
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly diastereoselective entry into chiral spirooxindole-based 4-methyleneazetidines via formal [2+2] annulation reaction†
G.
Rainoldi
,
M.
Faltracco
,
L.
Lo Presti
,
A.
Silvani
 * and
G.
Lesma
* and
G.
Lesma
Dipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, Milano, 20133, Italy. E-mail: alessandra.silvani@unimi.it
First published on 26th August 2016
Abstract
A diastereoselective, DABCO-catalyzed reaction of isatin-derived ketimines with allenoates is described. Exploiting the stereodirecting effect of the bulky tert-butanesulfinyl chiral auxiliary, the method provides an efficient access to single diastereoisomers of unprecedented spirocyclic oxindoles, bearing a 4-methyleneazetidine ring at the spiro junction. The versatility of the method is fully demonstrated by further transformations including the conversion to relevant spirocyclic oxindolo-β-lactams.
The strained four-membered ring system of azetidines1 is present as a partial structure of a number of natural products2 and pharmaceutical agents3 (Fig. 1). Owing to their strong molecular rigidity and, at the same time, to their reasonable stability, azetidines are of special interest, being able to potentially provide improved physicochemical properties in their interaction with biological systems.
 | ||
| Fig. 1 Examples of biologically relevant compounds containing azetidine and azetidin-2-one moieties. | ||
They have had enormous application in medicinal chemistry in the form of azetidin-2-ones (β-lactams), which are the key components of many biologically active compounds showing antibacterial, antifungal, and anti-inflammatory properties.4 Various multifunctional spiro β-lactam derivatives have also been reported recently,5 among which spirooxindolyl β-lactams are particularly relevant.6 Despite this interest in azetidin-2-ones, in general azetidines have received much less attention with respect to their lower and higher small-ring homologues, and their application in the context of drug discovery is not so common. In particular, only a few novel spirocyclic azetidine scaffolds have been recently proposed,7 which are able to access unexplored chemical space and act as potential new lead compounds.8
Our long-standing interest in the asymmetric synthesis of 3,3-disubstituted oxindole derivatives and related spiro compounds,9 combined with the growing interest in hybrid drugs as therapeutic agents, inspired us to combine the two biologically active oxindole and azetidine moieties, by means of a spiro arrangement of the two ring systems.10 We conceived of the synthesis of chiral spirooxindole-based 4-methyleneazetidines as an unprecedented, intriguing combination of pharmacologically relevant motifs. Such methyleneazetidines can also be seen as key intermediates for the synthesis of particularly relevant spirooxindolyl β-lactam derivatives.
Relying on our previous experience with isatin-derived ketimines, we looked at the formal [2+2] annulation reactions of such compounds with allenoates, as a practical and direct strategy to obtain highly functionalized target compounds, with a good level of atom-economy. Since Shi's pioneer work11 disclosing an abnormal aza-Baylis–Hillman reaction of N-tosylated imines with allenoates to give azetidine derivatives in the presence of the strong Lewis base DABCO (1,4-diazabicyclo[2.2.2]octane), additional examples of such [2+2] annulation were reported, on both electron-deficient aldimines and ketimines. A detailed investigation on the effect of different Lewis bases in the reaction of allenoates with cyclic trifluoromethyl ketimines was accomplished by Ma and co-workers.12
Also remarkable is the application of this strategy by Ye and co-workers,13 affording functionalized biologically relevant sultam-fused azetidines. The asymmetric approach to this [2+2] annulation is more challenging, with only two studies reporting enantioselective, organocatalytic versions employing cinchona alkaloid-derived catalysts.14 Finally, a diastereoselective synthesis of 2-azetidines was exploited by Santos and co-workers,15 based on allenoates bearing a chiral auxiliary on the ester moiety.
To the best of our knowledge, no diastereoselective [2+2] annulation-based strategies employing chiral imines have been described for the preparation of methyleneazetidines. Herein we first demonstrate the suitability of chiral, isatin-derived tert-butanesulfinyl ketimines for reaction with allenoates, applying this reaction to the synthesis of unprecedented, enantiopure spirooxindole-based 4-methyleneazetidines.
