DOI:
10.1039/C5CE01769F
(Paper)
CrystEngComm, 2016,
18, 123-129
A systematic study on ternary inclusion crystals consisting of dianilines and three positional isomers of ditoluoyl-L-tartaric acid†
Received
4th September 2015
, Accepted 17th November 2015
First published on 30th November 2015
Introduction
Considerable research has been conducted on the design and synthesis of functional organic crystals. The functions and physical properties of organic crystals depend not only on the structure of the organic molecules, but also on their arrangement in the crystalline state.1 Extensive research in the field of crystal engineering has led to the development of diverse supramolecular synthons to control the molecular arrangement; however, it is still difficult to predict the exact molecular arrangement in the crystalline state.2
Lattice inclusion crystals have attracted much interest because of their applications in fluorescent materials,3 gas sorption,4 and chiral separations.5 Several excellent host molecules have been reported; however, a slight structural modification of a host molecule may result in a different molecular arrangement and loss of inclusion ability. Therefore, it is necessary to investigate a series of molecules by trial and error to find a suitable host molecule for a target guest molecule. Much time and effort are required to prepare good host molecules. On the other hand, supramolecular hosts consisting of two components can be systematically investigated because their inclusion ability can be easily tuned by changing their combination.6 In such cases, the inclusion of guest molecules affords ternary inclusion crystals; however, the rational preparation of such ternary inclusion crystals is still challenging because single and binary crystals can be produced.7
Carboxylic acid–primary amine supramolecular synthons have been used for constructing diverse hydrogen-bonding networks from zero-dimensional (0D) clusters to three-dimensional (3D) extended networks.8 We have reported the synthesis of ternary inclusion crystals from a combination of carboxylic acids, primary amines, and alcohols.9 In particular, the salt of dibenzoyl-L-tartaric acid (1b) and 4,4′-diaminodiphenylmethane (2M) formed a 3D rigid hydrogen-bonding network for the enantioselective inclusion of aromatic and aliphatic alcohols.10 The two aromatic moieties of 1b with a twisted conformation were oriented in a gauche position, and the rigid network generated with diamine 2M was probably responsible for the high inclusion ability of the salt for alcohols. To investigate the structural effect of each component on the supramolecular structure and inclusion ability in detail, three positional isomers of ditoluoyl-L-tartaric acid (o, m, and p-; 1o, 1m, 1p) with the same hydrogen-bonding ability were selected as analogs of 1b. Primary amine 2M and its oxygenated analog (2O) were selected as the amine components because their rotational flexibility around the pivotal central atom is advantageous for high inclusion ability (Scheme 1). Although tartaric acid derivatives 1 have been commonly used for the diastereomeric resolution of amines via crystallization, the crystal structures of their salts have been less explored.11 V-shaped aromatic diamines 2 have been used for constructing various hydrogen-bonding networks by mixing with acidic counterparts.12 Herein, the crystal structures and inclusion abilities of eight combinations of salts obtained from 1 and 2 were investigated, and the effects of their structural changes were evaluated.
 |
| | Scheme 1 Structures of chiral diaroyl-L-tartaric acids (1) and achiral diamines (2). | |
Experimental
Materials and methods
Dibenzoyl-L-tartaric acid (1b), di-(p-toluoyl)-L-tartaric acid (1p), 4,4′-diaminodiphenylmethane (2M), 4,4′-oxydianiline (2O), and aliphatic alcohols were purchased and used as received without further purification. Di-(o-toluoyl)-L-tartaric acid (1o) and di-(m-toluoyl)-L-tartaric acid (1m) were prepared from L-tartaric acid and the corresponding toluoyl chlorides.13 The 1H NMR spectra of the inclusion crystals in CDCl3/CD3OD were recorded using a 300 or 500 MHz Bruker AVANCE 300 or AVANCE 500 spectrometer. The TG-DTA analysis was carried out using an SII EXSTAR 6000 system at a heating rate of 10 °C min−1 up to 200 °C. Powder X-ray diffraction patterns were recorded using a Rigaku RINT 2000 with graphite monochromated CuKα radiation.
Preparation and analysis of inclusion crystals
Equimolar amounts of acid 1 and amine 2 were dissolved in 2-butanol or 2-pentanol, and the solution was left to crystallize until the solvent was evaporated. The crystals thus obtained were collected and washed with hexane. The crystals were characterized by 1H NMR spectroscopy.
