
Hydrogen-bonding networks of purine derivatives and their bilayers for guest intercalation†
Yoona
Jang
a,
Seo Yeon
Yoo
b,
Hye Rin
Gu
b,
Yu Jin
Lee
b,
Young Shin
Cha
b,
Laekyeong
You
a,
Kyungkyou
Noh
a and
Jaheon
Kim
*a
aDepartment of Chemistry, Soongsil University, Seoul 06978, Korea. E-mail: jaheon@ssu.ac.kr; Fax: +82 2 824 4383; Tel: +82 2 820 0459
bHankuk Academy of Foreign Studies, Yongin 17035, Korea
First published on 9th November 2015
Abstract
Two purine derivatives, 6-chloro-9-propyl-purin-2-amine (pr-GCl) and 6-chloro-9-pentyl-purin-2-amine (pt-GCl) have been synthesized and their crystal structures were determined by single crystal X-ray diffraction analyses. The purine rings in pr-GCl or pt-GCl form unique two-dimensional hydrogen-bonding networks which in turn stack via π–π interactions to give bilayers covered with alkyl chains. In pt-GCl, the pentyl groups interact effectively among themselves so that void space for guest molecules is not available. In contrast, pr-GCl can form host–guest co-crystals. Nuclear magnetic resonance (NMR) analyses of the pr-GCl crystals immersed in various solvents for up to 60 min indicate that aromatic molecules (benzene, xylene isomers) are better guests than aliphatic ones (n-hexane, cyclohexane, isooctane) in terms of their inclusion time and amount. Powder X-ray diffraction (PXRD) patterns for the guest-included pr-GCl crystals are quite different from the simulated one, supporting the guest diffusion into the pr-GCl crystals. The crystal structure of p-xylene@pr-GCl reveals that p-xylene molecules are intercalated between the characteristic pr-GCl bilayers that are shown in both pr-GCl and pt-GCl crystals.
Introduction
Self-assembly of organic molecules via hydrogen bonding is a reliable route to achieve well-defined structural architectures.1 A prerequisite for this process is to devise appropriate building blocks which have complementary hydrogen-bonding donor and/or acceptor pairs. A molecule itself can act as both a hydrogen donor and acceptor like trimesic acid.2 In many situations, however, it is hard to predict what type of hydrogen-bonding network will be formed in a crystalline lattice; for example, anhydrous guanine forms eight hydrogen bonds with three nearest neighbours to produce a flat two-dimensional network.3 In this regard, symmetrical and reliable donor/acceptor pairs are preferred as building blocks, as demonstrated in cyanuric acid/melamine,4 guanidinium/organic sulfonate (GS),5 and so on.6 It is notable that the GS pairs developed by Ward and co-workers form robust two-dimensional hydrogen-bonding networks with alternating arrays of positive hydrogen bond donors and negative acceptors, and can accommodate guest molecules between the hydrogen-bonding bilayers to form numerous host–guest complex crystals by introducing various pendant groups into organic sulfonates. Thus far, systems like the GS pairs, which can retain their global architecture during guest-inclusion processes, are rare.5aRecently, it has been shown that nucleobases such as uracil and thymine derivatives can form similar bilayers to the GS pairs.7 For example, N1-hexyluracil or N1-hexylthymine molecules by themselves form flat two-dimensional hydrogen-bonding networks which in turn stack with other layers via π–π interactions and their hexyl chains fill the space between the bilayers, which resembles a lipid bilayer.7a,b The overall feature could be retained when fluorouracil derivatives are crystallized;7c that is, the uracil ring is responsible for building hydrogen-bonding networks whose patterns are affected by the alkyl chains with or without terminal functional groups and concomitant various weak interactions. Interestingly, in the case of cytosine,7d its N1-hexyl derivative does not show the characteristic bilayer structure observed in both N1-hexylthymine and N1-hexyluracil, implying that the type of nucleobase may affect the formation of bilayer structures.
