
Solvent-mediated supramolecular templated assembly of a metal organophosphonate via a crystal–amorphous–crystal transformation†
Shikha
Narang
a,
Udai P.
Singh
*a and
P.
Venugopalan
b
aDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee – 247 667, India. E-mail: udaipfcy@iitr.ac.in
bDepartment of Chemistry, Panjab University, Chandigarh 160014, India
First published on 24th November 2015
Abstract
Three low-dimensional Cu(II)-based organophosphonates were synthesized: two molecular cationic complexes, [Cu2(μ2-L)2(2,2′BPy)2(H2O)2](NO3)2·S (1) and [Cu(L)(2,2′BPy)2]·NO3·(H2O)2 (3), and one neutral complex, [Cu2(μ2-L)2(2,2′BPy)2(NO3)2]·MeOH (2), assembled by the phosphonomonoester i.e., ethylhydrogen(anthracen-9-ylmethyl)phosphonate and an auxillary bidendate ligand (2,2′-bipyridine). Single-crystal X-ray diffraction analysis showed that complex 1 consisted of a dimer unit arranged supramolecularly with a nitrate anion and that these layers were further assembled via π⋯π interactions, which resulted in the formation of a porous supramolecular architecture. The cationic complexes 1 and 3 showed supramolecular templated assembly by simply altering the solvent, which resulted in a change in architecture. The remarkable change from a cationic complex to a neutral complex via the formation of an amorphous solid (1a) (a crystal–amorphous–crystal transformation) was observed when crystals of 1 were heated at 90 °C under vacuum. It is hypothesized that the uncoordinated nitrate ion moved from the lattice to the metal, resulting in the transformation of the cationic metal complex into a neutral metal complex. These complexes also showed luminescent behaviour.
Introduction
There has been much interest in the synthesis of metal organophosphonates bearing a variety of different organic groups for potential applications in sorption,1 catalysis,2 proton conductivity3 and magnetic materials.4 The rational method of controlling the functions and dimensions of the channels in the open framework is to select a suitable interlinking organic group on the phosphonic acid.5,6 The coordination geometry and the charge on the framework is chiefly controlled by the inorganic groups present.7 These dynamic and flexible modular coordination compounds change their structure in response to external stimuli – such as heat, light or the solvent used – and thus exhibit solid state transformation.8,9 Such materials have potential applications10–12 in molecular capture, sensing and switches as a result of the presence of intermolecular interactions, including hydrogen bonds, π–π stacking and van der Waals forces.The rational design and controlled synthesis of supramolecular complexes with similar compositions and supramolecular templated assembly facilitated by a solvent have attracted the attention of researchers.13–15 It is well known that solvent molecules are important structure-directing agents. The use of different solvents may give different coordination metal complexes, resulting in the formation of different crystalline solids that may also show solid state transformation. Such solid state transformations have led to a strategy of switching one material to another by exposure to external stimuli.16–19 This dynamic behaviour of the system is caused by non-covalent interactions, such as π⋯π interactions, and has led to the synthesis of adaptive materials that respond to external stimuli.20–22 The use of this strategy with solid structures has led to reversible solid state transformations, such as single crystal–single crystal, single crystal–amorphous or amorphous–crystal transformations.23–25 There have been many reports on the transformation of 1D, 2D and 3D coordination complexes in the solid state26–28 and many on Cu(II) phosphonates and their molecular analogues.29–34 However, to the best of our knowledge, there have been few reports of the reversible transformation of low-dimensional (0D) materials involving phosphonomonoesters.
The predilection of organophosphonates to show multi-denticity in metal complexes has often resulted in obstacles to the crystallization of the phosphonate due to the formation of insoluble compounds. Various strategies35–41 have been used to overcome this solubility issue in tailoring molecular metal phosphonates, including: (1) the use of sterically hindered phosphonic acid; (2) the use of a chelating ligand; and (3) control of the degree of protonation, i.e. the exploitation of phosphonic acid as a source of monoanions in the bidentate chelation mode. However, reports of the formation of molecular metal complexes of phosphonates are still scarce compared with reports of carboxylates.42–46 We therefore constructed a molecular metal phosphonate from a newly synthesized anthracene-based phosphonomonoester and a chelating ligand (2,2′-bipyridine (2,2′BPy)) keeping these strategies in mind. This is the first reported use of a monoester in the formation of a molecular phosphonate. The monoester reduces the possibility of coordination with the metal ion and increases the solubility of the compound, which, in turn, enhances the probability of later crystallization.