Single-crystal X-ray analysis
Single crystals of the inclusion complexes were obtained by evaporating a methanol solution of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of acid 1 and amine 2 followed by their recrystallization from an appropriate solvent. X-ray crystallographic data were collected using a Bruker Smart APEX II diffractometer with graphite monochromated MoKα radiation. The structures were solved by a direct method using SIR 2004 (ref. 14) and refined by SHELXL-2013 program.15 The crystallographic parameters of the inclusion crystals are summarized in Table S1† and the hydrogen bond metrics are summarized in Tables S2 and S3.†
:1 mixture of acid 1 and amine 2 followed by their recrystallization from an appropriate solvent. X-ray crystallographic data were collected using a Bruker Smart APEX II diffractometer with graphite monochromated MoKα radiation. The structures were solved by a direct method using SIR 2004 (ref. 14) and refined by SHELXL-2013 program.15 The crystallographic parameters of the inclusion crystals are summarized in Table S1† and the hydrogen bond metrics are summarized in Tables S2 and S3.†
Results and discussion
Inclusion crystals of 1b·2M (A1) and 1b·2O (A2)
As previously reported, the salt of acid 1b and amine 2M recrystallized from 2-butanol not only incorporated one 2-butanol molecule, but also incorporated two water molecules, thus affording the 1b·2M·2-BuOH·2H2O quaternary inclusion crystal (Crystal A1) with the space group P21.10 However, the recrystallization product of a mixture of equimolar amounts of acid 1b and amine 2O from 2-butanol showed no inclusion of 2-butanol in the salt crystal. The single-crystal X-ray structure analysis showed that the salt was crystallized in the same space group P21 and incorporated only water molecules, affording the 1b·2O·2H2O ternary inclusion crystal (Crystal A2, Fig. 1). Except for the absence of 2-butanol, the inclusion crystal A2 has common structural features with crystal A1: first, the two carboxylate groups of 1b were positioned in an anti-configuration, and the two benzoyloxy groups occupied the gauche position. Water inclusion can be attributed to the less dense molecular packing arising from the skewed conformation of the two benzoyloxy groups of 1b. Second, acid 1b and amine 2O along with two water molecules (w1 and w2) formed 2D sheet-like hydrogen-bonding networks parallel to the ab plane (Fig. 1a), which was further covalently linked by 2O along the c axis to construct a 3D network (Fig. 1b). On the other hand, a slight structural difference between the methylene group and oxygen atom of diamines 2M and 2O changed their molecular conformation and crystal packing. The torsion angles between each phenyl ring and the plane including the central three atoms (C–C–C or C–O–C) of amine 2 (defined as θ) were measured to evaluate the thickness of the dianilines. The angles in crystal A1 (θ = 81.0(9)° and 50.9(9)°) were larger than the corresponding angles in crystal A2 (θ = 46.5(6)° and 33.6(6)°). In crystal A1, 2-butanol formed hydrogen bonds between w2 and a nitrogen atom; however, the thinner molecular structure of diamine 2O allows dense molecular packing without incorporating 2-butanol (Fig. 1a).
 |
| | Fig. 1 Crystal structure of 1b·2O·2H2O (A2). a) Viewed from the c axis. Hydrogen-bonding networks were assisted by two water molecules (w1 and w2). b) Viewed from the b axis. 2D hydrogen-bonding networks are shaded. The dotted lines show hydrogen bonds. Oxygen and nitrogen atoms are represented by red and blue balls, respectively. | |
Inclusion crystals of 1o·2M (B1) and 1o·2O (B2)
To elucidate the effect of a methyl substituent on the resulting crystals, equimolar amounts of di(o-toluoyl)-L-tartaric acid (1o) containing a methyl group and each diamine 2 were mixed and recrystallized from 2-butanol. Although needle-like fine crystals were obtained, neither 1o·2M nor 1o·2O afforded ternary inclusion crystals with 2-butanol as the guest molecule. The X-ray crystallographic analysis of 1o·2M and 1o·2O showed that three water molecules were incorporated in both the crystals. The X-ray crystal structures of ternary inclusion crystals 1o·2M·3H2O (B1) and 1o·2O·3H2O (B2) are shown in Fig. 2. Despite the difference in the central atom of diamines 2M and 2O, these two ternary inclusion crystals were isostructural and crystallized in the C2 space group (θ = 52.0(5)° for B1 and θ = 47.6(3)° for B2). The components 1o, 2M or 2O, and one of the water molecules lie about twofold axes. The two o-toluoyloxy groups of 1o were twisted and positioned in a gauche conformation. The molecular packing of B1 and B2 was similar to that of A2, and the 2D networks along the ab plane combined with diamine 2 constructed a rigid 3D hydrogen-bonding network. Compared to crystal A2, the two methyl groups at the ortho-position of acid 1o were directed toward the a axis, and the cell constant a of crystals B1 and B2 increased from 13.029(3) Å (A2) to 13.604(2) Å (B1) and 13.524(2) Å (B2). In addition to the two symmetrically related water molecules (w1), the third water molecule (denoted as w2 in Fig. 2a) was included to compensate for the expanded space created by the methyl groups. Thus, the salts of acid 1o and amine 2 also afforded ternary inclusion crystals, whereas the methyl groups introduced at the ortho-position of 1b filled the space used for incorporating 2-butanol, and thus only small water molecules were incorporated.