Here, we report our serendipitous finding that two alkylated purine derivatives, 6-chloro-9-propyl-purin-2-amine (termed pr-GCl) and 6-chloro-9-pentyl-purin-2-amine (termed pr-GCl), which are synthetic precursors of their respective guanine derivatives (Scheme 1), can be also assembled into bilayer structures by forming unique hydrogen-bonding networks. Interestingly, pr-GCl can accommodate various guest molecules in the crystals, while pt-GCl does not. When pr-GCl is recrystallized in p-xylene, its crystal structure reveals that p-xylene molecules are intercalated in the bilayers of host pr-GCl molecules. This feature is similar to that observed in the host–guest co-crystals of a GS system.5
 | ||
| Scheme 1 Purine derivatives and host bilayers in this work. | ||
Experimental section
Synthetic procedures
The organic molecules in this work method were synthesized by a literature method with slight modifications.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as eluent (0.92 g, 4.36 mmol, 75% yield). For further analyses, pr-GCl was recrystallized in CH2Cl2 by a simple evaporation method at room temperature. EA: calcd. for C8H10ClN5·(H2O)0.25(CH2Cl2)0.08: C, 43.35; H, 4.84; N, 31.29%. Found: C, 43.45; H, 4.61; N, 31.32%.
:1 v/v) as eluent (0.92 g, 4.36 mmol, 75% yield). For further analyses, pt-GCl was recrystallized in CH2Cl2 by a simple evaporation method at room temperature. EA: calcd. for C10H14N5Cl: C, 50.11; H, 5.89; N, 29.22%. Found: C, 49.95; H, 5.86; N, 28.78%.
:1 v/v) as eluent (0.92 g, 4.36 mmol, 75% yield). For further analyses, pr-GCl was recrystallized in CH2Cl2 by a simple evaporation method at room temperature. EA: calcd. for C8H10ClN5·(H2O)0.25(CH2Cl2)0.08: C, 43.35; H, 4.84; N, 31.29%. Found: C, 43.45; H, 4.61; N, 31.32%.
:1 v/v) as eluent (0.92 g, 4.36 mmol, 75% yield). For further analyses, pt-GCl was recrystallized in CH2Cl2 by a simple evaporation method at room temperature. EA: calcd. for C10H14N5Cl: C, 50.11; H, 5.89; N, 29.22%. Found: C, 49.95; H, 5.86; N, 28.78%.
X-ray crystallography
X-ray diffraction intensities were collected on a Bruker APEX CCD diffractometer using Mo Kα radiation (λ = 0.71075 Å) at 173 K for pr-GCl and 298 K for pt-GCl. Due to its very thin crystal morphology, the X-ray data for p-xylene@pt-GCl was obtained at 173 K with a synchrotron light source (λ = 0.71000 Å) on an ADSC Quantum-210 detector at 2D-SMC at Pohang Accelerator Laboratory (PAL), Korea. Both pr-GCl and pt-GCl crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), while p-xylene@pr-GCl belongs to the orthorhombic space group Ccca (no. 68, origin choice 2). The initial structures were solved by direct methods using SHELX-S and refined by full-matrix least-squares techniques against F2 with SHELXL-2013.9 Four and eight independent molecules were respectively identified in pr-GCl and pt-GCl. In p-xylene@pr-GCl, two pr-GCl and two p-xylene molecules were defined as an asymmetric unit. One of the two pr-GCl molecules was disordered over two general sites with the same probability. The p-xylene molecules were also disordered over two sites at different inversion centres; that is, two independent molecules sit on different special positions (0.5, 0.5, 0.5) and (0.75, 0.75, 0.5), respectively. Disorder models for each p-xylene were built based on electron densities, avoiding possible bad guest–guest and guest–host contacts. Due to the very weak intensity data, the final R values for p-xylene@pr-GCl were quite high but the refined structure gave reasonable crystal packing and bond geometries. For pr-GCl, the occluded solvent molecules could not be identified due to diffuse electron densities. Therefore, the structural refinement was conducted by employing the SQUEEZE treatment within the PLATON software package.10 All non-H atoms were refined anisotropically, and all H atoms were generated in ideal positions and included for the refinement processes. The space groups were checked by the ADDSYM routine of PLATON.10 Crystal and refinement data are listed in Tables S1 (pr-GCl), S3 (pt-GCl), and S5 (p-xylene@pr-GCl).† The ORTEP drawings are displayed in Fig. S12 (pr-GCl), S13 (pt-GCl), and S14 (p-xylene@pr-GCl).†
