of compound 4 has been previously reported, it needs triethylamine and HNO3 as raw reaction materials.16a Herein, the framework of 4 can be prepared by slow evaporation of the filtrate of 3 or by using DMA as the reaction solvent at 95 °C. Compound 4 crystallizes in an orthorhombic system with the space group Pbcn. The asymmetric unit contains one Zn2+ ion and two tba ligands. Being similar to 3, the Zn(II) atom is also four-coordinated to two oxygen atoms from two symmetry-related 4-tba ligands and two nitrogen atoms from two other 4-tba ligands (Fig. 5a). Although the 4-tba ligands also act as bidentate, linear linkers, they adopt different coordination geometries (Fig. 2d) with the oxygen and nitrogen in different orientations. Each Zn(II) atom is connected to four adjacent Zn(II) atoms through four linear 4-tba linkers resulting in a 3D diamond-like framework with the point symbol 66 and the long symbol 62·62·62·62·62·62, typical of a dia topology, containing large adamantanoid cages (Fig. 5b). The Zn⋯Zn distances separated by 4-tba ligands are 12.21 and 12.28 Å, and the Zn⋯Zn⋯Zn angles range from 89.77° to 119.60°, which is a significant deviation from the value of 109.5° expected for an idealized diamond network. A single adamantanoid framework possesses a large cavity and causes four independent equivalent cages to interpenetrate with each other (Fig. 5c and d). The interpenetration mode of the four independent nets is the so-called “normal” type for diamondoid frames, which belongs to class Ia. Despite even 4-fold interpenetration, 4 displays a 1D porous framework with rhombic channels along the b-axis (Fig. 5e and S14,† effective aperture is 2.2 × 2.9 Å2). The total void volume of the channel is estimated to be 2136.8 Å3, approximately 39.6% of the total crystal volume of 5391.6 Å3.
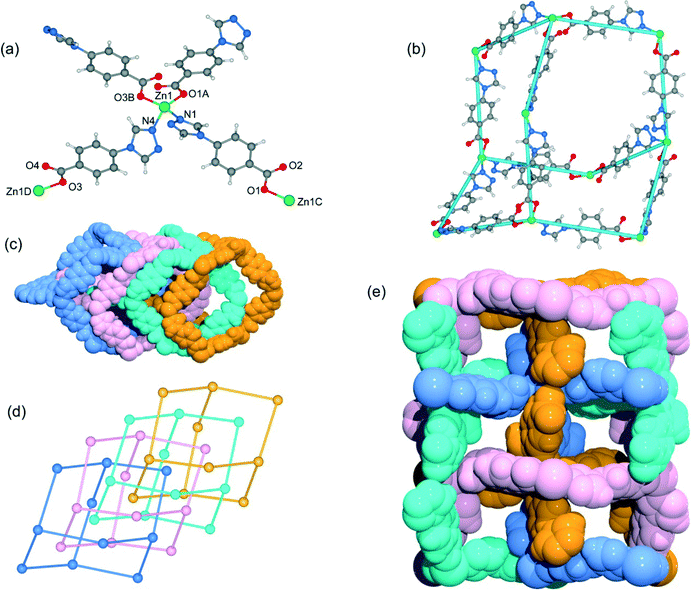 |
| | Fig. 5 (a) The coordination environment of the zinc atoms and 4-tba ligands in 4. Symmetry codes: A: x − 1/2, −y + 1/2, −z + 2; B: −x + 1/2, −y − 1/2, z + 1/2; C: x + 1/2, −y + 1/2, −z + 2; D: −x + 1/2, −y − 1/2, z − 1/2. (b) A single diamond motif in 4. (c) Space-filling diagram of the 4-fold interpenetrating adamantanoid cages in 4. (d) Topological representation of the 4-fold interpenetration in 4. (e) Space-filling diagram of the 3D framework of 4 viewed along the b-axis. | |
3.5. Crystal structure of [Cd(4-tba)2]·DMF·3.6H2O (5)
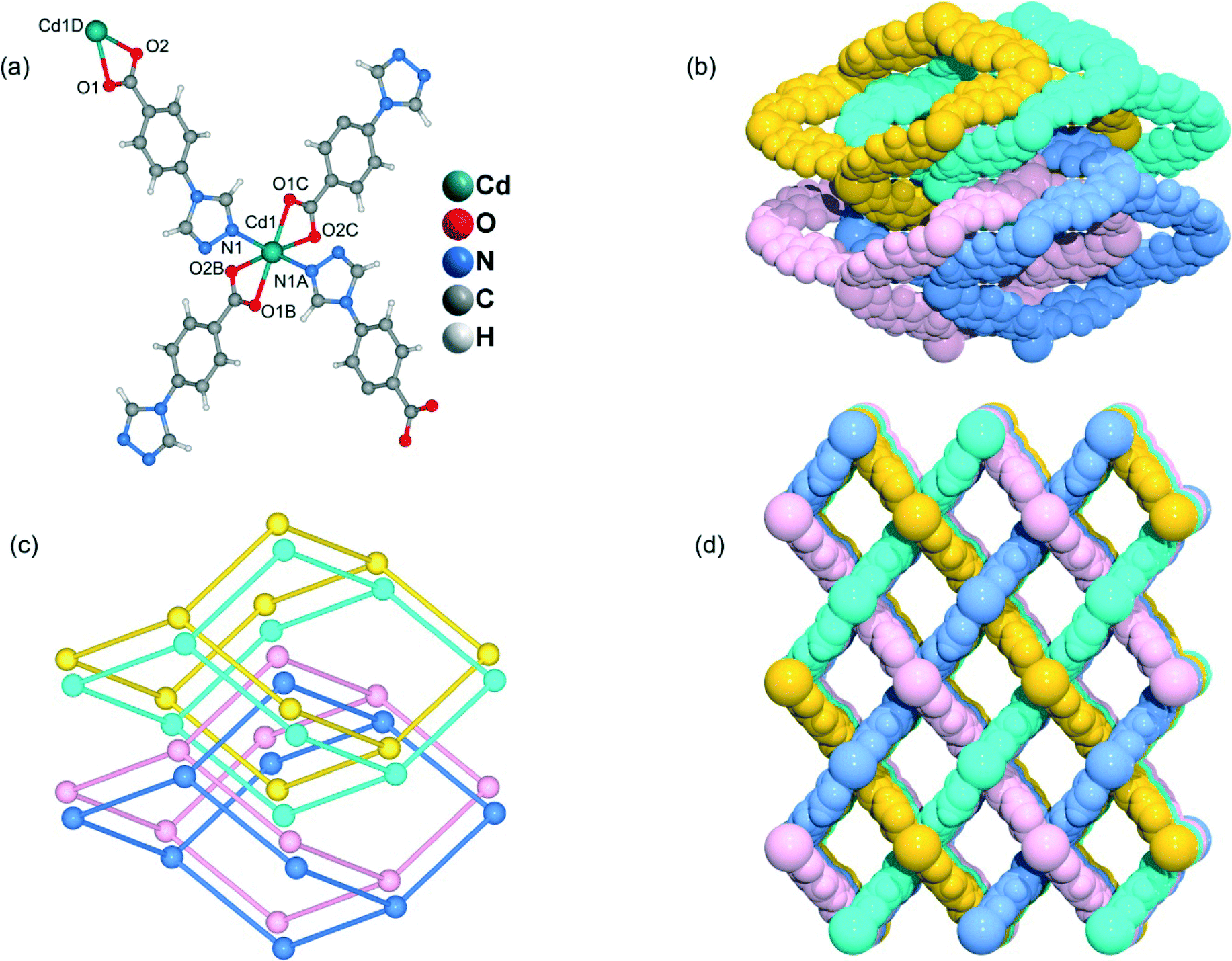
Heating a mixture of Cd(NO3)2·4H2O and 4-Htba in DMF (5 mL) at 95 °C for 72 h afforded pure-phase 5 as colorless block crystals. Compound 5 crystallizes in an orthorhombic system with the space group Pnc2, and the asymmetric unit contains one half of a Cd2+ ion and one tba ligand as shown in Fig. 6a. In 5, the Cd1 center with the O4N2 binding set is coordinated by two pairs of oxygen atoms of chelating carboxylate groups from two symmetry-related 4-tba ligands and two N atoms from two other 4-tba ligands (Fig. 6a), displaying a highly distorted octahedral coordination sphere. Each 4-tba ligand exhibits μ2-coordination modes to connect two cadmium(II) ions through one N atom from the triazolyl group and two O atoms from one carboxylate group with a μ1-η1:η1 chelating mode (Fig. 2e). Given that the chelating carboxylates are treated as one connecting point, each Cd(II) atom is connected to four adjacent Cd(II) atoms through four linear 4-tba linkers resulting in a uninodal 