 |
| | Fig. 2 Crystal structures of 1o·2M·3H2O (B1) and 1o·2O·3H2O (B2). a) 2D hydrogen-bonding network of 1o·2M·3H2O viewed from the c axis. b) 1o·2M·3H2O viewed from the b axis. c) 1o·2O·3H2O viewed from the b axis. 2D hydrogen-bonding networks are shaded. The dotted lines show hydrogen bonds. Oxygen and nitrogen atoms are represented by red and blue balls, respectively. | |
Inclusion crystals of 1m·2M (C1) and 1m·2O (C2)
Similar experiments were carried out with di(m-toluoyl)-L-tartaric acid (1m); two types of salt with diamines 2 were recrystallized from 2-butanol. The crystals were analyzed by 1H NMR spectroscopy and found to contain 20% 2-butanol for 1m·2M and 160% 2-butanol for 1m·2O. The X-ray structure of the crystal obtained from the 2-butanol solution of an equimolar mixture of 1m and 2M is shown in Fig. 3. Crystal C1 (2(1m)·2M·2-BuOH) consisted of acid 1m, amine 2M, and 2-butanol in a ratio of 2:1:1, and a ternary inclusion crystal incorporating 2-butanol was formed. The components 1m, 2M, and 2-butanol lie about twofold axes. Notably, only one of the two carboxyl groups of acid 1m was ionized and the other was neutral, while two m-toluoyloxy groups of 1m adopted a gauche conformation. A 1D ribbon-like hydrogen-bonding network of acid 1m was constructed along the c axis by intermolecular hydrogen bonds between carboxyl hydrogen atoms and carboxylate oxygen atoms (Fig. 3a). Two pairs of the ribbon structure were linked by amine 2M, thus affording a 2D zigzag sheet-like network parallel to the ac plane (Fig. 3b). Such a hydrogen-bonding motif has been reported in other salts of acid 1b and amines.16 The 2D sheets were further stacked in parallel with sliding toward the a axis, forming a channel-like cavity along the bent structure of acid 2M. The 2D hydrogen-bonding network was reinforced by π–π stacking between the m-tolyl groups of 1m (Fig. 3a). Although all the methyl groups were oriented toward the cavity to achieve close packing, the guest 2-butanol was incorporated in the remaining channel due to the slightly thick conformation of 2M (θ = 81(1)°). However, no strong intermolecular interactions such as hydrogen bonds between 2-butanol and the host molecules were observed, which would have decreased the inclusion ratio of 2-butanol in the bulk crystals.