It was of particular interest to study the structural dynamism resulting from the change in the chemical environment available to the framework. We predicted that the synthesized ligand, with the introduction of π⋯π interactions in the extended supramolecular architecture,47–53 might be responsible for the porosity of the structure. We also thought that the complex might display emission properties as a result of the presence of the fluorescent organic group on the phosphonomonoester. We report here three 0D Cu(II) complexes (1–3) based on a newly synthesized ethyl hydrogen(anthracen-9-ylmethyl)phosphonate (L) in the presence of a chelating agent 2,2′BPy (Scheme 1). The cationic framework (1) with extra framework nitrate ions was held together via dynamic non-covalent interactions and showed a crystal–amorphous transformation on desorption of the solvent. Resorption in methanol resulted in a different complex, [Cu2(μ2-L)2(2,2′BPy)2(NO3)2]·MeOH (2). However, the latter complex was a neutral framework with methanol molecules present in the lattice, as deduced by thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD) and single-crystal XRD analyses.
 | ||
| Scheme 1 Representation of complexes 1–4. | ||
Results and discussion
Three metal phosphonate supramolecular solids (1–3) were produced with monophosphonate, 2,2′BPy and Cu(NO3)2 in addition to a supramolecular solid (4) formed by the movement of the phosphonomonoester (L) from the solid complex to the mother liquor. The blue-coloured solid complex 4 was detected when the green crystals of 1 in the mother liquor were allowed to stand at room temperature for 10–15 days. These metal organophosphonates had a different structure when characterized by IR, PXRD and single-crystal XRD analyses. Complex 1 showed a reversible transformation from a crystalline to amorphous structure, which remodelled itself back into the crystalline phase 1′, as characterized by single-crystal XRD analysis.The presence of triethylamine was essential in the synthesis of this system, otherwise it might have been possible to acquire a slightly modified molecular system. The nature of the framework was altered by slight changes in the reaction conditions.32,54 Dinuclear molecular systems were obtained with a change in the solvent. As a result, the solvent-mediated supramolecular templated assembly of metal organophosphonates was achieved with π⋯π interactions between the organic moiety and the auxiliary ligand stacked over each other, resulting in a porous/non-porous extended 3D supramolecular framework.
Description of crystal structure 1
Single-crystal X-ray diffraction analysis revealed that complex 1 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetrical unit contained one crystallographic Cu(II) ion, one molecule each of the monoester and auxiliary ligand 2,2′BPy, one coordinated water molecule and an uncoordinated distorted nitrate anion together with distorted solvent molecules (Fig. 1a). The five-coordinated Cu(II) ion lay on a crystallographic two-fold axis, resulting in the formation of a dinuclear assembly. This dimer assembly acted as a composite building unit for the resulting supramolecular network and was located at an inversion centre in the solid state. The coordination environment around the Cu(II) ion (Fig. S1a†) showed that the Cu atom formed a distorted square pyramidal coordination with the N2O3 donor set. The equatorial plane contained two nitrogen-coordinating sites from same bidentate ligand; these two sites were occupied by