
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric aerobic oxidative NHC-catalysed synthesis of dihydropyranones utilising a system of electron transfer mediators†
A.
Axelsson
,
E.
Hammarvid
,
L.
Ta
and
H.
Sundén
 *
*
Chemistry and Chemical Engineering, Chalmers University of Technology, Kemivägen 10 412 96 Göteborg, Sweden. E-mail: sundenh@chalmers.se
First published on 7th September 2016
Abstract
In the context of green chemistry, the replacement of high molecular weight stoichiometric oxidants with O2 is most desirable but difficult. Here, we report the asymmetric aerobic oxidative synthesis of dihydropyranones. The oxidation is aided by a system of electron transfer mediators and is selective toward the homoenolate. The dihydropyranones can be isolated in high to excellent yields, with high ee (up to 95%).
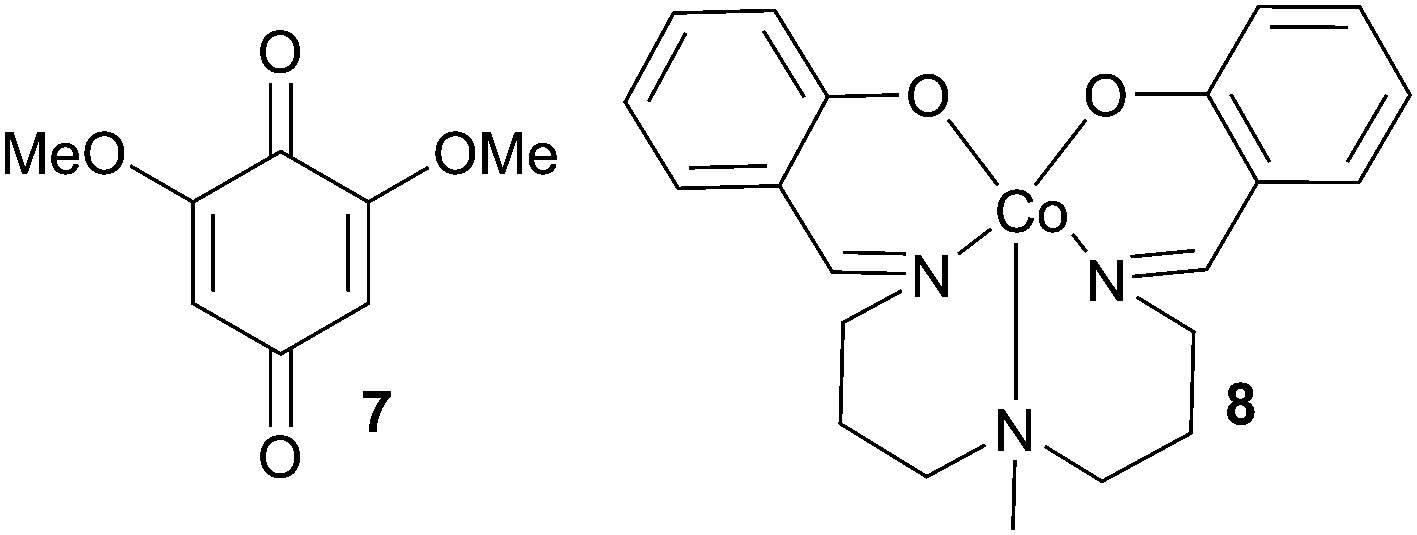
Within the field of asymmetric N-heterocyclic carbene (NHC) catalysis,1–8 oxidative reaction paths have emerged as an attractive way to activate α,β-unsaturated aldehydes towards nucleophiles.9–11 Mechanistically, this strategy includes a selective in situ oxidation of the homoenolate to the α,β-unsaturated acyl azolium (Scheme 1a).12 The oxidatively derived acyl azolium is a well-exploited reaction intermediate and a prominent starting point to introduce stereodiscrimination by using chiral catalysts. Kharasch oxidant (1, Scheme 1) is the most frequently used oxidant in NHC catalysis.13 However, it suffers from a high molecular weight, resulting in a high E factor for these reactions,14 thus severely restricting the scalability of these reactions.