3D 4-connected dia (66) topology (Fig. S15†). The Cd⋯Cd distances separated by 4-tba ligands are 12.58 Å, and the Cd⋯Cd⋯Cd angles range from 96.37° to 125.61°, which is a significant deviation from the value of 109.5° expected for an idealized diamond network. The single 3D network also consists of large windows, which are filled via mutual interpenetration of four independent equivalent networks (Fig. 6b). Interestingly, their mode of interpenetration is different from the normal mode and may be described as two sets of normal 2-fold nets, that is, an unusual [2 + 2] mode of interpenetration (Fig. 6c). Analysis of the topology of interpenetration according to recent classification reveals that this interpenetration in 5 belongs to class IIIa.23 To the best of our knowledge, among those unusual interpenetrating examples, 4-fold interpenetrating dia MOFs with a [2 + 2] mode are relatively scarce.24 Despite even 4-fold interpenetration, 5 still possesses large square channels with an effective pore diameter of 6.4 Å along the a direction (Fig. 6d and S16†). The total void volume of the channel is estimated to be 628.7 Å3, approximately 44.1% of the total crystal volume of 1427.1 Å3.
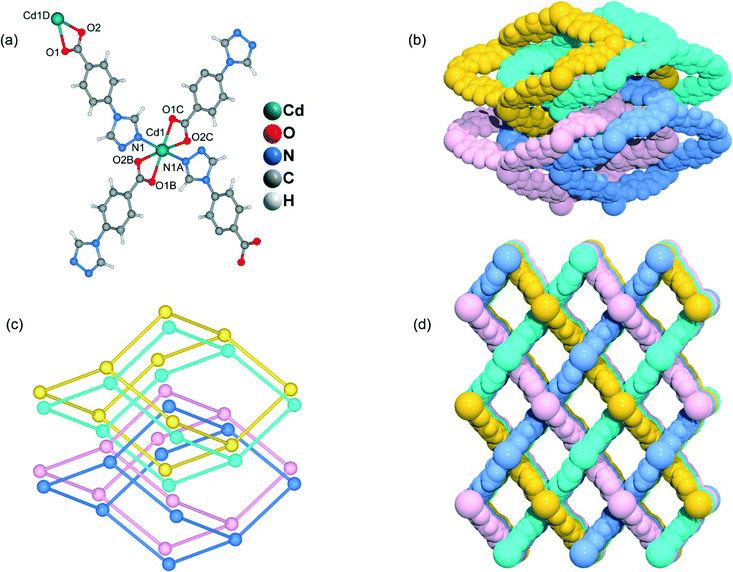 |
| | Fig. 6 (a) The coordination environment of the cadmium atoms and 4-tba ligands in 5. Symmetry codes: A: −x + 1, −y, z; B: x + 1, −y − 1/2, z − 1/2; C: −x, y + 1/2, z − 1/2; D: x − 1, −y − 1/2, z + 1/2. (b) Space-filling diagram of the 4-fold interpenetrating adamantanoid cages in 5. (c) Topological representation of the 4-fold interpenetration in 5. (d) Space-filling diagram of the 3D framework of 5 viewed along the b-axis. | |
3.6. Crystal structure of [Cd(4-tba)2] (6)
Heating a mixture of Cd(NO3)2·4H2O, 4-Htba and 4,4′-bipyridine in the mixed solvent of DMF/MeOH at 120 °C afforded colorless block crystals of 6. Compound 6 exhibits a 5-fold interpenetrated dia topological net. In contrast to 5, 6 crystallizes in the orthorhombic space group Fdd2 rather than Pnc2 in 5 (Table 1). Being similar to 5, the asymmetric unit also contains one half of a Cd2+ ion and one tba ligand (Fig. 7a) and each Cd atom is also coordinated by two pairs of oxygen atoms of chelating carboxylate groups from two symmetry-related 4-tba ligands and two N atoms from two other 4-tba ligands. Although each 4-tba ligand in 6 adopts coordination modes (Fig. 2e) similar to those in 5, the dihedral angle between the benzene and triazole rings in 6 (46.13°) is much larger than that in 5 (19.98°). As shown in Fig. S17,† each Cd(II) atom is also connected to four adjacent Cd(II) atoms through four linear 4-tba linkers resulting in a 3D 4-connected dia (66) topology. Notably, the large adamantanoid cages in a single dia network are more distorted than those in 4 and 5, in which the Cd⋯Cd distances separated by 4-tba ligands are 12.31 Å and the Cd⋯Cd⋯Cd angles range from 96.58° to 140.41°. As illustrated in Fig. 7b, the single framework of 6 is interpenetrated by four identical dia nets, giving rise to the final 5-fold interpenetrating dia framework. Being different from 5, the interpenetrating fashion in 6 determined by TOPOS suggests the so-called ‘normal’ type for diamondoid frames, which belongs to class Ia with the interpenetration vector being equal to the c-axis (6.55 Å). Due to 5-fold interpenetration, 6 is a dense structure with no residual solvent-accessible area in the unit cell.