two oxygens from the monoester and the fifth site was occupied by the oxygen of the water molecule, which was nearly perpendicular to the plane. The Cu(II)–N1, Cu(II)–N2, Cu(II)–O1, Cu(II)–O3 and Cu(II)–O4(water) distances (2.004(15), 2.015(9), 1.945(13), 1.932(14) and 2.245(13) Å, respectively) were in agreement with those reported for other Cu(II) phosphonate compounds.32,55 Each of two bipyridine moieties and the monoester held the dinuclear assembly together; the latter was also bridged in an isobidentate manner to the metal atom, resulting in the formation of a single chair-shaped eight-membered ring (Cu2O4P2). This had a remarkable resemblance to the S8R building units of zeolites.56 Crystallographic data and other pertinent information for complexes 1–4 and ligand L are given in Table S1.† The selected bond lengths and angles for complexes 1–4 are listed in Table S2.†
. The asymmetrical unit contained one crystallographic Cu(II) ion, one molecule each of the monoester and auxiliary ligand 2,2′BPy, one coordinated water molecule and an uncoordinated distorted nitrate anion together with distorted solvent molecules (Fig. 1a). The five-coordinated Cu(II) ion lay on a crystallographic two-fold axis, resulting in the formation of a dinuclear assembly. This dimer assembly acted as a composite building unit for the resulting supramolecular network and was located at an inversion centre in the solid state. The coordination environment around the Cu(II) ion (Fig. S1a†) showed that the Cu atom formed a distorted square pyramidal coordination with the N2O3 donor set. The equatorial plane contained two nitrogen-coordinating sites from same bidentate ligand; these two sites were occupied by two oxygens from the monoester and the fifth site was occupied by the oxygen of the water molecule, which was nearly perpendicular to the plane. The Cu(II)–N1, Cu(II)–N2, Cu(II)–O1, Cu(II)–O3 and Cu(II)–O4(water) distances (2.004(15), 2.015(9), 1.945(13), 1.932(14) and 2.245(13) Å, respectively) were in agreement with those reported for other Cu(II) phosphonate compounds.32,55 Each of two bipyridine moieties and the monoester held the dinuclear assembly together; the latter was also bridged in an isobidentate manner to the metal atom, resulting in the formation of a single chair-shaped eight-membered ring (Cu2O4P2). This had a remarkable resemblance to the S8R building units of zeolites.56 Crystallographic data and other pertinent information for complexes 1–4 and ligand L are given in Table S1.† The selected bond lengths and angles for complexes 1–4 are listed in Table S2.†
 | ||
| Fig. 1 Crystal structures of (a) complex 1; (b) complex 2; (c) complex 3; and (d) complex 4. C–H bonds are omitted for clarity. | ||
The composite building units were further assembled through π–π stacking interactions, which were mainly responsible for the extension of the 0D structure into a 3D supramolecular network. On scrutiny, it was observed that the 1D chain of the composite unit and the uncoordinated nitrate ion ran parallel to the b–c plane and was linked via hydrogen bonds between the coordinated water molecule and the uncoordinated nitrate ion (O4–H4A⋯O5; 1.932 Å). These 1D chains ran parallel to each other and were glued together via C–H⋯O and C–H⋯N interactions along the a–c plane, resulting in an interdigitated structure (Fig. S2†). These discrete 2D sheets were stacked over one another along the b-axis, resulting in a porous supramolecular framework ending in a 1D channel along the a-axis, accommodated by disordered nitrate and disordered solvent molecules (Fig. 2b). The strength of the complete framework was based on an array of π–π stacking between the π electron cloud of the anthryl group and the 2,2′Bpy.