 |
| | Fig. 3 Crystal structure of 2(1m)·2M·2-BuOH (C1). a) 1D ribbon-like structures along the c axis. b) 2D network viewed from the c axis. The guest 2-BuOH is shown using the CPK model. | |
The X-ray structure of the inclusion crystal of 1m·2O was different from that of C1 (Fig. 4). The crystals obtained from the combination of acid 1m and amine 2O incorporated both 2-butanol and water molecules, similar to crystal A1. The composition ratio was 1m:2O:2-BuOH:H2O = 1:1:2:1, and the quaternary inclusion crystal C2 (1m·2O·2(2-BuOH)·H2O) was formed. The conformation of acid 1m in the crystal was similarly twisted, and the two m-toluoyloxy groups occupied a gauche position. Further, the torsional angles of 2O (θ = 51.6(9)° and 30(1)°) were smaller than those of 2M. The space group was P212121, and wavy 2D hydrogen-bonding networks parallel to the ac plane were observed in the crystals prepared from acid 1m and amine 2O. The neighboring wavy sheets were linked by hydrogen bonds between water (w1) and 2-butanol (G1) molecules. Another 2-butanol molecule (G2) was incorporated in the remaining void space between the two sheets. The hydroxy group of G2 formed two hydrogen bonds with ammonium hydrogen and carboxylate oxygen atoms. As in the case of crystal C1, the decreased amounts of 2-butanol in the bulk inclusion crystals compared to the single crystal C2 can be attributed to the facile desorption of 2-butanol from the crystals. Thus, the methyl groups introduced to the meta-position of acid 1b changed the crystal packing of the host molecules to construct sheet-like wavy structures, and 2-butanol was incorporated between the sheet-like hydrogen-bonding networks.
 |
| | Fig. 4 Crystal structure of 1m·2O·2(2-BuOH)·H2O (C2) viewed from the a axis. | |
Inclusion crystals of 1p·2M (D1) and 1p·2O (D2)
Finally, the mixtures of di(p-toluoyl)-L-tartaric acid (1p) and diamines 2 were recrystallized from 2-butanol. The obtained bulk solid 1p·2M contained 2-butanol, but with a low inclusion ratio of 20%. Although the single crystal prepared from the 2-butanol solution of 1p·2M could not be fully refined due to the disordered structure, the same molecular assembly was observed in the crystal obtained from a methanol solution, affording a better result. The crystal D1 (1p·2M, Fig. 5a) belonged to the highly symmetrical chiral space group I41, and a characteristic 20 Å square-shaped tube-like structure was formed along the c axis. Each side of the square contained hydrogen-bonding sites, and the bent structure of 2M played the role of a vertex. The wide torsion angles of 2M (θ = 75(1)° and 74(1)°) were suitable for the formation of a walled tube structure. Similar to salt crystal C1 described above, one of the carboxyl groups of acid 1p was neutral, and the other ionized to form a salt with 2M. Only one of the amino groups of 2M was ionized, resulting in a 1:1 salt formation between acid 1p and amine 2M. All the functional groups of acid 1p and amine 2M remained outside of the tubes to form a 1D hydrogen-bonding network and the hydrophobic p-tolyl groups remained inside, similar to a micellar rod. The neighboring tubes were connected by complementary hydrogen bonds between amino and ammonium nitrogen atoms (the N⋯N distance was 2.68 Å). A hydrophobic channel with a diameter of ~6.6 Å (the closest distance between the two corresponding hydrogen atoms of acid 1p at opposite positions) was observed inside the tube. Although residual electron density was observed in the channel, the amount of incorporated solvent was too small to determine the atomic positions because of its narrow size and the absence of any hydrogen-bonding sites within the channel. The two p-toluoyloxy groups were oriented in a gauche position, and the phenyl planes were aligned almost perpendicular to the tube structure. They are alternately stacked along the channel with an inclination to form a right-handed helix (Fig. 5b).