We began our investigation by catalyst screening and reaction optimization using the known 1-benzyl-isatin-derived N-tert-butanesulfinyl ketimine 1a and ethyl buta-2,3-dienoate 2a (Table 1). A number of amines such as diazabicyclo[2.2.2]octane (DABCO), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), N,N-dimethyl-4-aminopyridine (DMAP) and pyridine were screened at room temperature in THF for 24 hours (entries 1–4). Only DABCO promoted the reaction efficiently to give the desired azetidine 3a. DBU proved to be quite ineffective; but with pyridine or DMAP the reaction afforded complex mixtures, from which only traces of the probable aza-Morita–Baylis–Hillman adduct could be tentatively identified by NMR. Next, solvent screening revealed that the reaction with DABCO worked also in toluene, dioxane and dichloromethane, but in slightly lower yields with respect to THF (entries 5–7). Using a greater excess of ethyl buta-2,3-dienoate 2a had positive effects on the chemical yields, with a value of 2.0 equivalents performing best (entries 8 and 9). Finally, decreasing the substrate concentration prolonged the reaction time reducing the conversion, while increasing the concentration gave a less clean reaction, however with a good isolated yield for 3a (entries 10 and 11). With respect to the stereochemical outcome, we were delighted to realize that, in all reactions, only the E-double bond isomer was formed, bearing, at the best 1H NMR sensitivity, only a single C-3 relative configuration with respect to the chiral auxiliary.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Equiv. 2a | Conc. [M] | Time [h] | Yieldb (%) |
| a All reactions were performed with 0.15 mmol of 1a. b Isolated yields. nr = no reaction. | ||||||
| 1 | DABCO | THF | 1.5 | 0.1 | 24 | 76 |
| 2 | DBU | THF | 1.5 | 0.1 | 24 | nr |
| 3 | DMAP | THF | 1.5 | 0.1 | 24 | — |
| 4 | Pyridine | THF | 1.5 | 0.1 | 24 | — |
| 5 | DABCO | Toluene | 1.5 | 0.1 | 24 | 67 |
| 6 | DABCO | Dioxane | 1.5 | 0.1 | 24 | 57 |
| 7 | DABCO | CH2Cl2 | 1.5 | 0.1 | 24 | 72 |
| 8 | DABCO | THF | 2.0 | 0.1 | 8 | 83 |
| 9 | DABCO | THF | 2.5 | 0.1 | 8 | 69 |
| 10 | DABCO | THF | 2.0 | 0.05 | 24 | 48 |
| 11 | DABCO | THF | 2.0 | 0.2 | 8 | 79 |

With the optimized reaction conditions in hand, a variety of N-tert-butanesulfinyl ketimines 1 were next explored, to investigate the substrate scope of this novel, DABCO-catalyzed formal [2+2] annulation reaction. The generality was also evaluated with respect to allenoates 2 (Table 2). The protecting group on the oxindole nitrogen atom was found to have a moderate effect on the reactivity, with best yields obtained with R1 = Bn, Me, Trt and p-Br–Bn (entries 1–9). Surprisingly, ketimine 1c, bearing the N-trityl protecting group, afforded the corresponding azetidine 3c with the highest yield, but definitely lower stereocontrol, probably due to a steric mismatching effect of the bulky trityl group with the chiral auxiliary tert-butanesulfinyl moiety (entry 3). Unprotected ketimine 1d also gave the reaction, affording the unexpected compound 3d, deriving from both the [2+2] annulation and the competitive NH participation with allenoate (entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Compound 1 (R1/R2) | Compound 2 (R3/R4) | Yieldb (%) | drc |
| a Reactions were performed with 0.15 mmol of 1 and 0.30 mmol of 2. b Isolated yields. c Determined by 1H NMR analysis of the crude mixture. d Reaction performed with 4 equiv. of 2a. Trt = trityl; nd = not detected. | ||||
| 1 | 1a (Bn/H) | 2a (H/Et) | 3a, 83 | >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | 1b (Me/H) | 2a (H/Et) | 3b, 85 | >99:1 |
| 3 | 1c (Trt/H) | 2a (H/Et) | 3c, 97 | 74:26 |
| 4 | 1d (H/H) | 2a (H/Et) | 3d, 38 (75)d | >99:1 |
| 5 | 1e (p-NO2-Bn/H) | 2a (H/Et) | 3e, 36 | >99:1 |
| 6 | 1f (p-OMe-Bn/H) | 2a (H/Et) | 3f, 51 | >99:1 |
| 7 | 1g (p-Br-Bn/H) | 2a (H/Et) | 3g, 95 | >99:1 |
| 8 | 1h (Ac/H) | 2a (H/Et) | 3d, 31 (68)d | >99:1 |
| 9 | 1i (Allyl/H) | 2a (H/Et) | 3i, 66 | >99:1 |
| 10 | 1j (Bn/5-OMe) | 2a (H/Et) | 3j, 81 | >99:1 |
| 11 | 1k (Bn/5-Cl) | 2a (H/Et) | 3k, 59 | >99:1 |
| 12 | 1l (Bn/5-F) | 2a (H/Et) | 3l, 47 | >99:1 |
| 13 | 1m (Bn/6-Br) | 2a (H/Et) | 3m, 71 | >99:1 |
| 14 | 1a (Bn/H) | 2b (H/Bn) | 3n, 52 | >99:1 |
| 15 | 1a (Bn/H) | 2c (Me/Et) | nd | — |

The same compound 3d could also be recovered from the reaction of ketimine 1h (entry 8), probably due to the great lability of the acetyl protecting group.