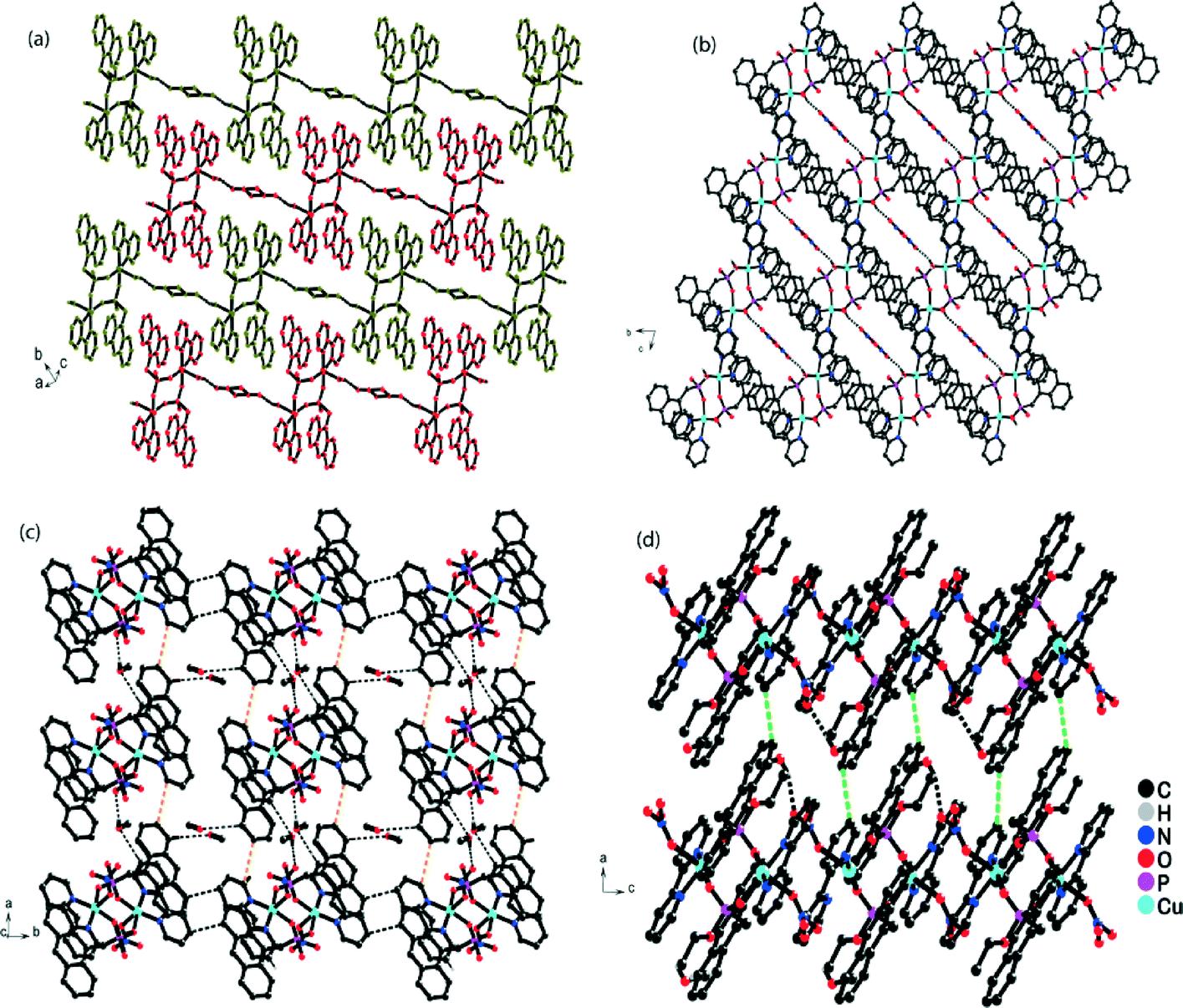 | ||
| Fig. 2 (a) View of the interdigitated parallel 1D chains of the composite unit and nitrate anion in the a–c plane. (b) 3D extended framework in the b–c plane displaying the porous framework with the uncoordinated nitrate anion inside the pores of complex 1. (c) View of 2D sheet representing the interaction of the methanol molecule with the framework along the c-axis in complex 2. (d) 3D representation of complex 2 in the a–c plane displaying the π⋯π interaction between the anthracene moiety and the 2,2′BPy units of neighboring 2D sheets. | ||
A disordered nitrate anion was present in the crystal lattice and some residual electron density was observed by X-ray crystallography, which validated the presence of few disordered solvent molecules. In addition, there was a potentially disordered solvent void of 153.1 Å3 containing 70 electrons per unit cell as determined by PLATON;57 the residual electron density was removed by the SQUEEZE program.
Description of crystal structure 2
The neutral complex [Cu2(μ2-C15H12PO3)2(2,2′BPy)2(NO3)2]·MeOH (2) was formed from the cationic complex 1via solid state transformation (crystal–amorphous–crystal). The coordination environment around the Cu(II) ion remained unchanged, with the same N2O3 donor set in both complexes. However, the crystal system was different in terms of both the unit cell parameters and the space group, which changed from triclinic (P) to monoclinic (P21/c). Structurally, the coordinated water molecule in complex 1 was substituted by a nitrate ion in complex 2, changing the cationic framework to a neutral framework. The bond lengths and angles around the Cu(II) ion were observed to be close to the bond lengths found in complex 1. However, its asymmetrical unit consisted of a neutral composite unit together with an extra framework solvent molecule (methanol) (Fig. 1b). Similar to complex 1, the dimeric unit in 2 lay about an inversion centre in the solid state and formed an S8R ring (Fig. S1b†) with extension to a 2D framework. This assembled further to generate the 3D network, which remained unchanged. The intermolecular non-covalent interactions such as π–π stacking were responsible for increasing the dimensionality of the framework from a 0D structure to a 3D network.
The structural study showed that the composite unit of compound 2 formed a 1D chain along the b-axis, which was then stitched together by π–π stacking between the 2,2′BPy and the anthracene ring of the two different composite units. This 1D chain was extended to 2D via a C12–H12⋯O6 interaction between the anthranyl C–H and the nitrate anion of another layer along the a-axis, leading to the formation of a sheet (Fig. 2c). The 2D sheets were further assembled into a 3D supramolecular polymer through π–π interactions in the a–c plane (Fig. 2d). The offset π–π (centroid–centroid) distance in the interlayer and intralayer were 3.202 and 3.625 Å, respectively, and the π-rings were displaced by 1.253 Å in the interlayer and 1.375 Å in the intralayer (Fig. S3†), as calculated by Olex2 (version 1.2.2).58 Various non-classical interactions such as C–O and C–N were involved in the interlayer sheets (see ESI†). The crystallographic data and selected bond distances of complex 2 are given in Tables S1 and S2,† respectively.