 |
| | Fig. 5 Crystal structure of 1p·2M (D1). a) Tube-like structures viewed from the c axis. b) Helical arrangement of phenyl groups along the tube-like structure. | |
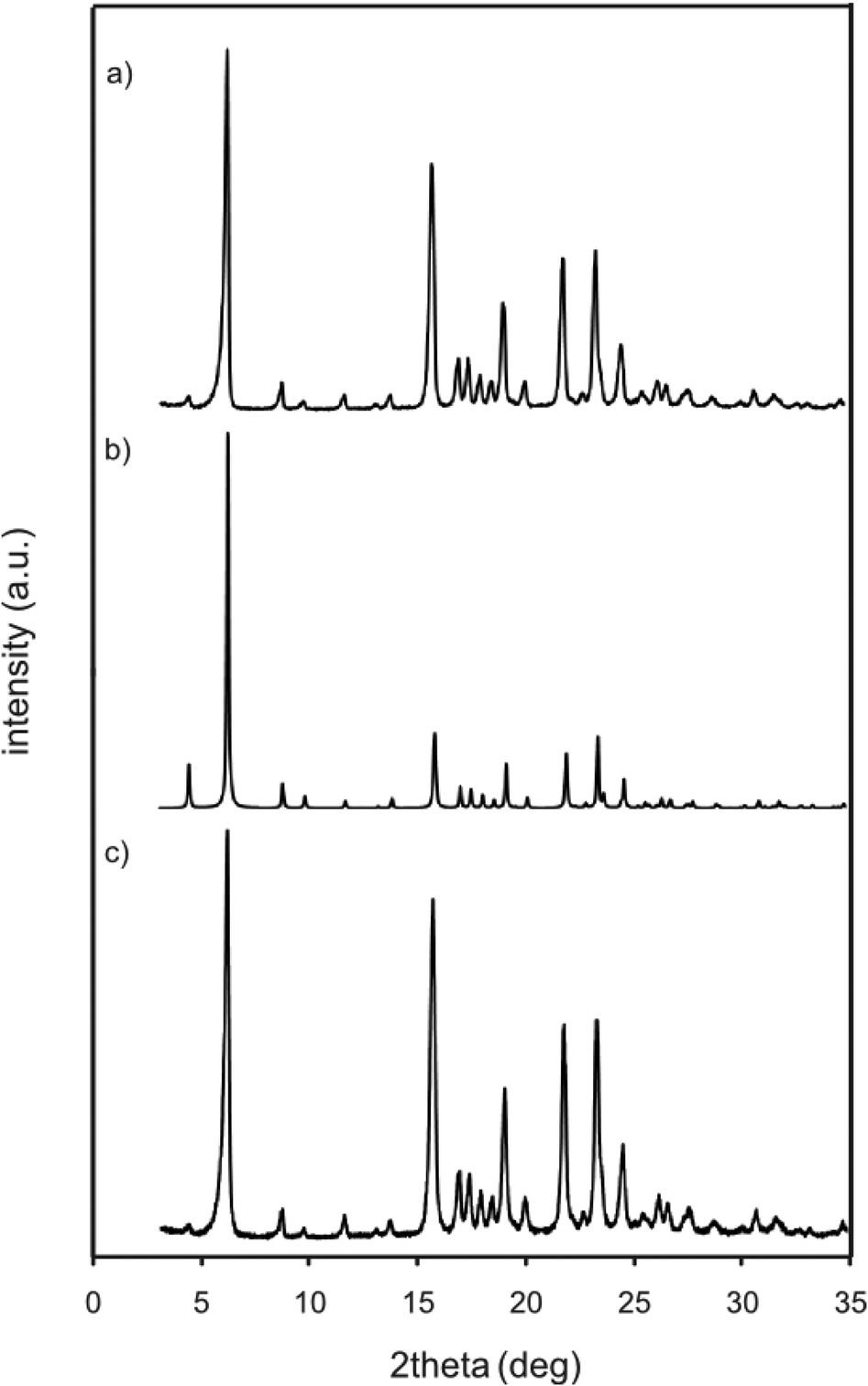
The powder X-ray diffraction pattern of the solid prepared by evaporating a methanol solution of 1p·2M followed by drying under air was almost identical to that simulated from a single crystal of D1 (Fig. 6), indicating that such a tube-like structure was formed in the bulk solid prepared by fast evaporation. Moreover, the TG analysis of the solid showed no weight loss up to 200 °C before its decomposition. Notably, the powder X-ray diffraction pattern remained unchanged even after heating the salt to up to 100 °C, indicating that the tube-like structure was maintained without incorporating any guest molecules inside the tube.
 |
| | Fig. 6 X-ray diffraction patterns of the salt 1p·2M. a) Bulk solid prepared from methanol solution. b) Simulated from the crystal structure of D1. c) Bulk solid after heating at 100 °C. | |
On the other hand, the 1H NMR spectrum of salt 1p·2O recrystallized from 2-butanol showed that as many as 2 molar equivalents of 2-butanol were incorporated in the bulk solid. The X-ray structure of crystal 1p·2O obtained from a 2-butanol solution indicated that 2-butanol was incorporated in the salt, however, the R-factor was not satisfactory because of the highly disordered structure of 2-butanol. When 1p·2O was recrystallized from 2-pentanol instead of 2-butanol, a similar inclusion crystal D2 was obtained. 2D sheet-like wavy hydrogen-bonding networks were formed from acid 1p and amine 2O in D2. The molar ratio was 1p:2O:2-pentanol = 1:1:2, and the 1p·2O·2(2-PenOH) ternary crystal was obtained (Fig. 7). The space group of D2 was P212121, and two neighboring sheets were stacked in an antiparallel fashion. Two guest 2-pentanol molecules (G1 and G2) were incorporated between two neighboring sheets, and they were bound by two hydrogen bonds with the carboxylate oxygen atom of acid 1p and ammonium nitrogen atom of amine 2O. The wavy hydrogen-bonding networks created by the bent nature of amine 2O (θ = 52.5(7)° and 28.9(7)°) together with the twisted conformation of acid 1p may contribute to the high inclusion ability of the salt.