(no. 2), while p-xylene@pr-GCl belongs to the orthorhombic space group Ccca (no. 68, origin choice 2). The initial structures were solved by direct methods using SHELX-S and refined by full-matrix least-squares techniques against F2 with SHELXL-2013.9 Four and eight independent molecules were respectively identified in pr-GCl and pt-GCl. In p-xylene@pr-GCl, two pr-GCl and two p-xylene molecules were defined as an asymmetric unit. One of the two pr-GCl molecules was disordered over two general sites with the same probability. The p-xylene molecules were also disordered over two sites at different inversion centres; that is, two independent molecules sit on different special positions (0.5, 0.5, 0.5) and (0.75, 0.75, 0.5), respectively. Disorder models for each p-xylene were built based on electron densities, avoiding possible bad guest–guest and guest–host contacts. Due to the very weak intensity data, the final R values for p-xylene@pr-GCl were quite high but the refined structure gave reasonable crystal packing and bond geometries. For pr-GCl, the occluded solvent molecules could not be identified due to diffuse electron densities. Therefore, the structural refinement was conducted by employing the SQUEEZE treatment within the PLATON software package.10 All non-H atoms were refined anisotropically, and all H atoms were generated in ideal positions and included for the refinement processes. The space groups were checked by the ADDSYM routine of PLATON.10 Crystal and refinement data are listed in Tables S1 (pr-GCl), S3 (pt-GCl), and S5 (p-xylene@pr-GCl).† The ORTEP drawings are displayed in Fig. S12 (pr-GCl), S13 (pt-GCl), and S14 (p-xylene@pr-GCl).†
Results and discussion
Crystal structures of pr-GCl and pt-GCl
The alkylated purine derivatives in this work are prepared as precursors to their guanine forms as shown in Scheme 1. Guanine (G), a base in DNA, can form stacked layers of two-dimensional hydrogen-bonding networks via π–π interactions.3 The layered structure is lost when G is functionalized with an ethyl group and instead assembles into hydrogen-bonding tapes.11 Thus, our intention was to introduce longer alkyl chains, propyl or pentyl groups to G (abbreviated as pr-G or pt-G, respectively), and to investigate their crystal packing by single crystal X-ray diffraction (SCXRD) analyses. However, the obtained small microcrystals of R-Gs were not suitable for structure elucidation. Instead, we focused on the crystal structures of pr-GCl and pt-GCl because they resemble their guanine forms in terms of shapes and potential hydrogen bonding ability.While pt-GCl crystallizes without void space, pr-GCl forms co-crystals with dichloromethane (CH2Cl2) which was used as a solvent and detected in the 1H-NMR spectrum measured with dissolved crystals in DMSO-d6 (Fig. S1†). It was not possible to identify the included dichloromethane based on the current X-ray data due to their severe disorder, which requires a SQUEEZE treatment.10 In spite of this difference, the overall crystal packing is similar to each other. In both crystals, the purine rings form hydrogen bonds with adjacent molecules to form almost flat layers which stack via π–π