Description of crystal structure 3
X-ray diffraction analysis revealed that complex 3 was also a 0D dinuclear structure with the asymmetrical unit consisting of one Cu(II) centre coordinated by one and two molecules of phosphonomonoester and 2,2′BPy, respectively, along with one nitrate and two water molecules in its lattice (Fig. 1c). The metal ion was penta-coordinated by two bidentate 2,2′BPy ligands and one phosphonomonoester ligand and displayed a distorted square pyramidal coordination geometry with an N4O donor set. Unlike complexes 1 and 2, complex 3 consisted of a mononuclear unit linked supramolecularly to another unit via O–H⋯O H–bonding between the nitrate anion and a water molecule (Fig. 3a and S4†). This H-bonded unit was extended to a 2D sheet via C–H⋯O and C–H⋯π interactions (Fig. 3b and S5†). | ||
| Fig. 3 (a) Discrete chain of the dimer unit with the nitrate anion and water molecule in complex 3. (b) Representation of 2D sheet in complex 3. | ||
The green-coloured crystals of complex 1 changed to a blue colour after 10–15 days of exposure to air as a result of the loss of the phosphonomonoester and were deformed into complex 4. This complex consisted of two bidentate 2,2′BPy ligands and a nitrate anion coordinated with a metal ion, together with one molecule each of a nitrate ion and water in the lattice with an N4O donor set in a distorted square pyramidal geometry (Fig. S6†). Structural analysis revealed that a 1D chain was formed through continuous H-bonding between the nitrate anion and the water molecule (Fig. S7†) and was extended to a 2D sheet via C–H⋯O weak interactions (Fig. S8†).
Solid state transformation (crystal–amorphous–crystal)
We have prepared a porous supramolecular solid (complex 1) by the supramolecular assembly of a dinuclear unit and nitrate anion in the presence of a polar aprotic solvent (acetonitrile). A remarkable effect was observed when crystals of 1 transformed into a green solid powder, 1a, which lost crystallinity on heating under vacuum for 1 day. Complex 1a did not show any X-ray diffraction pattern. Complex 1a was later characterized by PXRD and elemental analysis and found to be amorphous (calculated analysis C 55.81, H 4.16, N 7.23%; found C 55.96, H 4.30, N 7.15%). Solvent-mediated structural transformation was accomplished when the amorphous solid 1a was stored in methanol; it deformed into a different dimer unit, complex 2, in the crystalline form as confirmed by single-crystal analysis. Complex 2 had different cell parameters from complex 1 and structural analysis revealed that the new compound 2 was formed by replacing one coordinated water molecule for each Cu(II) ion by an extra framework nitrate anion. Hence a new non-porous supramolecular polymer was established from a porous supramolecular polymer. Although the geometry of the metal centre remained unchanged, the size and shape of the cavity was altered and hence the overall structural change suggested a dynamic behaviour for complex 1. We tried unsuccessfully to synthesize complex 2 directly from the phosphomonoester and 2,2′BPy. However, when the amorphous solid 1a was immersed in a methanol and acetonitrile mixture, it changed back to the original crystalline form (1′).Another interesting observation was that the green crystals of 1 transmuted to the blue crystals of complex 4 [Cu(2,2′BPy)2·(NO3)]·NO3·H2O on exposure to air for 10–15 days.59 This resulted from the loss of the phosphonomonoester unit from the complex to the mother liquor, as evidenced by single-crystal XRD analysis. Although the destabilization of complex 1 in the presence of moisture is not yet clearly understood, the structural transformation resulting from the destabilization of the architecture has been confirmed by the change in colour and by single-crystal XRD studies.60–62 Another supramolecular solid, complex 3, with a mononuclear unit was observed in the absence of acetonitrile and was sustained by weak interactions, such as H-bonding and π⋯π and C–H⋯π interactions.
On investigating the supramolecular complexes 1–3, it was found that the solvent played an important role in the synthesis and transformation of these supramolecular solids as it controlled the crystal structure, dynamics and flexibility of the network in the crystal–amorphous–crystal transformation. It was inferred that acetonitrile, as the solvent, played an important role in synthesizing the dimer complex of the phosphonomonoester, otherwise a monomeric unit would be formed. The stability of these flexible complexes was studied by TGA, elemental analysis, PXRD and IR spectrometry. Complex 1 lost methanol and a coordinated water molecule in the temperature range 80–110 °C (analysis calculated 7.22%; found 6.77%), as deduced by the TGA profile, and produced the dehydrated complex 1a [Cu2(C15H12PO3)2(2,2′BPy)2]·(NO3)2 (Fig. 4b). As the removal of water breaks the interlayer H-bonding, it was anticipated that these molecules would arrange themselves to form an amorphous state.63 This was also supported by IR spectrometry, which showed that the peak for coordinated water was absent in the IR spectra of complex 1a (Fig. 4a). The characteristic medium-intensity IR absorption band of M–OH in complex 2 appeared at 3765 cm−1 and that of N–O at 1370 cm−1, corresponding to the coordinated water molecule and free nitrate anion, respectively. The broad absorption band at 2920 cm−1 in complex 2 showed the presence of the H-bonded O–H in the methanol molecule and the corresponding absorption band for coordinated N–O from nitrate was seen at 1320 cm−1. PXRD clearly showed that complexes 1 and 1′ were the same because the observed peaks were well matched. However, the absence of peaks in the PXRD pattern of complex 1a demonstrated that complex 1a was amorphous (Fig. 5a).