 |
| | Fig. 7 Crystal structure of 1p·2O·2(2-PenOH) (D2) viewed from the a axis. | |
General discussion of the eight salt crystals A1–D2
Recrystallization of equimolar mixtures of dicarboxylic acids 1 and diamines 2 from 2-butanol afforded the corresponding 1:1 salt crystals except for crystal C1. The conformation of acids 1 in the salt crystals was almost identical: the two carboxyl groups oriented in a trans position, whereas the two bulky aroyloxy groups oriented in a gauche position. Such a twisted molecular shape generated a vacant space in the salt crystal, resulting in solvent inclusion in all the eight salt crystals even though the stoichiometry was not clear for crystal D1. Diverse hydrogen-bonding networks were obtained from the combination of acids 1 and amines 2: 3D hydrogen-bonding networks for crystals A1, A2, B1, and B2; 2D sheet-like hydrogen-bonding networks for crystals C1, C2, and D2; and highly symmetric 1D tube-like structures for crystal D1. Notably, the dimensionality of the networks mainly changed with the position (o/m/p) of the methyl group of acids 1. As the position of the methyl groups changed from ortho to meta to para, and the distance between them and the hydrogen-bonded carboxylate groups increased, the dimensionality of the hydrogen-bonding networks decreased from 3D to 2D to 1D. As discussed in our previous paper, one of the factors that determines the dimensionality of the hydrogen-bonding networks is the volume fraction of the hydrophilic/hydrophobic regions of the constituent molecules.9f Crystals A1, A2, B1, and B2 probably have comparable hydrophilic/hydrophobic regions due to the multiple water molecules incorporated; on the other hand, the increased molecular length of acids 1m and 1p expanded the hydrophobic region that disturbed the hydrophilic/hydrophobic balance and changed the hydrogen-bonding network to achieve more dense molecular packing. Also, the difference in the central atoms of diamines 2 changed their conformation: the twist angles of amine 2M in crystals A1–D1 (50° < θ < 81°) were larger than those of amine 2O in crystals A2–D2 (28° < θ < 53°), and the structure of amine 2M was thicker than that of amine 2O, probably because of the smaller C–C–C bond angles of amine 2M (112–116°) compared to the C–O–C bond angles of amine 2O (117–121°) and more steric repulsion between the methylene hydrogen atoms and phenyl groups of amine 2M. Thus, only a slight structural modification of amine 2 effectively controlled the molecular packing and inclusion ability of the resulting supramolecular hosts.
Conclusions
In this study, eight combinations of salts were prepared from dibenzoyl-L-tartaric acid (1b)/three positional isomers of ditoluoyl-L-tartaric acid (1o, 1m, 1p) and two bent diamines (2M, 2O), and their X-ray crystal structures were investigated. Diverse hydrogen-bonding networks—1D robust tubes, 2D sheets, and 3D connected networks—were formed, depending on the selected acid/amine components. In particular, the position of the methyl substituents on acids 1 significantly affected the dimensionality of the hydrogen-bonding networks. All the salts showed inclusion ability, and some of them successfully embedded 2-butanol molecules between their sheet-like hydrogen-bonding networks. This study demonstrated the variation in supramolecular networks comprising carboxylate–ammonium salts and the applications of diaroyl tartaric acids as a component of supramolecular hosts. Further design of multicomponent crystals from carboxylate–ammonium salts is underway.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 22750119 (to K. K.).
Notes and references
-
(a)
Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. J. Vittal, Wiley, Chichester, West Sussex, England, 2006 Search PubMed;
(b) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed;
(c) C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
-
(a) A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309–314 CrossRef CAS;
(b) J. D. Dunitz, Chem. Commun., 2003, 545–548 RSC;
(c) G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed;
(d) S. L. Price, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 313–328 CrossRef CAS PubMed;
(e) S. L. Price, Chem. Soc. Rev., 2014, 43, 2098–2111 RSC.