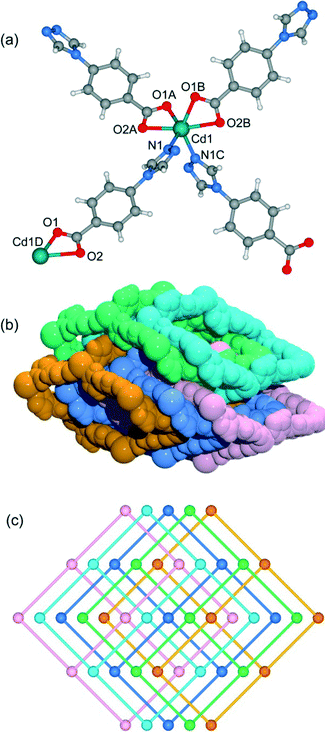 |
| | Fig. 7 (a) The coordination environment of the cadmium atoms and 4-tba ligands in 6. Symmetry codes: A: x − 1/4, −y + 1/4, z − 5/4; B: −x + 1/4, y − 1/4, z − 5/4; C: −x, −y, z; D: x + 1/4, −y + 1/4, z + 5/4. (b) Space-filling diagram of the 5-fold interpenetrating adamantanoid cages in 6. (c) Topological representation of the 5-fold interpenetration in 6. | |
3.7. Crystal structure of [Cd5Cl4(4-tba)6]·1.5DMF·4H2O (7)
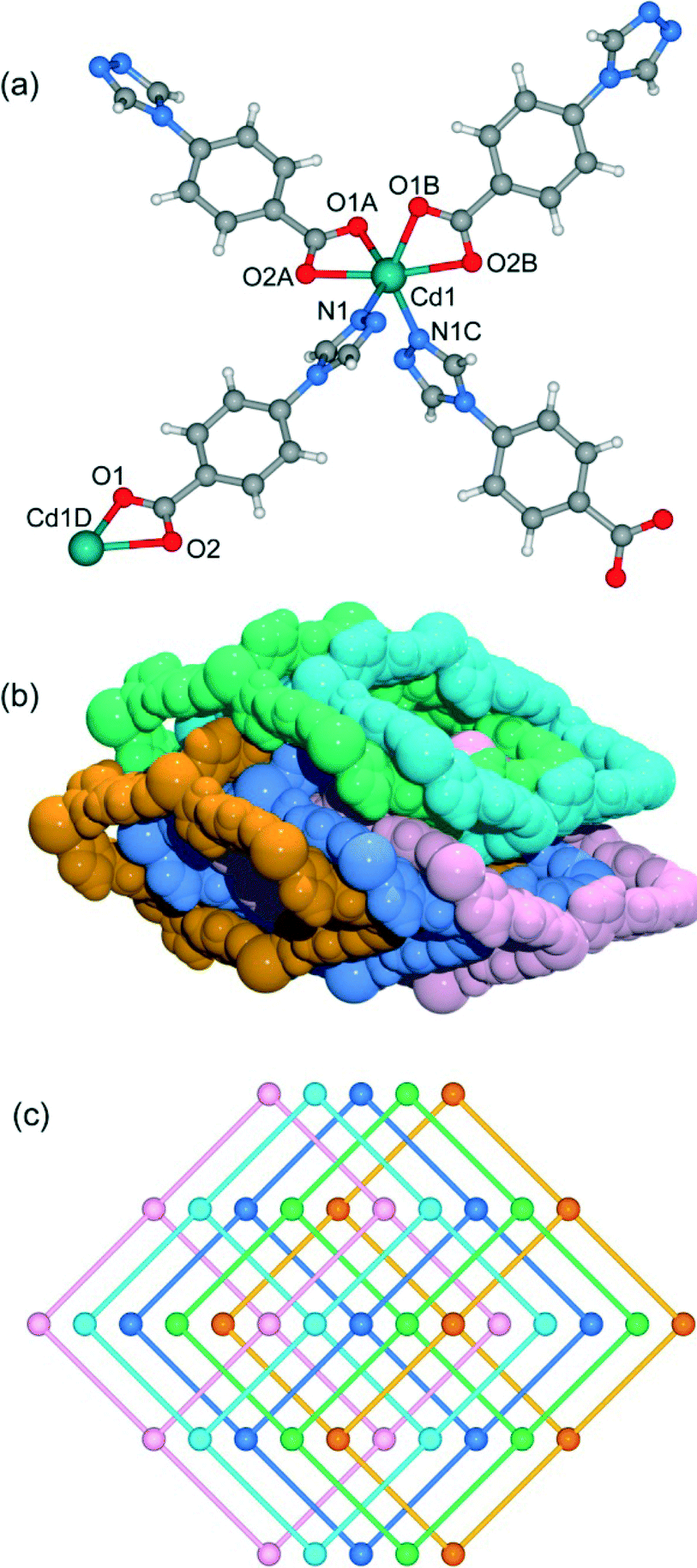
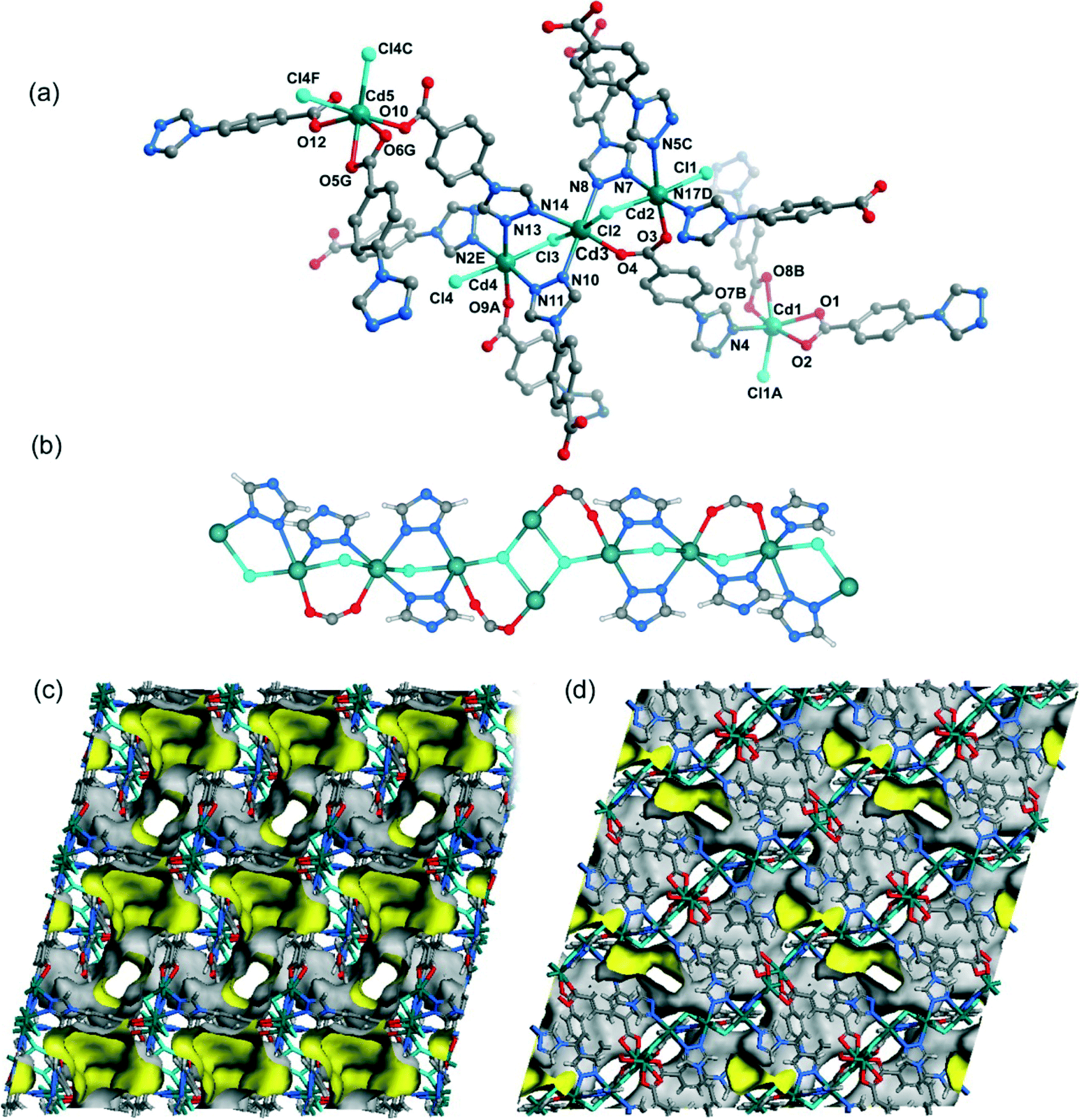
When CdCl2·2.5H2O instead of Cd(NO3)2·4H2O and the mixed solvent of DMF/CH3CN in place of DMF were utilized under similar synthetic conditions to those for 5, a different structure was formed (7). Compound 7 crystallizes in the triclinic space group P. As shown in Fig. 8a, there are five crystallographically independent Cd(II) ions in the asymmetric unit with distorted octahedral coordination geometries. A notable feature of the structure of 7 is that the 4-tba ligands exhibit four coordination modes existing together in one compound: (i) they act as bidentate linkers, using one N atom and one oxygen atom to bridge two Cd(II) ions (Fig. 2d); (ii) they also act as bidentate linkers, using one N atom to bridge one Cd(II) ion and two oxygen atoms to chelate one Cd(II) ion (Fig. 2e); (iii) they adopt μ3-bridging modes, using two N atoms to bridge two Cd(II) ions and two oxygen atoms to chelate one Cd(II) ion (Fig. 2b); (iv) they exhibit μ4-bridging modes to bridge four Cd(II) ions where the carboxylate group bridges two adjacent Cd(II) ions in bis-monodentate syn–syn mode (Fig. 2f). Also interesting is the decanuclear cadmium–chloride chain unit linked by μ2-chloride and μ3-chloride (Fig. 8b). As shown in Fig. 8c and d, the decanuclear cadmium–chloride units are linked by 4-tba ligands with four different coordination modes into a 3D framework with 2D channels in which DMF and water solvates reside. The channels along the a-axis are rectangular with the size of 2.01 × 4.52 Å2, whereas the channels along the c-axis are dumbbell-shaped with the effective aperture of 2.45 × 6.38 Å2. The cavity volume is estimated to be ca. 38.4% of the total crystal volume, calculated using the PLATON program after guest removal. When μ2-chloride and μ3-chloride are regarded as rod and 3-connected nodes, respectively, and the 4-tba ligands are regarded as 2-, 3- and 4-connected nodes, the 3D structure of 7 can be rationalized as an unusual 10-nodal (3,3,3,4,4,4,5,6,6,6)-connected topology (Fig. S18†).
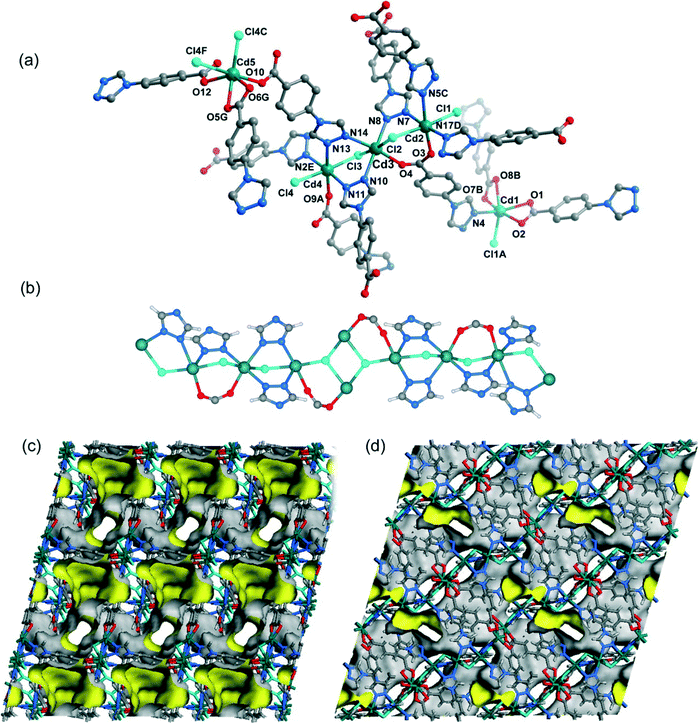 |