 | ||
| Fig. 4 (a) IR spectra of complexes 1, 1a, 1′ and 2 confirming the reversibility of the complexes. (b) TGA plots for complexes 1–3. | ||
 | ||
| Fig. 5 (a) PXRD patterns of supramolecular isomers. (b) Emission spectra of ligand L and complexes 1–3 in the solid state at room temperature. | ||
Luminescence properties
The solid state emission of the ligand and complexes 1–3 was investigated at room temperature (Fig. 5b). The fluorescent spectra of these complexes in the solid state displayed the maximum emission at 516, 482 and 492 nm, respectively, with an excitation wavelength of 215 nm. However, the ligand showed an emission at 433 nm on excitation at 350 nm, which can be assigned to a π–π* transition.64 We suggest that the emission of complexes 1–3 in the solid state was mainly due to a metal-to-ligand charge transfer transition. The fluorescence spectra of the Cu(II) organophosphonate complexes exhibited a red shift compared with that of the ligand, probably due to back-coupling of the π-bond between the metal and the ligand, which further increased the mobility of the electron transition and enhanced the π–π* conjugation length of the coordination of the ligand with the metal attributable to the five-membered ring. This resulted in conformational co-planarity and further reduced the energy gap between the π and π* molecular orbitals of the ligand.65 The increase in conformational rigidity in the molecular structure with the introduction of the metal ion caused the metal complexes to fluoresce.Conclusion
It is hypothesized that the uncoordinated nitrate anion moved from the lattice to the metal, resulting in modification of the cationic molecular complex to a neutral complex via an amorphous state. The dynamic nature of supramolecular complex 1 means that it can reversibly switch to another supramolecular solid 2 by passing through the amorphous state and can also convert back to its original form in the presence of a mixture of CH3OH/CH3CN. However, the importance of acetonitrile was observed when different crystalline forms were seen in methanol and were characterized by IR, PXRD and single-crystal XRD. The emission of the three complexes 1–3 in the solid state was studied at room temperature and the results suggest that the complexes show good luminescence properties as a result of a metal-to-ligand charge transfer transition.Acknowledgements
The authors gratefully acknowledge CSIR, New Delhi, India for financial assistance in the form of SRF to SN.References
- M. Taddei, F. Costantino, A. Ienco, A. Comotti, P. V. Dau and S. M. Cohen, Chem. Commun., 2013, 49, 1315–1317 RSC.
- T.-B. Liao, Y. Ling, Z.-X. Chen, Y.-M. Zhou and L.-H. Weng, Chem. Commun., 2010, 46, 1100–1102 RSC.
- G. Alberti, M. Casciola, E. D'Alessandro and M. Pica, J. Mater. Chem., 2004, 14, 1910–1914 RSC.
- J. A. Sheikh, A. Adhikary, H. S. Jena, S. Biswas and S. Konar, Inorg. Chem., 2014, 53, 1606–1613 CrossRef CAS PubMed.
- R. Vivani, F. Costantino, U. Costantino and M. Nocchetti, Inorg. Chem., 2006, 45, 2388–2390 CrossRef CAS PubMed.
- R. Vivani, G. Alberti, F. Costantino and M. Nocchetti, Microporous Mesoporous Mater., 2008, 107, 58–70 CrossRef CAS.
- J. Goura and V. Chandrasekhar, Chem. Rev., 2015, 115, 6854–6965 CrossRef CAS PubMed.
- G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 21, 1755–1775 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- S. K. Ghosh, W. Kaneko, D. Kiriya, M. Ohba and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 8843–8847 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., 2004, 116, 2388–2430 CrossRef.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed.
- Z. Ju and D. Yuan, CrystEngComm, 2013, 15, 9513–9520 RSC.
- G. Aromí, P. Gamez, O. Roubeau, P. C. Berzal, H. Kooijman, A. L. Spek, W. L. Driessen and J. Reedijk, Inorg. Chem., 2002, 41, 3673–3683 CrossRef.