interactions to give bilayers running parallel to the crystallographic ac-plane for pr-GCl and bc-plane for pt-GCl, respectively (Fig. 1a). In the pr-GCl crystal, a terminal ethyl moiety of the propyl group is positioned almost perpendicularly to the purine ring and the remaining ethylene (–CH2–) group attached to N is located nearly at the molecular plane (Fig. 1). The pentyl group in pt-GCl also behaves in a similar way. Thus, a bilayer is covered with alkyl chains on both sides, which is reminiscent of the bilayer of GS systems having pendant groups in organic sulfonates.5b
 | ||
| Fig. 1 Crystal structures of (a) pr-GCl and pt-GCl are displayed with stick models. Colour codes: C, grey; H, white; N, blue; Cl, green. (b) Hydrogen-bonding network in a layer of pr-GCl or pt-GCl is shown with possible hydrogen bonds as dotted red lines. Only the –CH2 groups in the propyl chains are drawn for simplicity. A hexamer unit is highlighted with coloured molecules. (c) The hydrogen bonds in pr-GCl (or pt-GCl) are compared with those in anhydrous guanine. | ||
A hydrogen-bonding layer can be described as fused pseudo-hexagonal tiles, and a pr-GCl or pt-GCl molecule interacts with three neighbouring molecules via six possible hydrogen bonds (Fig. 1b). An amino group (–NH2) in the middle molecule is engaged in hydrogen bonds with two sp2 N acceptors located in two different molecules; indeed, two N donor atoms in the middle pr-GCl molecule in turn interact with two –NH2 groups in the neighbouring molecules. The H atom attached to the C atom in an imidazole ring interacts with a pyrimidine N atom via weak hydrogen bonding. Interestingly, this hydrogen bonding pattern is similar to that observed in G; a central G also interacts with three nearest neighbours via hydrogen bonds in the crystal structure of anhydrous G (Fig. 1c).3 However, G forms a total of eight hydrogen bonds, whereas pr-GCl or pt-GCl forms six because the Cl atom of R-GCl is not involved in hydrogen bonding and the introduced alkyl groups prevent their bonded N atoms from participating in hydrogen bonding. It is notable that two Cl atoms are facing each other in the hexamer unit with an average Cl⋯Cl distance of 3.50 (pr-GCl) or 3.30 Å (pt-GCl) and an average bond angle of ~160° around two Cl atoms (∠C–Cl⋯Cl). This arrangement is classified as type I halogen bonding.12 However, at a Cl⋯Cl distance of 3.5 Å, the interaction energy is calculated to be ~5 kJ mol−1,13 which is small and thus not considered as a true halogen bonding.12
The local hydrogen-bonding fashion is almost the same for pr-GCl and pt-GCl, wherein the two layers in pr-GCl are less displaced from each other than those in pt-GCl: the distances between the centres of the hexamer units are 3.56 and 5.46 Å in pr-GCl and pt-GCl, respectively (Fig. 2a). This different displacement is mainly attributed to the guest included in pr-GCl. Calculations of Hirshfeld surfaces14 indicate that only 73% of the molecular surface of pr-GCl is in contact with neighbouring pr-GCl, whereas a pt-GCl molecule is completely enclosed (100%) by other pt-GCl molecules. That is, the remaining 27% of the pr-GCl surface does not touch its neighbours but the ‘missing’ dichloromethane in the crystal structure. In both structures, the H(a selected molecule)–H(other molecules) contacts are calculated to have a dominant contribution to the crystal packing: 28/73 and 45/100% for pr-GCl and pt-GCl, respectively (not shown). This indicates that the displacement of two π-stacked layers in a bilayer should be affected by the interactions among the alkyl chains or the interactions with included guest molecules if R-GCl forms host–guest co-crystals.