| | Fig. 8 (a) The coordination environment of the cadmium atoms in 7. The hydrogen atoms are omitted for clarity. Symmetry codes: A: x + 1, y, z; B: x + 1, y − 1, z; C: x − 1, y, z; D: x + 1, y, z − 1; E: x − 1, y, z + 1; F: −x − 1, −y + 2, −z + 1; G: x − 1, y + 1, z. (b) The decanuclear cadmium–chloride chain unit linked by μ2-chloride and μ3-chloride in 7. (c) Views of the 3D framework and channel structure of 7 along the c-axis. (d) Views of the 3D framework and channel structure of 7 along the a-axis. | |
3.8. Synthesis of the complexes and comparison of the structures
Compounds 1–7 were synthesized under hydro/solvothermal conditions. In general, the reaction variables (solvent, metal salt species, concentration, temperature, pH value, anion, etc.) can influence the reaction process and the final products. Solvent as the template is one of the key factors for the construction of metal complexes because solvents can act as space-filling molecules.9 Complexes 1–3 with different structures were obtained under the same reaction conditions by using different reaction solvents. Besides the solvent effect, reaction temperature is another key factor in controlling the topology and dimensionality of the frameworks by thermal desolvation and changing the coordination modes. The mononuclear structure of 1 and dense structures of 2, 3 and 6 were obtained at 120 °C, while the porous structures of 4, 5 and 7 were crystallized at a low temperature of 95 °C. Moreover, the framework of 4 can also be obtained by slow evaporation of the filtrate of 3 (verified by PXRD, Fig. S4†), which demonstrates that the concentration also influences the formation of 3 and 4. It should be pointed out that 4,4′-bipyridine plays a key role in the synthesis of 6, and it cannot be obtained without 4,4′-bipyridine. Thus, 4,4′-bipyridine may act as a weak base to deprotonate 4-Htba and provide automatic control of the acid/base balance of the solution. Comparing 5 with 7, we find that the anion also influences the formation of the network besides the solvent effect, and the chloride anion from CdCl2·2.5H2O can be involved in coordinating metal ions. Also, the nature of different metal centers is a key factor for the construction of diverse frameworks, and different coordination geometries around ZnII/CdII centers resulted in completely different structures for the two series of ZnII/CdII complexes 1–7. Compared to the ionic radius of the Zn(II) ion (74 pm), that of the disparate Cd(II) metal ion (97 pm) is relatively larger; as a result, it has a tendency to adopt higher coordination numbers to accommodate more bulkiness in its coordination sphere, thereby forming complicated or higher-dimensional structures.