-
(a) Y. Ooyama, S. Nagano, M. Okamura and K. Yoshida, Eur. J. Org. Chem., 2008, 5899–5906 CrossRef CAS;
(b) T. Hinoue, M. Miyata, I. Hisaki and N. Tohnai, Angew. Chem., Int. Ed., 2012, 51, 155–158 CrossRef CAS PubMed;
(c) N. Nishiguchi, T. Kinuta, T. Sato, Y. Nakano, T. Harada, N. Tajima, M. Fujiki, R. Kuroda, Y. Matsubara and Y. Imai, Cryst. Growth Des., 2012, 12, 1859–1864 CrossRef CAS;
(d) M. Sugino, K. Hatanaka, Y. Araki, I. Hisaki, M. Miyata and N. Tohnai, Chem. – Eur. J., 2014, 20, 3069–3076 CrossRef CAS PubMed.
-
(a) M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed;
(b) X.-Z. Luo, X.-J. Jia, J.-H. Deng, J.-L. Zhong, H.-J. Liu, K.-J. Wang and D.-C. Zhong, J. Am. Chem. Soc., 2013, 135, 11684–11687 CrossRef CAS PubMed;
(c) A. Yamamoto, T. Hirukawa, I. Hisaki, M. Miyata and N. Tohnai, Tetrahedron Lett., 2013, 54, 1268–1273 CrossRef CAS;
(d) T. Jacobs, V. J. Smith, L. H. Thomas and L. J. Barbour, Chem. Commun., 2014, 50, 85–87 RSC.
-
(a) M. Akazome, S. Toma, T. Horiguchi, K. Megumi and S. Matsumoto, Tetrahedron, 2011, 67, 2844–2848 CrossRef CAS;
(b) L. R. Nassimbeni, H. Su and T.-L. Curtin, Chem. Commun., 2012, 48, 8526–8528 RSC;
(c) C. S. Frampton, K. A. Ketuly, A. H. A. Hadi, J. H. Gall and D. D. MacNicol, Chem. Commun., 2013, 49, 7198–7200 RSC;
(d) K. Megumi, S. Yokota, S. Matsumoto and M. Akazome, Tetrahedron Lett., 2013, 54, 707–710 CrossRef CAS;
(e) P. Li, Y. He, J. Guang, L. Weng, J. C. Zhao, S. Xiang and B. Chen, J. Am. Chem. Soc., 2014, 136, 547–549 CrossRef CAS PubMed.
-
(a) Y. Imai, T. Sato and R. Kuroda, Chem. Commun., 2005, 3289–3291 RSC;
(b) Y. Imai, K. Kawaguchi, T. Sato, R. Kuroda and Y. Matsubara, Tetrahedron Lett., 2006, 47, 7885–7888 CrossRef CAS;
(c) R. Santra and K. Biradha, Cryst. Growth Des., 2009, 9, 4969–4978 CrossRef CAS;
(d) S. Roy and K. Biradha, Cryst. Growth Des., 2011, 11, 4120–4128 CrossRef CAS;
(e) K. Megumi, F. N. B. M. Arif, S. Matsumoto and M. Akazome, Cryst. Growth Des., 2012, 12, 5680–5685 CrossRef CAS;
(f) Y. Manjare, V. Nagarajan and V. R. Pedireddi, Cryst. Growth Des., 2014, 14, 723–729 CrossRef CAS.
-
(a) T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546–7550 CrossRef PubMed;
(b) B. R. Bhogala and A. Nangia, New J. Chem., 2008, 32, 800–807 RSC;
(c) C. B. Aakeröy, J. Desper, M. Fasulo, I. Hussain, B. Levin and N. Schultheiss, CrystEngComm, 2008, 10, 1816–1821 RSC;
(d) S. Tothadi and G. R. Desiraju, Chem. Commun., 2013, 49, 7791–7793 RSC.