 | ||
| Fig. 2 (a) The relative displacement of the hexamer units in the π–π stacked layers is compared: the upper and bottom layers are coloured with yellow and blue, respectively, and the centre of each hexamer is marked as a coloured dot. (b) Hirshfeld surfaces of pr-GCl and pt-GCl are shown. For simplicity, neighbouring molecules are not drawn. | ||
Guest inclusion experiments in pr-GCl crystals
We conducted guest inclusion experiments with dried pr-GCl crystals. A typical procedure is as follows. Dried pr-GCl crystals (ca. 10 mg) were immersed in a 4 mL vial containing n-hexane (1.0 mL). A total of seven vials were prepared in this way. After 5, 10, 15, 20, 30, 45, and 60 min, the crystals in each batch were collected by filtration, washed quickly with n-hexane, and dried for 1 min in a gentle flow of N2 stream. Each sample was dissolved in DMSO-d6 (0.50 mL) to measure the 1H-NMR spectrum, and the ratio of n-hexane/pr-GCl in moles was calculated based on the integration of the identified signals (Fig. 3 and S15†). The potential guest molecules selected were aliphatic and aromatic solvents: n-hexane, cyclohexane, isooctane, benzene, o-xylene, m-xylene, and p-xylene. The host crystals in aromatic solvent were dissolved at ~25 °C. In the case of cyclohexane, the solvent became frozen due to its high melting point, 6.5 °C. Therefore, the experimental temperature was maintained at 20 °C by employing a circulating chiller; at this temperature, the crystals were stable. | ||
| Fig. 3 Guest inclusion plot based on the 1H-NMR analyses of the pr-GCl crystals immersed in solvent. Each point is the average value of two independent measurements. | ||
The plot in Fig. 3 gives some information on the inclusion properties of the guests in a semi-quantitative manner. First, the aromatic solvents are better guests in pr-GCl crystals than the aliphatic ones in terms of inclusion time and amount. For instance, the amount of benzene included in 5 min was greater than that of cyclohexane by more than ten folds. Within each class of guests, the small ones such as benzene and cyclohexane are favoured than the larger ones. It is also notable that p-xylene is included faster than the other isomers, o- and m-xylenes, indicating that the molecular shape can affect the inclusion process. However, it is not possible to explain at the molecular level why certain guests are favoured over others. Thus, we measured the powder X-ray diffraction (PXRD) patterns of the sample crystals employed in the NMR measurements (60 min) in order to compare their patterns with that of pr-GCl host crystals (Fig. 4).
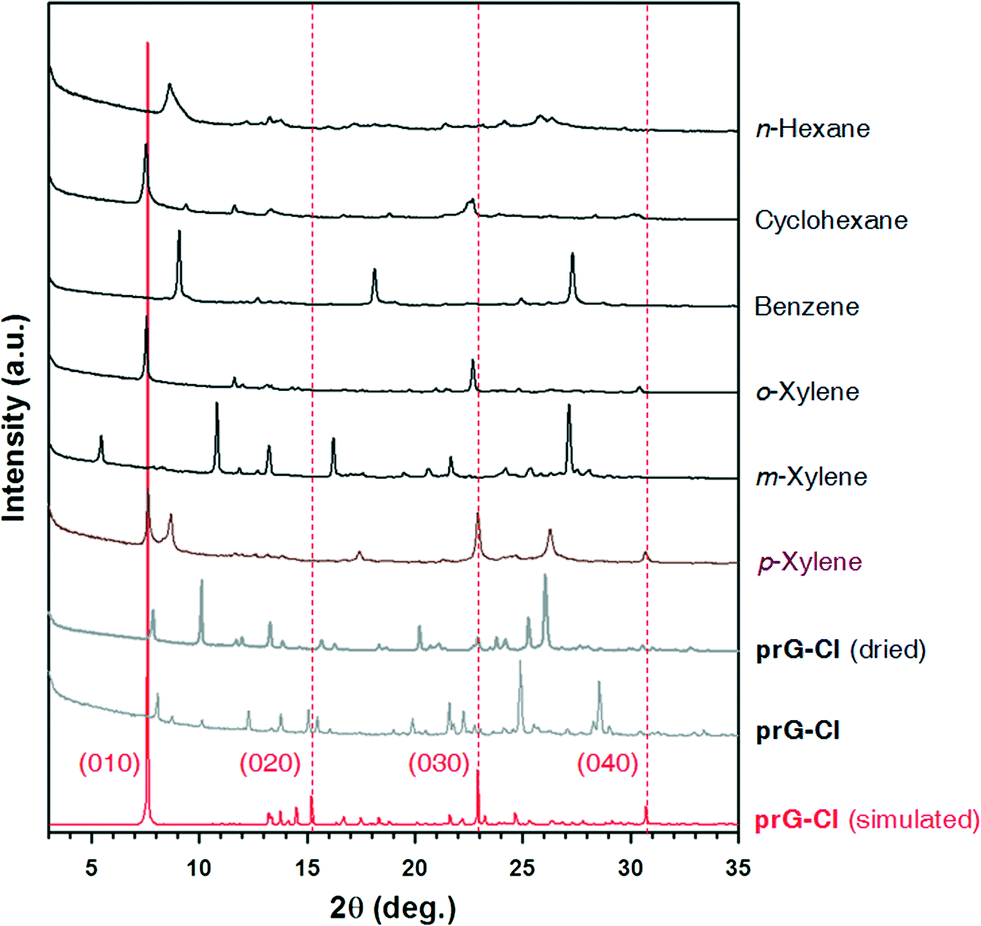 | ||
| Fig. 4 PXRD patterns for pr-GCl and its guest-inclusion crystals are compared with the simulated one generated from the crystal structure of pr-GCl. The pr-GCl crystals were immersed for 1 h in each solvent listed on the plot before PXRD measurement. | ||