On the other hand, the 4-Htba ligand involving bifunctional carboxylate and triazolyl groups can exhibit diverse coordination modes as shown in Fig. 2. The carboxylate group of 4-Htba in all complexes is deprotonated to give a 4-tba anion to ligate with a bivalent metal ion. The benzene and triazole rings in the 4-tba anion ligand could freely rotate along the C–N bonds to adjust themselves to match with the coordination preferences. The dihedral angles α between the benzene rings and triazole rings were calculated and are compiled in Table 2. As is well known, the different coordination modes of the 4-tba ligand always bring about different dihedral angles α. For example, the dihedral angles α in complexes 1–4 can range from 24.84 to 42.35°. However, although the 4-tba ligands in 5 and 6 take similar coordination modes (Fig. 2e), the dihedral angles α can be largely different due to the different crystal packing modes: 5 is a [2 + 2] interpenetrating dia net belonging to class IIIa, whereas 6 is a 5-fold interpenetrating dia net of class Ia. Compound 5 shows the smallest dihedral angles α among all the complexes 1–7. Furthermore, the π–π stacking interactions in these compounds also vary differently. Compounds 1–3 possess significant offset face-to-face π–π stacking interactions, whereas there are almost negligible aromatic stacking interactions existing in 4–7 because their long centroid−centroid distances are beyond 4.0 Å (Table S2†).
Table 2 Dihedral angles between the benzene and triazole rings in 1–7
| Compound |
α/° |
Coordination modes |
|
1
|
24.84 |
(a) |
|
2
|
41.70 |
(b) |
|
3
|
40.34 |
(c) |
|
4
|
39.56, 42.35 |
(d) |
|
5
|
19.98 |
(e) |
|
6
|
46.13 |
(e) |
|
7
|
28.26–50.53 |
(b), (d)–(f) |
3.9. Powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA)
In order to confirm the phase purity of these compounds, powder X-ray diffraction (PXRD) patterns of 1–7 were obtained at room temperature. As shown in Fig. S1–S7,† the peak positions of the theoretical and experimental PXRD patterns are in agreement with each other, indicating that single phases of 1–7 are formed.
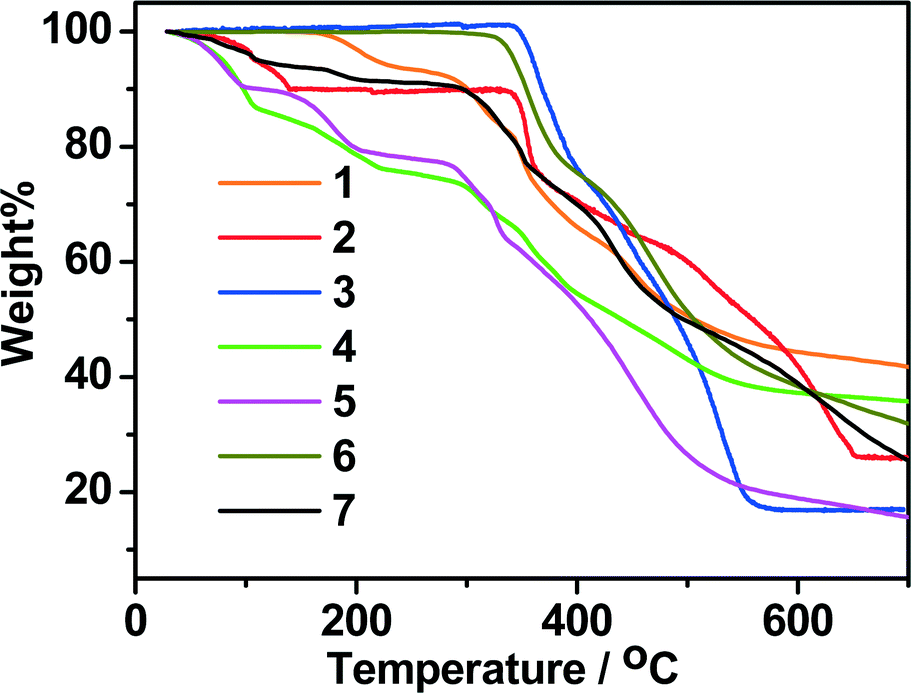
Thermogravimetric analyses (TGA) were carried out to study the thermal stabilities of 1–7. As shown in Fig. 9, in general, the mononuclear structure of 1 and the porous structures of 4, 5 and 7 can be stable below 280 °C. The 2D structure of 2 and the dense structures of 3 and 6 can be stable up to 315 °C, and thus are much more stable than the mononuclear structure (1) and the porous structures (4, 5 and 7). In the TG curve of 1, coordinated water molecules were lost from 100 to 275 °C (calcd.: 7.54%, found: 7.49%). Above 275 °C, the sample suffered an abrupt weight loss, indicating the collapse of the remaining substance. For 2, the lattice methanol molecules were released from 40 to 145 °C (calcd.: 9.98%, found: 10.17%). The framework was stable upon heating to 325 °C, and then the gradual and rapid weight decrease occurred. For 3, it can be stable under 335 °C and decomposed at higher temperature. For 4, the weight loss of 25.32% at temperatures below 270 °C is consistent with the removal of one molecule of DMA and 3.5 molecules of H2O per formula unit (calcd.: 5.37%), and further weight loss indicates the decomposition of the framework from 280 °C. For 5, the weight loss of 22.05% at temperatures below 250 °C is consistent with the removal of one molecule of DMF and 3.6 molecules of H2O per formula unit (calcd.: 22.01%). In the case of compound 6, no obvious weight loss was observed until the temperature rose to 315 °C. The TG curve of 7 shows a steady weight loss involving water and DMF molecules below 250 °C (observed: 8.90%, calculated: 9.02%), and the remaining substance begins to decompose from 274 °C.