Next, N-benzylisatin ketimines with various substituents on the aromatic ring were explored. Moderate to good yields of the corresponding azetidines were achieved in the presence of a variety of substituents including electron-donating groups (entry 10), and halogen substituents (entries 11–13) at the 5- or 6-position on the oxindole aromatic ring.
Finally, we investigated the formal [2+2] annulation with two different allenoates, namely benzyl buta-2,3-dienoate 2b and ethyl 2-methylbuta-2,3-dienoate 2c. Employing 2b, the corresponding azetidine 3n could be easily obtained, albeit in moderate yield (entry 14). On the other hand, α-substituted allenoate 2c proved to be completely unreactive under the reaction conditions and the corresponding azetidine could not be detected, even after a longer reaction time (entry 15). This result can be ascribed both to the stereoelectronic influence of the methyl group on the allenoate 2c, and to the high steric challenge inherent in the formation of the tetrasubstituted double bond, jointed to the spiroazetidine ring.
As usual for such kinds of studies, we also proceeded to a scale-up experiment, employing 1 mmol of ketimine 1a (reaction conditions as reported in Table 2). After 24 hours, the desired azetidine 3a could be obtained in a yield quite similar to that obtained on the small scale, still as a single diastereoisomer (yield 80%, dr >99:1).
To determine the absolute stereochemistry of the oxindole C-3 spiro center, a single crystal of 4-methyleneazetidine 3a was subjected to X-ray crystallographic analysis. The experiment unambiguously confirms the E-double bond configuration and assigns the stereochemistry of the spiro center to the (S)-configuration, derived from (R)-tert-butanesulfinyl ketimine 1a (Fig. 2).
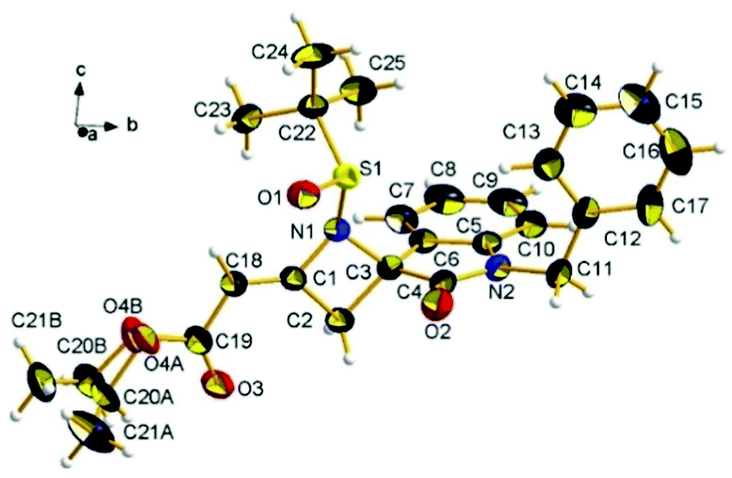 | ||
| Fig. 2 ORTEP view of compound 3a, with the atom-numbering scheme and the crystallographic reference system. Thermal ellipsoids of non-H atoms were drawn at the 25% probability level. The two equiprobable conformations of the disordered ethoxy chain of the ester moiety are shown (see ESI†). | ||
In order to explain this stereochemical outcome, we refer to the established mechanism of Lewis base-catalysed similar formal [2+2] annulations.11 Accordingly, the nitrogen base DABCO acts as a nucleophile activator, producing a zwitterionic allylic carbanion from the allenoate reagent. Such an intermediate undergoes a γ-addition to the ketimine, followed by intramolecular nucleophilic attack to give the azetidine ring with regeneration of DABCO. On this basis, a plausible transition state for the formation of (3,S)-3a is illustrated in Fig. 3. In the working model, the sterochemical outcome can be explained assuming a syn-periplanar relationship between the imine and the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond,16 which favours the shown conformation for starting ketimine 1a. The excellent diastereoselectivity observed in the azetidine formation may be due to the high stereodirecting effect of the bulky tert-butanesulfinyl group. According to this, the delivery of the DABCO-activated nucleophile would occur from the less hindered Re-face, resulting in the formation of the (3,S)-azetidine.