The PXRD pattern of the pr-GCl host crystals grown in dichloromethane were measured as a reference and compared to the simulated one from the crystal structure (Fig. 4). Confusingly, pr-GCl crystals gave a different PXRD pattern from the simulated one from a crystal structure. In the simulated PXRD pattern, the peak at 2θ = 7.58° corresponding to the crystallographic (010) plane is extremely dominant in terms of intensity. However, in the PXRD pattern of pr-GCl crystals, the dominant peak was absent; instead other peaks appeared which were not found in the simulated pattern. Even the fully ‘dried pr-GCl’ crystals gave a new pattern which does not match that of pr-GCl or the simulated pattern. The dominant peak in the simulated pattern is indicative of the well-ordered bilayers in crystals with a calculated d-spacing of (010) 11.643 Å. Therefore, the disappearance of the peak in the measured patterns means that the bilayer stacking is not present any more in both dried and as-grown crystals, which is probably due to the fast escape of the dichloromethane guests when exposed to air.
Compared to host crystals, all solvent-included crystals showed simpler PXRD patterns (Fig. 4). In some crystals such as the pr-GCl crystals immersed in cyclohexane, o-xylene, or p-xylene relatively strong peaks whose diffraction angles correspond to the (010) and (030) reflections are observed in the simulated pattern. As these peaks are related to the regular separation of bilayers, it is suggested that the host–guest crystals would have similar packing modes to that observed in the pr-GCl crystal structure. In the benzene-included crystals, the strong peaks shifted to higher diffraction angles. In contrast, the m-xylene-included crystals produced a peak at a lower angle, 2θ = 5°. Unfortunately, when pr-GCl crystals are immersed in solvent, they cracked and became unsuitable for SCXRD studies (Fig. S11†). Nevertheless, the changes in the PXRD patterns of the dried pr-GCl crystals upon guest inclusion strongly indicate that the pr-GCl host can have an ability to accommodate those guests by adjusting their relative locations in the crystal lattice. However, without their crystal structures, it is hard to describe in detail the crystal packing of host and guest molecules in each case.
Indirect evidence for this suggestion is that the measured PXRD pattern for dried pt-GCl crystals nicely matched the simulated one from the crystal structure, indicating that the pt-GCl layers are quite tightly packed and hardly disrupted: the strongest peak is attributed to (002) at 2θ = 8.30° with a d-spacing value of 10.650 Å (Fig. 5). In order to further verify the dense packing nature of pt-GCl, guest-inclusion experiments were carried out using the same solvents used for pr-GCl: pt-GCl crystals (ca. 10 mg) were immersed at room temperature in each solvent (2.0 mL) for 1 day, and the collected samples were analysed. As shown in the PXRD patterns in Fig. 5, all samples immersed in different solvents exhibited the same pattern, indicating that in pt-GCl crystals, no available void space is present or the pentyl chains effectively packed among themselves do not allow any guest molecules to diffuse into the crystal lattice. This implies that the host properties of the alkylated purine derivatives can be controlled by changing the length of the side chains while keeping the unique bilayer structures.