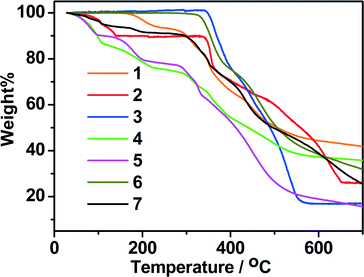 |
| | Fig. 9 TG curves of 1–7 in N2. | |
3.10. Photoluminescence properties
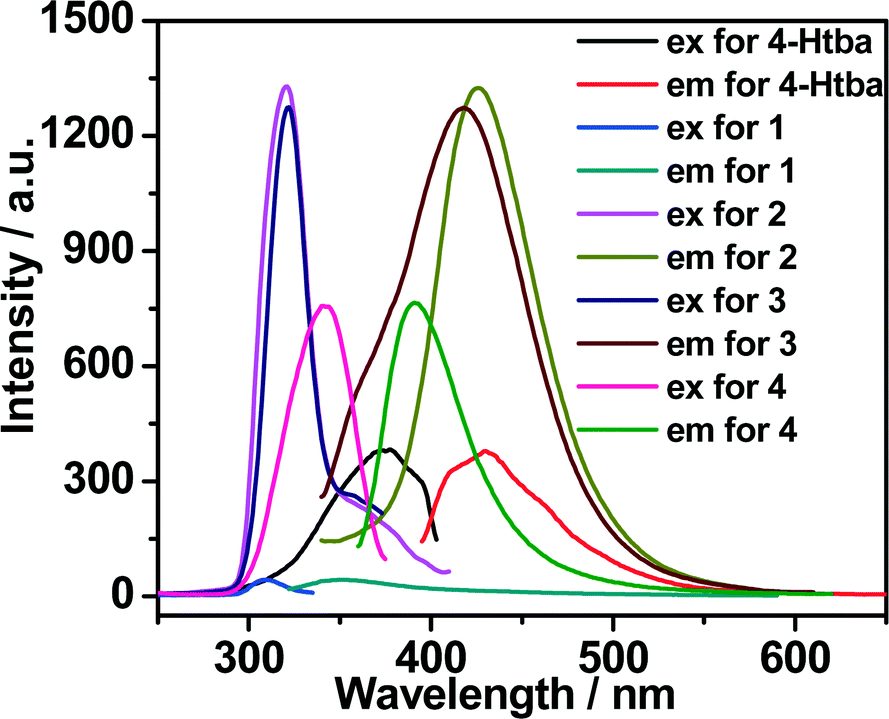
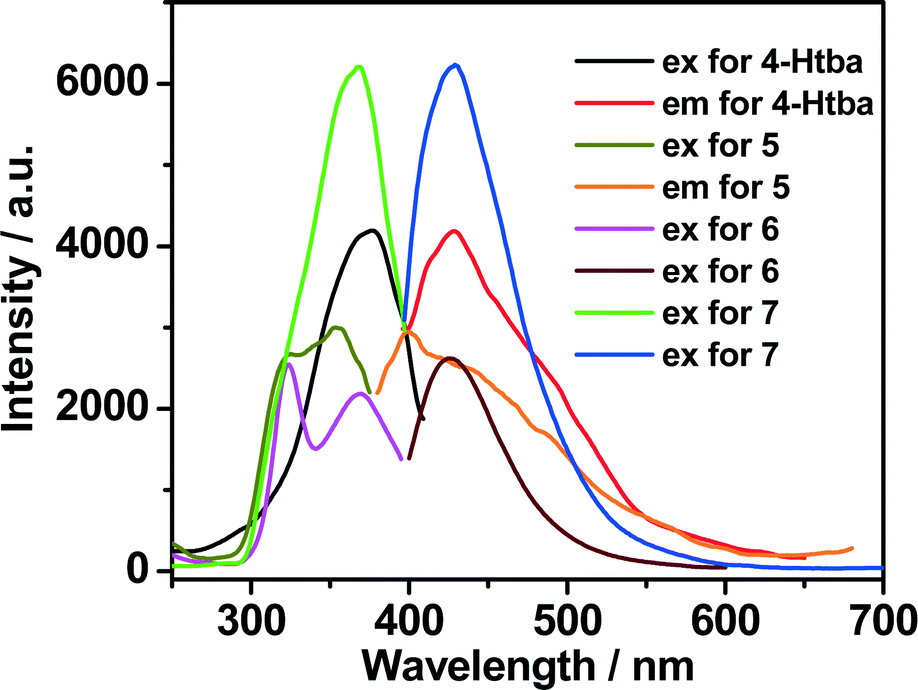
Luminescent MOFs with d10 metal ions are of great interest due to their various applications in photochemistry, chemical sensors and light emitting diodes. Therefore, the solid-state luminescence of the free ligand Htba and compounds 1–7 is investigated at room temperature, as depicted in Fig. 10, 11 and Table 3. The main emission peaks of 4-Htba were observed at 427 nm (λex = 377 nm), which can be assigned to π* → n or π* → π transitions.24 Compounds 1–7 emit purple or bluish-purple emission with maximum emission peaks at 351 nm, 426 nm, 418 nm, 391 nm, 400 nm, 425 nm and 429 nm if excited at 309 nm, 321 nm, 322 nm, 343 nm, 353 nm, 324 nm and 369 nm, respectively. Since the Zn(II) and Cd(II) ions are difficult to oxidize or reduce due to their d10 configurations, the emission of these complexes is neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature. Thus, the emission of 1–7 is probably attributed to the intraligand transitions modified by metal coordination. In contrast to the free ligand (4-Htba), the fluorescence spectra of compounds 2–4 and 7 show a significant enhancement in intensity, and those of 1 and 3–5 exhibit blue shifts of 76, 9, 36 and 27 nm, respectively. Their different emission behaviours may mainly originate from the different metal centers, the coordination modes of the ligands, the coordination angles of the ligands, the dihedral angles between the benzene and triazole rings as well as the weak interactions in the network lattice, which have close relationships to the photoluminescence behaviour. Furthermore, 1 displays a very weak emission intensity in comparison to the organic ligand and other zinc complexes, which can be assigned to the occurrence of fluorescence quenching in 1. The fluorescence quenching may be due to the coordinated water molecules which may efficiently quench the fluorescence of compounds through high-energy O–H oscillators.25 Besides, the crystal densities of dense structures 2 (1.784 g cm−3), 3 (1.660 g cm−3) and 6 (1.813 g cm−3) are also markedly higher than those of porous structures 4 (1.458 g cm−3), 5 (1.458 g cm−3) and 7 (1.508 g cm−3), which implies interligand contacts (Table S2†) and/or network rigidities in the order of dense structure > porous structure. Therefore, the larger Stokes shifts of dense structures 2 (105 nm), 3 (96 nm) and 6 (101 nm) may be ascribed to the strong interligand contacts and/or the network rigidities changing the HOMO–LUMO gaps of intraligand transitions, which increase the excited state distortion and nonradiative transitions.