 | ||
| Scheme 1 (a) Asymmetric oxidative carbene-catalysed formation of dihydropyranones. (b) Stoichiometric oxidation with the Kharasch oxidant. (c) Direct aerobic oxidation with O2. (d) Aerobic oxidation via multistep electron transfer. | ||
For reasons of atom economy and waste prevention, the replacement of high molecular weight stoichiometric oxidants with oxygen (O2) is most desirable. However, oxidations using O2 are problematic, due to a high energy barrier for the reaction between oxygen and the substrate leading to unselective reactions.15 In oxidative NHC catalysis direct usage of oxygen results in carboxylic acid formation as the main product16–18 or as a stoichiometric byproduct.19
In synthesis, a common method of addressing the issue of the poor reactivity of O2 has been to mimic the respiratory chain20 by introducing a system of electron transfer mediators (ETMs).15 ETMs provide a low-energy path for the electrons to flow from the substrate to a suitable terminal oxidant such as O2. A prime example within transition-metal catalysis, where ETMs enable the use of O2 as the terminal oxidant, is the Wacker oxidation in which CuCl2 serves as the ETM between Pd0 and O2.21–24 In this respect, Bäckvall and co-workers have shown that coupled ETMs are applicable to a plethora of reactions, for instance in the aerobic 1,4-oxidations of 1,3-dienes,25,26 aerobic oxidative carbocyclisations27,28 and aerobic oxidations of alcohols.29–31
Dihydropyranones are an interesting class of compounds, as they can be found in several natural products32,33 and can be used in the synthesis of, for example, 1,5-diketones,34 2-cyclohexanones,35 dihydro-2-pyridones,36 and pyrones.37–39 Thus, several enantioselective NHC-catalysed19,40–45 and transition-metal-catalysed46,47 strategies have been developed. However, the majority suffer from poor atom economy (as in the use of stoichiometric oxidants),40,43,45 incorporated leaving groups,42,44,48 sacrificial reagents19 and coupling reagents.49 Clearly, an enantioselective, mild and environmentally benign entry to this scaffold would be of great interest.
Here, as a continuation of our interest in aerobic multistep electron transfer NHC catalysis,50 we present an asymmetric synthesis of dihydropyranones.
Initial screening was focused on identifying reaction conditions compatible with open reaction vessels and wet solvents, using the synthesis of dihydropyranone 6 from acetylacetone 5 and cinnamaldehyde (Table 1) as the model reaction. The screening was performed with stoichiometric amounts of 1 and under these constraints NHC catalyst 3, LiOAc dihydrate as a base and toluene as the reaction solvent proved to be vital for generating reactions with high yield and ee (see ESI† for details). We then tested whether the enantioselective synthesis of dihydropyranones would be compatible with a coupled system of electron transfer mediators (Table 1), enabling the use of aerial O2 as the terminal oxidant. With the ETM/ETM′ combination bis(salicylideniminato-3-propyl)methylaminocobalt(II) (8) and 1, the reaction delivered 6, albeit at poor yield with 94% ee (entry 1). Changing the quinone from 1 to 7 improved the yield (58%, entry 2). A small improvement was achieved when using iron(II) phthalocyanine (FePc) and 7 as the ETM couple and dihydropyranone 6 could be isolated in 62% after 72 h (entry 3). The reaction time could be shortened by using couple 1 and FePc as ETMs, yielding 6 at 62% yield with 95% ee after 48 h. Heating the reaction at 40 °C was key to shortening the reaction times to acceptable levels (25 h, entry 5). Interestingly, the reaction conducted in an atmosphere of pure O2 did not render any product (entry 6). A reasonable explanation could be that an excessive concentration of O2 promoted the inactivation of the FePc. This inactivation could be due to formation of the oxygen-bridged FePc dimer, which is catalytically inactive under our reaction conditions.51 This result prompted us to investigate whether the FePc was stable during the course of the reaction. Following 1 and its reduced phenolic