 | ||
| Fig. 5 PXRD patterns for pt-GCl samples compared with the simulated one generated from the crystal structure of pt-GCl. The pr-GCl crystals were immersed for 1 day in each solvent listed on the plot before PXRD measurement. For clear comparison, the measured patterns were slightly shifted to lower angles. | ||
Structure of recrystallized pt-GCl in p-xylene
In order to obtain direct evidence of the above speculation, we tried to grow host–guest crystals directly in an aromatic solvent. When the aromatic solvent dissolving the host crystals at about 25 °C was cooled to about 20 °C, guest-included pr-GCl crystals were obtained as very thin plates (Fig. S11†). Except for the case of p-xylene, their measured PXRD patterns matched well to those of the host–guest crystals prepared for the NMR experiments, that is, the pr-GCl crystals immersed in solvent (Fig. S16†). As the crystals are so thin and aggregated, they became easily broken into parts when separated for crystal mounting for SCXRD experiments. After many trials, we managed to collect X-ray data for a single crystal obtained in p-xylene (p-xylene@pr-GCl) using a synchrotron facility. Unfortunately, the data quality was not sufficient (R1 = 26%) but both pr-GCl and disordered p-xylene molecules could be identified successfully (Fig. S14†). As seen in Fig. 6, pr-GCl molecules form bilayers, between which p-xylene molecules are occluded. The bilayers are separated by 11.035 Å which is between 11.643 Å (CH2Cl2@pr-GCl) and 10.650 Å (pt-GCl). It is not understood why the simulated PXRD pattern did not matched the measured one, as with the case observed for pr-GCl (Fig. S16†). Nevertheless, the crystal structure of p-xylene@pr-GCl shows that the hydrogen-bonding networks formed by pr-GCl can be maintained and the characteristic bilayers can also work for intercalating guest molecules. This type of assembly may be regarded as an LSAM (long range synthon Aufbau module) or a large synthon.12a | ||
| Fig. 6 (a) Unit cell of recrystallized pr-GCl in p-xylene (p-xylene@pr-GCl) is shown with van der Waals surfaces of pr-GCl molecules. The empty space is occupied by disordered p-xylene molecules in crystals. (b) Selected pr-GCl layers are displayed with included p-xylene molecules. (c) A top view of (b) shows an array of the guest p-xylene molecules; p-xylene is disordered over two sites in the crystal structure, and one of the possible ordered arrays is presented. | ||
Conclusions
In this work, 6-chloro-9-propyl-purin-2-amine (pr-GCl) molecules are assembled into two-dimensional hydrogen-bonding networks which in turn form bilayers via π–π stacking. The bilayers are coated with alkyl chains, and can intercalate various solvent molecules in crystalline states while keeping the unique hydrogen-bonding networks. However, a similar molecule, 6-chloro-9-pentyl-purin-2-amine (pt-GCl), does not play a host role due to the dense packing of the pentyl chains between the bilayers. Thus, it is anticipated that R-GCl, which is functionalised with other alkyl or aryl groups, will show new guest-inclusion behaviours or recognise specific guest molecules not observed in pr-GCl.Acknowledgements
This research was supported by the Soongsil University Research Fund (No-201110000507). Single-crystal X-ray diffraction data for p-xylene@pr-GCl were collected using a synchrotron radiation facility at the beamline 2D-SMC of Pohang Accelerator Laboratory (PAL).Notes and references
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS PubMed.
- S. V. Kolotuchin, E. E. Fenlon, S. R. Wilson, C. J. Loweth and S. C. Zimmerman, Angew. Chem., Int. Ed. Engl., 1995, 34, 2654 CrossRef CAS.