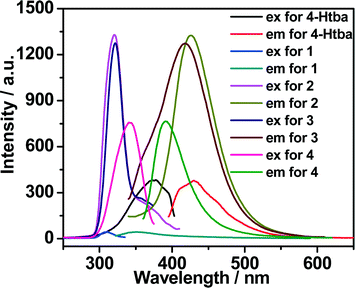 |
| | Fig. 10 The excitation and emission spectra of Zn(II) complexes 1–4 in the solid state at room temperature (excitation slit: 2.5, emission slit: 1.0). | |
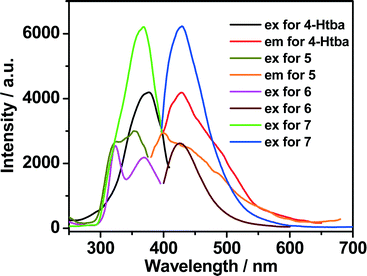 |
| | Fig. 11 The excitation and emission spectra of Cd(II) complexes 5–7 in the solid state at room temperature (excitation slit: 5.0, emission slit: 2.5). | |
Table 3 Summary of solid-state photophysical data for 1–7
| Compound |
Crystal density/g cm−3 |
λ
ex/nm |
λ
max/nm |
Blue shifts compared to 4-Htba/nm |
Stokes shifts/nm |
| 4-Htba |
— |
377 |
427 |
— |
50 |
|
1
|
1.754 |
309 |
351 |
76 |
53 |
|
2
|
1.784 |
321 |
426 |
1 |
105 |
|
3
|
1.660 |
322 |
418 |
9 |
96 |
|
4
|
1.458 |
343 |
391 |
36 |
48 |
|
5
|
1.458 |
353 |
400 |
27 |
47 |
|
6
|
1.813 |
324 |
425 |
2 |
101 |
|
7
|
1.508 |
369 |
429 |
−2 |
60 |
Conclusions
In conclusion, a series of Zn(II)/Cd(II) complexes have been synthesized under hydro/solvothermal conditions by using a triazolate–carboxylate bifunctional ligand, and their structures vary from a discrete mononuclear structure, a two-dimensional (2D) non-interpenetrating layer, a 2D 2-fold interpenetrating layer, a 4-fold interpenetrating dia network and a 5-fold interpenetrating dia framework to a 3D, decanuclear cadmium–chloride chain-based porous framework. The diverse structures of the compounds are caused by the different coordination modes of the central metal ions and the organic ligands as well as the different reaction conditions. In addition, the thermal stability and photoluminescence properties of these complexes have been investigated, and it was revealed that the dense structures 2, 3 and 6 display higher thermal stability and bluish-purple emissions with larger Stokes shifts, which may serve as candidates for luminescent materials.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21261028, 21162043 and 21367026) as well as the Science and Technology Planning Project of Yunnan Province (2010ZC070, 2011FZ080 and 2012FB141). We also thank Professor Xiao-Ming Chen at Sun Yat-sen University for kindly providing laboratory assistance.
Notes and references
-
(a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed;
(b) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed;
(c) M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 CrossRef CAS PubMed.
-
(a) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed;
(b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed;
(c) R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703 CrossRef CAS PubMed;
(d) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657 RSC;
(e) H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836 CrossRef CAS PubMed.
-
(a) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC;
(b) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed;
(c) B. V. Voorde, B. Bueken, J. Denayerb and D. D. Vos, Chem. Soc. Rev., 2014, 43, 5766 RSC.
-
(a) T. Maji, R. Matsuda and S. Kitagawa, Nature, 2007, 6, 142 CrossRef CAS PubMed;
(b) C. K. Brozek and M. Dinč, Chem. Soc. Rev., 2014, 43, 5456 RSC;
(c) B. Manna, S. Singh, A. Karmakar, A. V. Desai and S. K. Ghosh, Inorg. Chem., 2015, 54, 110 CrossRef CAS PubMed.
-
(a) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC;
(b) A. Corma, H. García and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed;
(c) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed;
(d) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750 RSC;
(e) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC.
-
(a) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC;
(b) P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249 RSC;
(c) E. Coronado and G. M. Espallargas, Chem. Soc. Rev., 2013, 42, 1525 RSC;
(d) W.-X. Zhang, P.-Q. Liao, R.-B. Lin, Y.-S. Wei, M.-H. Zeng and X.-M. Chen, Coord. Chem. Rev., 2015, 293–294, 263 CrossRef CAS.
-
(a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed;
(b) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(c) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
-
(a) M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282 CrossRef CAS;
(b) F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088 RSC;
(c) W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561 RSC.
-
(a) H.-L. Jiang, T. A. Makal and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232 CrossRef CAS;
(b) A. Santra and P. K. Bharadwaj, Cryst. Growth Des., 2014, 14, 1476 CrossRef CAS;
(c) N. Wei, M.-Y. Zhang, X.-N. Zhang, G.-M. Li, X.-D. Zhang and Z.-B. Han, Cryst. Growth Des., 2014, 14, 3002 CrossRef CAS;
(d) Z. Ju and D. Yuan, CrystEngComm, 2013, 15, 9513 RSC;
(e) Y.-F. Hou, B. Liu, K.-F. Yue, C.-S. Zhou, Y.-M. Wang, N. Yan and Y.-Y. Wang, CrystEngComm, 2014, 16, 9560 RSC;
(f) J. Duan, M. Higuchi, C. Zou, W. Jin and S. Kitagawa, CrystEngComm, 2015, 17, 5609 RSC.
-
(a) B. Zheng, J. H. Luo, F. Wang, Y. Peng, G. H. Li, Q. S. Huo and Y. L. Liu, Cryst. Growth Des., 2013, 13, 1033 CrossRef CAS;
(b) J. Bai, H.-L. Zhou, P.-Q. Liao, W.-X. Zhang and X.-M. Chen, CrystEngComm, 2015, 17, 4462 RSC;
(c) Y.-F. Peng, S. Zhao, K. Li, L. Liu, B.-L. Li and B. Wu, CrystEngComm, 2015, 17, 2544 RSC.
-
(a) Y. Yu, L. Zhang, Y. Zhou and Z. Zuhra, Dalton Trans., 2015, 44, 4601 RSC;
(b) T.-W. Tseng, T.-T. Luo, C.-C. Su, H.-H. Hsu, C.-I. Yang and K.-L. Lu, CrystEngComm, 2014, 16, 2626 RSC;
(c) S. Yuan, S.-S. Liu and D. Sun, CrystEngComm, 2014, 16, 1927 RSC;
(d) L. Wang, Z.-H. Yan, Z. Xiao, D. Guo, W. Wang and Y. Yang, CrystEngComm, 2013, 15, 5552 RSC.
-
(a) H.-L. Jiang and Q. Xu, CrystEngComm, 2010, 12, 3815 RSC;
(b) Z. Su, Y. Zhao, M. Chen and W.-Y. Sun, CrystEngComm, 2011, 13, 1539 RSC;
(c) Z. Xue, T. Sheng, Y. Wen, Y. Wang, S. Hu, R. Fu and X. Wu, CrystEngComm, 2015, 17, 598 RSC;
(d) M. E. Carnes, N. R. Lindquist, L. N. Zakharov and D. W. Johnson, Cryst. Growth Des., 2012, 12, 1579 CrossRef CAS.
-
(a) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed;
(b) F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088 RSC;
(c) Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618 RSC;
(d) G. Aromí, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485 CrossRef;
(e) J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001 CrossRef CAS PubMed.