- I. S. Lee, D. M. Shin and Y. K. Chung, Chem. – Eur. J., 2004, 10, 3158–3165 CrossRef CAS PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed.
- D.-X. Xue, W.-X. Zhang, X.-M. Chen and H.-Z. Wang, Chem. Commun., 2008, 1551–1553 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. D. Patel and J. B. Benedict, Chem. Commun., 2013, 49, 8012–8014 RSC.
- T. K. Maji, G. Mostafa, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2005, 127, 17152–17153 CrossRef CAS PubMed.
- M. P. Suh, J. W. Ko and H. J. Choi, J. Am. Chem. Soc., 2002, 124, 10976–10977 CrossRef CAS PubMed.
- A. J. Fletcher, E. J. Cussen, D. Bradshaw, M. J. Rosseinsky and K. M. Thomas, J. Am. Chem. Soc., 2004, 126, 9750–9759 CrossRef CAS PubMed.
- J. Piromchom, N. Wannarit, J. Boonmak, C. Pakawatchai and S. Youngme, Inorg. Chem. Commun., 2014, 40, 59–61 CrossRef CAS.
- J. Piromchom, N. Wannarit, J. Boonmak, K. Chainok, C. Pakawatchai and S. Youngme, Inorg. Chem. Commun., 2014, 44, 111–113 CrossRef CAS.
- A. Cheansirisomboon, C. Pakawatchai and S. Youngme, Dalton Trans., 2012, 41, 10698–10706 RSC.
- P. Shen, W.-W. He, D.-Y. Du, H.-L. Jiang, S.-L. Li, Z.-L. Lang, Z.-M. Su, Q. Fu and Y.-Q. Lan, Chem. Sci., 2014, 5, 1368–1374 RSC.
- R. Medishetty, L. L. Koh, G. K. Kole and J. J. Vittal, Angew. Chem., Int. Ed., 2011, 50, 10949–10952 CrossRef CAS PubMed.
- M. Oh, L. Rajput, D. Kim, D. Moon and M. S. Lah, Inorg. Chem., 2013, 52, 3891–3899 CrossRef CAS PubMed.
- V. Chandrasekhar and S. Kingsley, Angew. Chem., Int. Ed., 2000, 39, 2320–2322 CrossRef CAS.
- K. D. Demadis, E. Barouda, N. Stavgianoudaki and H. Zhao, Cryst. Growth Des., 2009, 9, 1250–1253 CAS.
- L. Liu, Z.-G. Sun, N. Zhang, Y.-Y. Zhu, Y. Zhao, X. Lu, F. Tong, W.-N. Wang and C.-Y. Huang, Cryst. Growth Des., 2010, 10, 406–413 CAS.
- V. Chandrasekhar, T. Senapati, E. C. Sañudo and R. Clérac, Inorg. Chem., 2009, 48, 6192–6204 CrossRef CAS PubMed.
- C.-I. Yang, Y.-T. Song, Y.-J. Yeh, Y.-H. Liu, T.-W. Tseng and K.-L. Lu, CrystEngComm, 2011, 13, 2678–2686 RSC.
- V. Chandrasekhar, L. Nagarajan, S. Hossain, K. Gopal, S. Ghosh and S. Verma, Inorg. Chem., 2012, 51, 5605–5616 CrossRef CAS PubMed.
- V. Chandrasekhar, L. Nagarajan, R. Clèrac, S. Ghosh and S. Verma, Inorg. Chem., 2008, 47, 1067–1073 CrossRef CAS PubMed.
- V. Chandrasekhar, T. Senapati, A. Dey and E. C. Sañudo, Inorg. Chem., 2011, 50, 1420–1428 CrossRef CAS PubMed.
- Y. S. Ma, Y. Song, Y. Z. Li and L. M. Zheng, Inorg. Chem., 2007, 46, 5459–5461 CrossRef CAS PubMed.
- E. K. Brechin, R. A. Coxall, A. Parkin, S. Parsons, P. A. Tasker and R. E. P. Winpenny, Angew. Chem., 2001, 113, 2772–2775 ( Angew. Chem., Int. Ed. , 2001 , 40 , 2700–2703 ) CrossRef.
- V. Baskar, M. Shanmugam, E. C. Sañudo, D. Collison, E. J. L. McInnes, Q. Wei and R. E. P. Winpenny, Chem. Commun., 2007, 37–39 RSC.
- G. S. Teat, T. Mallah, R. Sessoli, W. Wernsdorfer and R. E. P. Winpenny, Angew. Chem., 2005, 117, 5172–5176 ( Angew. Chem., Int. Ed. , 2005 , 44 , 5044–5048 ) CrossRef.