form on GCMS revealed that after approximately 3 h, only the reduced form of 1 was present in the reaction mixture. This could be a direct result of the formation of the catalytically inactive dimer species. The argument is strengthened by the fact that an additional portion of FePc reforms the oxidised form of 1. Consequently, the optimal result was achieved with sequential addition of FePc and 6 could be isolated in 79% yield with 94% ee (entry 11). Moreover, the background oxidation was probed by systematically eliminating each of the involved redox species (Table 2). Without quinone 1 or FePc the reactions perform poorly (entries 7 and 8). With no ETMs the reaction completely shuts down (entry 9), and without any O2 present, only small amounts of product can be isolated (17%, entry 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Temp. (°C) | Time (h) | ETM′ (eq.) | ETM (eq.) | Yield (%) | ee (%) |
| a The reactions were performed in open reaction vessels at the indicated temperature (see table) in toluene with cinnamaldehyde 3 (1 eq.), acetylacetone 4 (3 eq.), 3 (0.1 eq.), LiOAc·2H2O (0.65 eq.) ETM (see table), ETM′ (see table). b The reaction was conducted in an atmosphere of pure O2. c Yield was determined with NMR against internal standard. d Reaction conducted under an atmosphere of nitrogen. e Sequential addition of FePc see ESI for details. ND = not determined. | ||||||
| 1 | r.t | 46 | 8 (0.02) | 1 (0.1) | 29 | 94 |
| 2 | r.t | 48 | 8 (0.02) | 7 (0.1) | 58 | ND |
| 3 | r.t | 72 | FePc (0.02) | 7 (0.1) | 62 | 95 |
| 4 | r.t | 48 | FePc (0.02) | 1 (0.1) | 62 | 95 |
| 5 | 40 | 22 | FePc (0.02) | 1 (0.1) | 69 | 95 |
| 6b | r.t | 48 | FePc (0.02) | 1 (0.1) | 0 | — |
| 7c | r.t | 48 | FePC (0.02) | — | 6 | ND |
| 8c | r.t | 48 | — | 1 (0.1) | 7 | ND |
| 9c | r.t | 48 | — | — | 0 | — |
| 10d | r.t | 48 | FePC (0.02) | 1 (0.1) | 17 | ND |
| 11e | 40 | 25 | FePc (0.04) | 1 (0.02) | 79 | 94 |
|
||||||

| a The reactions were performed in open reaction vessels at 40 °C in toluene (2 mL) with catalyst 3 (0.1 eq.), α,β-unsaturated aldehyde (1 eq.), 1,3-dicarbonyl (3 eq.), LiOAc·2H2O (1 eq.) FePc (0.006 eq.) and 2 (0.2 eq.). b Isolated yields after purification with silica gel chromatography. c Major isomer combined yield r.r. determined by 1H NMR of the crude reaction mixture. |
|---|

|
Having identified our optimal reaction conditions, the scope of the reaction was examined (Table 2). The reaction performs well for a wide range of cinnamaldehydes in combination with acetylacetone as the nucleophile, and the corresponding dihydropyranones can be isolated in generally high yields with ee values of 81–94%. For example, p-chloro-cinnamaldehyde can be converted to the corresponding dihydropyranone 9 in 80% yield and 91% ee. Aliphatic aldehydes are also viable reaction partners with this strategy and 14 was isolated in 65% yield and 83% ee. Ketoesters as the nucleophilic reaction partner are also permitted by the reaction, giving high to excellent yields of the corresponding dihydropyranones (61–86% yield; 85–95% ee, entries 15–22). For example, ethyl 3-oxobutanoate can be reacted with furan containing α,β-unsaturated aldehyde to give lactone 19 61% yield and 90% ee. Moreover, methyl 3-oxobutanoate reacts with cinnamaldehyde to deliver the annulated product 20 in 79% yield and 95% ee. Asymmetrical aryl ketones are also efficient nucleophiles in this reaction and dihydropyranone 23 can be obtained in 67% combined yield in a 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 regioisomeric ratio and 87% ee of the major isomer. Encouraged by the efficiency and selectivity of the system, we examined whether the dihydropyranones could be functionalised in a tractable manner. In this respect, a transesterification would be of particular importance, as the products of such reaction are difficult to form via direct Michael addition of acetylacetone to cinnamates.52 Accordingly, dihydropyranone 6 was subjected to an NHC/base-catalysed transesterification with methanol, achieving 95% yield of ester 24 with maintained optical purity (Scheme 2).