-
(a) Z. Su, Y. Zhao, M. Chen and W.-Y. Sun, CrystEngComm, 2011, 13, 1539 RSC;
(b) C.-T. He, J.-Y. Tian, S.-Y. Liu, G. Ouyang, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 351 RSC;
(c) P. Pachfule, Y. Chen, S. C. Sahoo, J. Jiang and R. Banerjee, Chem. Mater., 2011, 23, 2908 CrossRef CAS;
(d) F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833 CrossRef CAS PubMed;
(e) D.-X. Xue, A. J. Cairns, Y. Belmabkhout, L. Wojtas, Y. Liu, M. H. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2013, 135, 7660 CrossRef CAS PubMed;
(f) Q. Zhang, H. Zhang, S. Zeng, D. Sun and C. Zhang, Chem. – Asian J., 2013, 8, 1985 CrossRef CAS PubMed;
(g) Q. Zhang, A. Geng, H. Zhang, F. Hu, Z.-H. Lu, D. Sun, X. Wei and C. Ma, Chem. – Eur. J., 2014, 20, 4885 CrossRef CAS PubMed.
-
(a) M. Chen, S.-S. Chen, T.-A. Okamura, Z. Su, M.-S. Chen, Y. Zhao, W.-Y. Sun and N. Ueyama, Cryst. Growth Des., 2011, 11, 1901 CrossRef CAS;
(b) T. Panda, P. Pachfule and R. Banerjee, Chem. Commun., 2011, 47, 7674 RSC;
(c) T. Panda, T. Kundu and R. Banerjee, Chem. Commun., 2012, 48, 5464 RSC;
(d) J. Xiao, Y. Wu, M. Li, B.-Y. Liu, X.-C. Huang and D. Li, Chem. – Eur. J., 2013, 19, 1891 CrossRef CAS PubMed;
(e) T. Li, J. Yang, X.-J. Hong, Y.-J. Ou, Z.-G. Gu and Y.-P. Cai, CrystEngComm, 2014, 16, 3848 RSC;
(f) Y.-L. Wang, J.-H. Fu, J.-J. Wei, X. Xu, X.-F. Li and Q.-Y. Liu, Cryst. Growth Des., 2012, 12, 4663 CrossRef CAS;
(g) M. Du, C.-P. Li, M. Chen, Z.-W. Ge, X. Wang, L. Wang and C.-S. Liu, J. Am. Chem. Soc., 2014, 136, 10906 CrossRef CAS PubMed;
(h) W.-Y. Gao, R. Cai, L. Meng, L. Wojtas, W. Zhou, T. Yildirim, X. Shi and S. Ma, Chem. Commun., 2013, 49, 10516 RSC;
(i) S. I. Vasylevs'kyy, G. A. Senchyk, A. B. Lysenko, E. B. Rusanov, A. N. Chernega, J. Jezierska, H. Krautscheid, K. V. Domasevitch and A. Ozarowski, Inorg. Chem., 2014, 53, 3642 CrossRef PubMed;
(j) A.-X. Zhu, Z.-Z. Qiu, L.-B. Yang, X.-D. Fang, S.-J. Chen, Q.-Q. Xu and Q.-X. Li, CrystEngComm, 2015, 17, 4787 RSC.
-
(a) J.-F. Song, R.-S. Zhou, J. Zhang, C.-Y. Xu, Y.-B. Li and B.-B. Wang, Z. Anorg. Allg. Chem., 2011, 637, 589 CrossRef CAS;
(b) T. Aharen, F. Habib, I. Korobkov, T. J. Burchell, R. Guillet-Nicolas, F. Kleiz and M. Murugesu, Dalton Trans., 2013, 42, 7795 RSC;
(c) R. J. Holmberg, M. Kay, I. Korobkov, E. Kadantsev, P. G. Boyd, T. Aharen, S. Desgreniers, T. K. Woo and M. Murugesu, Chem. Commun., 2014, 50, 5333 RSC;
(d) D.-M. Chen, W. Shi and P. Cheng, Chem. Commun., 2015, 51, 370 RSC;
(e) D.-M. Chen, J.-G. Ma and P. Cheng, Dalton Trans., 2015, 44, 8926 RSC.
-
G. M. Sheldrick, SADABS, Program for Rigaku R-AXIS SPIDER Diffractometer Absorption Correction, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
-
T. Higashi, ABSCOR, Program for Bruker Area Detector Absorption Correction, Rigaku Corporation, Tokyo, Japan, 1995 Search PubMed.
-
G. M. Sheldrick, SHELXS-97 and SHELXL-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
-
(a) S. Xu, W. Shao, M. Yu and G. Gong, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m728 CAS;
(b) L. V. Lukashuk, A. B. Lysenko, E. B. Rusanov, A. N. Chernega and K. V. Domasevitch, Acta Crystallogr., Sect. C: Struct. Chem., 2007, 63, m140 CAS.
- The trigonality index τ (τ = 0 denotes ideal square pyramidal; τ = 1 denotes ideal trigonal bipyramidal) was calculated according to the literature, see: A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
-
(a) K.-H. Cui, S.-Y. Yao, H.-Q. Li, Y.-T. Li, H.-P. Zhao, C.-J. Jiang and Y.-Q. Tian, CrystEngComm, 2011, 13, 3432 RSC;
(b) S. N. Wang, R. R. Yun, Y. Q. Peng, Q. F. Zhang, J. Lu, J. M. Dou, J. F. Bai, D. C. Li and D. Q. Wang, Cryst. Growth Des., 2012, 12, 79 CrossRef CAS;
(c) S.-S. Chen, Q. Liu, Y. Zhao, R. Qiao, L.-Q. Sheng, Z.-D. Liu, S. Yang and C.-F. Song, Cryst. Growth Des., 2014, 14, 3727 CrossRef CAS.
-
(a) H. He, F. Sun, H. Su, J. Jia, Q. Li and G. Zhu, CrystEngComm, 2014, 16, 339 RSC;
(b) Y. Yang, J. Yang, P. Du, Y.-Y. Liu and J.-F. Ma, CrystEngComm, 2014, 16, 1136 RSC;
(c) K. Liu, Y. Peng, F. Yang, D. Ma, G. Li, Z. Shi and S. Feng, CrystEngComm, 2014, 16, 4382 RSC;
(d) J. Li, T. Sheng, S. Bai, S. Hu, Y. Wen, R. Fu, Y. Huang, Z. Xue and X. Wu, CrystEngComm, 2014, 16, 2188 RSC.
-
(a) D.-L. Yang, X. Zhang, Y.-G. Yao and J. Zhang, CrystEngComm, 2014, 16, 8047 RSC;
(b) B. Chen, Y. Yang, F. Zapata, G. Qian, Y. Luo, J. Zhang and E. B. Lobkovsky, Inorg. Chem., 2006, 45, 8882 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, simulated and experimental X-ray powder diffraction patterns, TGA curve for 4′, selected hydrogen bond parameters for 1 and 2, π–π interactions as well as the topological analysis of 7. CCDC 1413432 for 1, 1413435 for 2, 1413438 for 3, 1422293 for 4, 1413443 for 5, 1413445 for 6 and 1413448 for 7. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01815c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.