- K. Guille and W. Clegg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o515 Search PubMed.
- T. J. Prior, J. A. Armstrong, D. M. Benoit and K. L. Marshall, CrystEngComm, 2013, 15, 5838 RSC.
- (a) A. C. Soegiarto, A. Comotti and M. D. Ward, J. Am. Chem. Soc., 2010, 132, 14603 CrossRef CAS PubMed; (b) M. J. Horner, K. T. Holman and M. D. Ward, J. Am. Chem. Soc., 2007, 129, 14640 CrossRef CAS PubMed; (c) K. T. Holman, A. M. Pivovar and M. D. Ward, Science, 2001, 294, 1907 CrossRef CAS PubMed.
- (a) T. C. W. Mak, C.-K. Lam, J. Han, Q. Li and F. Xue, in Organic Crystal Engineering, ed. E. R. T. Tiekink, J. J. Vittal and M. J. Zaworotko, Jhon Wiley & Sons, Ltd, Wiley, 2010, pp. 239–312 Search PubMed; (b) T. C. W. Mak and F. Xue, J. Am. Chem. Soc., 2000, 122, 9860 CrossRef CAS; (c) S. Lie, T. Maris and J. D. Wuest, Cryst. Growth Des., 2014, 14, 3658 CrossRef CAS; (d) C. A. Zentner, H. W. H. Lai, J. T. Greenfield, R. A. Wiscons, M. Zeller, C. F. Campana, O. Talu, S. A. FitzGerald and J. L. C. Rowsell, Chem. Commun., 2015, 51, 11642 RSC; (e) M. E. Garah, R. C. Perone, A. S. Bonilla, S. Haar, M. Campitiello, R. Gutierrez, G. Cuniberti, S. Masiero, A. Ciesielski and P. Samorì, Chem. Commun., 2015, 51, 11677 RSC.
- (a) M. Barceló-Oliver, B. A. Baquero, A. Bauzá, A. García-Raso, A. Terrón, I. Mata, E. Molins and A. Frontera, CrystEngComm, 2012, 14, 5777 RSC; (b) M. Barceló-Oliver, C. Estarellas, A. García-Raso, A. Terrón, A. Frontera, D. Quiñonero, E. Molins and P. M. Deyà, CrystEngComm, 2010, 12, 362 RSC; (c) M. Barceló-Oliver, C. Estarellas, A. García-Raso, A. Terrón, A. Frontera, D. Quiñonero, I. Mata, E. Molins and P. M. Deyà, CrystEngComm, 2010, 12, 3758 RSC; (d) M. Barceló-Oliver, A. Bauzá, B. A. Baquero, A. García-Raso, A. Terrón, E. Molins and A. Frontera, Tetrahedron Lett., 2013, 54, 5355 CrossRef.
- Y.-L. Wu, K. E. Brown and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 13322 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, SHELXS-97, Program for crystal structure analysis, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-2013 (version 2013/4), Programs for crystal structure analysis, University of Göttingen, Göttingen, Germany, 2013 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2011 Search PubMed.
- R. Destro, T. J. Kistenmacher and R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 79 CrossRef CAS.
- (a) A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef CAS PubMed; (b) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS; (c) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- M. V. Vener, A. V. Shishkina, A. A. Rykounov and V. G. Tsirelson, J. Phys. Chem. A, 2013, 117, 8459 CrossRef CAS PubMed.
- (a) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC; (b) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC; (c) M. J. Białek, J. K. Zaręba, J. Janczak and J. Zoń, Cryst. Growth Des., 2013, 13, 4039 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and IR spectra, crystal pictures, crystal and refinement tables, ORTEP drawings, NMR spectra for guest@pr-GCl crystals, and PXRD patterns. CCDC 1417638 (pr-GCl), 1417639 (pt-GCl), and 1417640 (p-xylene@pr-GCl). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01611h |
| This journal is © The Royal Society of Chemistry 2016 |