:7 regioisomeric ratio and 87% ee of the major isomer. Encouraged by the efficiency and selectivity of the system, we examined whether the dihydropyranones could be functionalised in a tractable manner. In this respect, a transesterification would be of particular importance, as the products of such reaction are difficult to form via direct Michael addition of acetylacetone to cinnamates.52 Accordingly, dihydropyranone 6 was subjected to an NHC/base-catalysed transesterification with methanol, achieving 95% yield of ester 24 with maintained optical purity (Scheme 2).
 | ||
| Scheme 2 Transesterification of dihydropyranones to methyl ester 24. | ||
The catalytic cycle starts with deprotonation of the chiral triazolium salt to generate NHC 25 (Scheme 3). The NHC adds to the α,β-unsaturated aldehyde 26 to produce homoenolate 27. The homoenolate is oxidised to the unsaturated acyl azolium 28 by O2, via a multistep electron transfer, mediated by the systems of ETMs. In the selectivity determining step 1,3-keto compound 30 adds to the acyl azolium in a 1,4-fashion producing intermediate 31.45,53–55 Tautomerisation followed by cyclisation delivers the product 33 and regenerates the NHC.
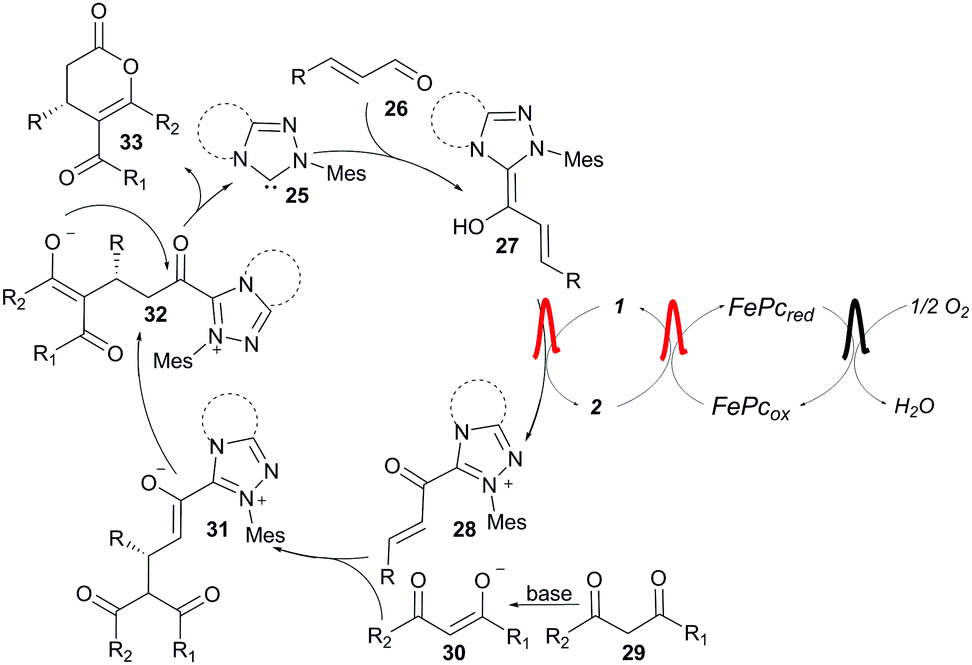 | ||
| Scheme 3 Catalytic cycle. | ||
In summary, we have developed an aerobic enantioselective synthesis of dihydropyranones relying on multistep electron transfer. We show that the homoenolate, derived from α,β-unsaturated aldehydes and a chiral catalyst, can be oxidised to the acyl azolium, by air, utilising a system of ETMs. The oxidation of the homoenolate is selective; consequently, no excess of aldehyde is required to compensate for the formation of carboxylic acid that normally accompanies aerobic oxidative NHC catalysis. Furthermore, the procedure demonstrates that stoichiometric use of high molecular weight oxidants such as the Kharasch oxidant (1) is not necessary in order to access important chemical processes in this field.