- Z.-Y. Du, X.-L. Li, Q.-Y. Liu and J.-G. Mao, Cryst. Growth Des., 2007, 7, 1501–1507 CAS.
- F. Wang, X. Jing, B. Zheng, G. Li, G. Zeng, Q. Huo and Y. Liu, Cryst. Growth Des., 2013, 13, 3522–3527 CAS.
- S. N. Zhao, S. Q. Su, X. Z. Song, M. Zhu, Z. M. Hao, X. Meng, S. Song and H. J. Zhang, Cryst. Growth Des., 2013, 13, 2756–2765 CAS.
- S. Sanda, S. Parshamoni, A. Adhikary and S. Konar, Cryst. Growth Des., 2013, 13, 5442–5449 CAS.
- Q. Han, C. He, M. Zhao, B. Qi, J. Niu and C. Duan, J. Am. Chem. Soc., 2013, 135, 10186–10189 CrossRef CAS PubMed.
- G. L. Liu, Y. J. Qin, L. Jing, G. Y. Wei and H. Li, Chem. Commun., 2013, 49, 1699–1701 RSC.
- A. Hazra, K. L. Gurunatha and T. K. Maji, Cryst. Growth Des., 2013, 13, 4824–4836 CAS.
- M. L. Cheney, G. J. McManus, J. A. Perman, Z. Wang and M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 616–617 CAS.
- G. R. Desiraju, Cryst. Growth Des., 2011, 11, 896–898 CAS.
- V. R. Hathwar, T. S. Thakur, R. Dubey, M. S. Pavan, T. N. G. Row and G. R. Desiraju, J. Phys. Chem. A, 2011, 115, 12852–12863 CrossRef CAS PubMed.
- S. Takamizawa, E. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 42, 4326–4331 CrossRef PubMed.
- R. Mondal, J. A. K. Howard, R. Banerjee and G. R. Desiraju, Cryst. Growth Des., 2006, 6, 2507–2516 CAS.
- R. Haldar, K. V. Rao, S. J. George and T. K. Maji, Chem. – Eur. J., 2012, 18, 5848–5852 CrossRef CAS PubMed.
- V. Chandrasekhar and L. Nagarajan, Inorg. Chem., 2012, 51, 8479–8487 CrossRef CAS PubMed.
- V. Chandrasekhar, P. Sasikumar, R. Boomishankar and G. Anantharaman, Inorg. Chem., 2006, 45, 3344–3351 CrossRef CAS PubMed.
- K. Yoshida, K. Toyoura, K. Matsunaga, A. Nakahira, H. Kurata, Y. H. Ikuhara and Y. Sasaki, Sci. Rep., 2013, 3, 2457 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. J. Fereday, P. Hodgson, S. Tyagi and B. J. Hathaway, J. Chem. Soc., Dalton Trans., 1981, 2070–2077 RSC.
- S. Supriya and S. K. Das, J. Am. Chem. Soc., 2007, 129, 3464–3465 CrossRef CAS PubMed.
- S. M. Mobin, A. K. Srivastava, P. Mathur and G. K. Lahiri, Inorg. Chem., 2009, 48, 4652–4654 CrossRef CAS PubMed.
- Z. Chen, S. Xiang, D. Zhao and B. Chen, Cryst. Growth Des., 2009, 9, 5293–5296 CAS.
- A. N. Khlobystov, N. R. Champness, C. J. Roberts, S. J. B. Tendler, C. Thompson and M. Schroder, CrystEngComm, 2002, 4, 426–431 RSC.
- C. K. Xia, C. Z. Lu, Q. Z. Zhang, X. He, J. J. Zhang and D. M. Wu, Cryst. Growth Des., 2005, 5, 1569–1574 Search PubMed; T. Yutaka, S. Obara, S. Ogawa, K. Nozaki, N. Ikeda and T. Ohno, Inorg. Chem., 2005, 44, 4737–4746 CrossRef CAS PubMed.
- M. W. Perkovic, Inorg. Chem., 2000, 39, 4962–4968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure solution and refinement procedures, experimental details for ligand synthesis and complexes 1–4, supplementary figures, including tables of crystallographic data, CIF files, and anisotropic thermal ellipsoids for complexes 1–4 reported in this paper. This material is available free of charge via the Internet at http://pubs.acs.org. Crystallographic data (excluding structure factors) for the structures and ligand reported in this paper have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as deposition no. CCDC 1042812–1042815, 1433403 and 1044127. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01594d |
| This journal is © The Royal Society of Chemistry 2016 |