-
(a) K. Kinbara, Y. Hashimoto, M. Sukegawa, H. Nohira and K. Saigo, J. Am. Chem. Soc., 1996, 118, 3441–3449 CrossRef CAS;
(b) K. Sada, K. Inoue, T. Tanaka, A. Tanaka, A. Epergyes, S. Nagahama, A. Matsumoto and M. Miyata, J. Am. Chem. Soc., 2004, 126, 1764–1771 CrossRef CAS PubMed;
(c) K. Sada, T. Watanabe, J. Miyamoto, T. Fukuda, N. Tohnai, M. Miyata, T. Kitayama, K. Maehara and K. Ute, Chem. Lett., 2004, 33, 160–161 CrossRef CAS;
(d) T. Yuge, M. Miyata and N. Tohnai, Cryst. Growth Des., 2006, 6, 1271–1273 CrossRef CAS;
(e) T. Yuge, T. Sakai, N. Kai, I. Hisaki, M. Miyata and N. Tohnai, Chem. – Eur. J, 2008, 14, 2984–2993 CrossRef CAS PubMed;
(f) P. Sahoo, D. K. Kumar, S. R. Raghavan and P. Dastidar, Chem. – Asian J., 2011, 6, 1038–1047 CrossRef CAS PubMed;
(g) T. Sasaki, I. Hisaki, T. Miyano, N. Tohnai, K. Morimoto, H. Sato, S. Tsuzuki and M. Miyata, Nat. Commun., 2013, 4, 1787 CrossRef PubMed;
(h) T. K. Adalder and P. Dastidar, Cryst. Growth Des., 2014, 14, 2254–2262 CrossRef CAS;
(i) T. Sasaki, Y. Ida, I. Hisaki, T. Yuge, Y. Uchida, N. Tohnai and M. Miyata, Chem. – Eur. J, 2014, 20, 2478–2487 CrossRef CAS PubMed.
-
(a) Y. Kobayashi, K. Kodama and K. Saigo, Org. Lett., 2004, 6, 2941–2944 CrossRef CAS PubMed;
(b) K. Kodama, Y. Kobayashi and K. Saigo, Chem. – Eur. J, 2007, 13, 2144–2152 CrossRef CAS PubMed;
(c) K. Kodama, Y. Kobayashi and K. Saigo, Cryst. Growth Des., 2007, 7, 935–939 CrossRef CAS;
(d) K. Kodama, A. Kanno, E. Sekine and T. Hirose, Org. Biomol. Chem., 2012, 10, 1877–1882 RSC;
(e) H. Shitara, T. Shintani, K. Kodama and T. Hirose, J. Org. Chem., 2013, 78, 9309–9316 CrossRef CAS PubMed;
(f) K. Kodama, H. Shitara and T. Hirose, Cryst. Growth Des., 2014, 14, 3549–3556 CrossRef CAS;
(g) K. Kodama, N. Kurozumi, H. Shitara and T. Hirose, Tetrahedron, 2014, 70, 7923–7928 CrossRef CAS.
- K. Kodama, E. Sekine and T. Hirose, Chem. – Eur. J., 2011, 17, 11527–11534 CrossRef CAS PubMed.
-
(a) K. Hatano, T. Takeda and R. Saito, J. Chem. Soc., Perkin Trans. 2, 1994, 579–584 RSC;
(b) T. Kohara, Y. Hashimoto, R. Shioya and K. Saigo, Tetrahedron: Asymmetry, 1999, 10, 4831–4840 CrossRef CAS;
(c) A. Berkessel, K. Roland, M. Schröder, J. M. Neudörfl and J. Lex, J. Org. Chem., 2006, 71, 9312–9318 CrossRef CAS PubMed;
(d) B. C. Gorske and H. E. Blackwell, J. Am. Chem. Soc., 2006, 128, 14378–14387 CrossRef CAS PubMed.
-
(a) V. R. Vangala, R. Mondal, C. K. Broder, J. A. K. Howard and G. R. Desiraju, Cryst. Growth Des., 2005, 5, 99–104 CrossRef CAS;
(b) S. Bhattacharya and B. K. Saha, Cryst. Growth Des., 2011, 11, 2194–2204 CrossRef CAS.
- H. Tohma, S. Takizawa, H. Watanabe, Y. Fukuoka, T. Maegawa and Y. Kita, J. Org. Chem., 1999, 64, 3519–3523 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. D. Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381 CrossRef CAS.
-
G. M. Sheldrick, SHELX-2013, Programs for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 2013 Search PubMed.
- H. Tomori, H. Yoshihara and K. Ogura, Bull. Chem. Soc. Jpn., 1996, 69, 3581–3590 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of crystallographic data, hydrogen bond metrics, and 1H NMR spectra. CCDC deposition numbers 1414396–1414401, and 1414403. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01769f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.