This work was generously supported by the Swedish Research Council VR and Formas.
Notes and references
- D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CAS.
- J. Mahatthananchai and J. W. Bode, in Contemporary Carbene Chemistry, John Wiley & Sons, Inc, 2013, ch. 9, pp. 237–273 DOI:10.1002/9781118730379.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- J. Vesely and R. Rios, Curr. Org. Chem., 2011, 15, 4046–4082 CrossRef CAS.
- S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem. – Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed.
- C. E. I. Knappke, A. Imami and A. J. von Wangelin, ChemCatChem, 2012, 4, 937–941 CrossRef CAS.
- J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef CAS PubMed.
- E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef CAS PubMed.
- M. S. Kharasch and B. S. Joshi, J. Org. Chem., 1957, 22, 1439–1443 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- P.-C. Chiang and J. W. Bode, Org. Lett., 2011, 13, 2422–2425 CrossRef CAS PubMed.
- B. Maji, S. Vedachalan, X. Ge, S. Cai and X.-W. Liu, J. Org. Chem., 2011, 76, 3016–3023 CrossRef CAS PubMed.
- Y.-C. Xin, S.-H. Shi, D.-D. Xie, X.-P. Hui and P.-F. Xu, Eur. J. Org. Chem., 2011, 6527–6531 CrossRef CAS.
- D. Xie, D. Shen, Q. Chen, J. Zhou, X. Zeng and G. Zhong, J. Org. Chem., 2016, 81, 6136–6141 CrossRef CAS PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, 4th edn, 2004 Search PubMed.
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Ruttinger and H. Kojer, Angew. Chem., 1959, 71, 176–182 CrossRef CAS.
- J. K. Stille and R. Divakaruni, J. Am. Chem. Soc., 1978, 100, 1303–1304 CrossRef CAS.
- J. E. Bäckvall, B. Åkermark and S. O. Ljunggren, J. Am. Chem. Soc., 1979, 101, 2411–2416 CrossRef.
- J.-E. Bäckvall and R. B. Hopkins, Tetrahedron Lett., 1988, 29, 2885–2888 CrossRef.
- J. E. Bäckvall, A. K. Awasthi and Z. D. Renko, J. Am. Chem. Soc., 1987, 109, 4750–4752 CrossRef.
- J.-E. Bäckvall, R. B. Hopkins, H. Grennberg, M. Mader and A. K. Awasthi, J. Am. Chem. Soc., 1990, 112, 5160–5166 CrossRef.
- J. Piera, K. Närhi and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2006, 45, 6914–6917 CrossRef CAS PubMed.
- E. V. Johnston, E. A. Karlsson, S. A. Lindberg, B. Åkermark and J.-E. Bäckvall, Chem. – Eur. J., 2009, 15, 6799–6801 CrossRef CAS PubMed.
- J.-E. Bäckvall, R. L. Chowdhury and U. Karlsson, J. Chem. Soc., Chem. Commun., 1991, 473–475 RSC.
- G.-Z. Wang, U. Andreasson and J.-E. Bäckvall, J. Chem. Soc., Chem. Commun., 1994, 1037–1038 RSC.
- G. Csjernyik, A. H. Ell, L. Fadini, B. Pugin and J.-E. Bäckvall, J. Org. Chem., 2002, 67, 1657–1662 CAS.
- T. Sugiyama, T. Murayama, K. Yamashita and T. Oritani, Biosci., Biotechnol., Biochem., 1995, 59, 1921–1924 CAS.
- L. Candish and D. W. Lupton, Org. Lett., 2010, 12, 4836–4839 CAS.
- D. C. Harrowven and J. C. Hannam, Tetrahedron, 1999, 55, 9333–9340 CrossRef CAS.
- F.-Y. Zhang and E. J. Corey, Org. Lett., 2000, 2, 1097–1100 CrossRef CAS PubMed.
- S. H. Thang and D. J. Rigg, Synth. Commun., 1993, 23, 2355–2361 CrossRef CAS.
- D. Rosenthal, P. Grabowich, E. F. Sabo and J. Fried, J. Am. Chem. Soc., 1963, 85, 3971–3979 CrossRef CAS.
- T. Kume, H. Iwasaki, Y. Yamamoto and K.-y. Akiba, Tetrahedron Lett., 1988, 29, 3825–3828 CrossRef CAS.
- A. K. Mandal and D. G. Jawalkar, J. Org. Chem., 1989, 54, 2364–2369 CrossRef CAS.
- Z.-Q. Zhu, X.-L. Zheng, N.-F. Jiang, X. Wan and J.-C. Xiao, Chem. Commun., 2011, 47, 8670–8672 RSC.
- Z.-Q. Rong, M.-Q. Jia and S.-L. You, Org. Lett., 2011, 13, 4080–4083 CrossRef CAS PubMed.
- G. Wang, X. Chen, G. Miao, W. Yao and C. Ma, J. Org. Chem., 2013, 78, 6223–6232 CrossRef CAS PubMed.
- J. Mo, L. Shen and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 8588–8591 CrossRef CAS PubMed.
- Q. Ni, J. Xiong, X. Song, G. Raabe and D. Enders, Chem. Commun., 2015, 51, 14628–14631 RSC.
- S. De Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266–9269 CrossRef CAS PubMed.
- D. A. Evans, R. J. Thomson and F. Franco, J. Am. Chem. Soc., 2005, 127, 10816–10817 CrossRef CAS PubMed.
- K. Itoh, M. Hasegawa, J. Tanaka and S. Kanemasa, Org. Lett., 2005, 7, 979–981 CrossRef CAS PubMed.
- F.-G. Sun, L.-H. Sun and S. Ye, Adv. Synth. Catal., 2011, 353, 3134–3138 CrossRef CAS.
- Y. Que, Y. Lu, W. Wang, Y. Wang, H. Wang, C. Yu, T. Li, X.-S. Wang, S. Shen and C. Yao, Chem. – Asian J., 2016, 11, 626 CrossRef CAS.
- L. Ta, A. Axelsson and H. Sundén, Green Chem., 2016, 18, 686–690 CAS.
- C. Ercolani, M. Gardini, F. Monacelli, G. Pennesi and G. Rossi, Inorg. Chem., 1983, 22, 2584–2589 CrossRef CAS.
- For a catalytic asymmetric isothiourea promoted formation of dihydropyranones and in situ ring opening process see, E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193–2200 RSC.
- S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176–14177 CrossRef CAS PubMed.
- R. C. Samanta, B. Maji, S. De Sarkar, K. Bergander, R. Froehlich, C. Mueck-Lichtenfeld, H. Mayr and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 5234–5238 CrossRef CAS PubMed.
- The selectivity determining step have been debated and another possible approach is an oxo 1,2-addition to the acyl azolium followed by a Claisen rearrangement to intermediate 31. See, J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810–8812 CrossRef CAS PubMed and B. Wanner, J. Mahatthananchai and J. W. Bode, Org. Lett., 2011, 13, 5378–5381 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H NMR, 13C NMR and 19F NMR data. See DOI: 10.1039/c6cc06060a |
| This journal is © The Royal Society of Chemistry 2016 |