O double bond,16 which favours the shown conformation for starting ketimine 1a. The excellent diastereoselectivity observed in the azetidine formation may be due to the high stereodirecting effect of the bulky tert-butanesulfinyl group. According to this, the delivery of the DABCO-activated nucleophile would occur from the less hindered Re-face, resulting in the formation of the (3,S)-azetidine.
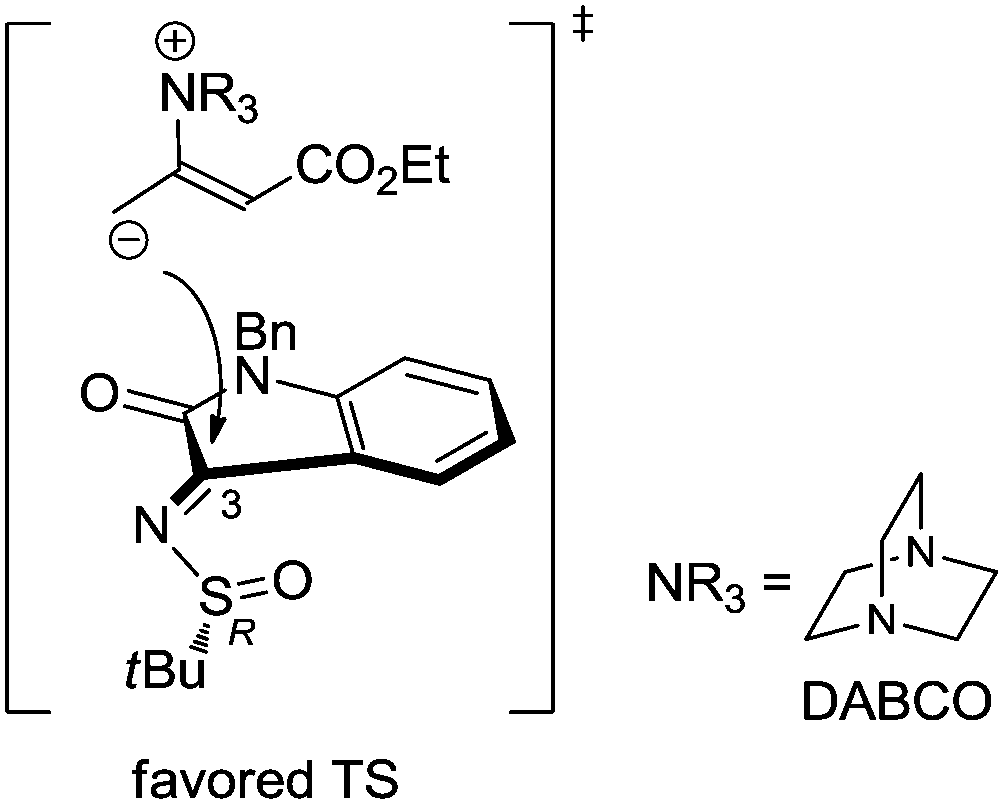 | ||
| Fig. 3 Plausible explanation of the stereochemical outcome. | ||
To demonstrate the synthetic utility of highly functionalized azetidines, we subjected compound 3a to cleavage of the N-tert-butanesulfinyl chiral auxiliary, to sulfur oxidation and to ozonolysis (Scheme 1). Treatment of sulfinyl amide 3a with anhydrous HCl in dioxane at room temperature afforded the secondary amine 4 in acceptable yield. On the other hand, by action of m-chloroperbenzoic acid at room temperature, the sulfinyl amide 3a was readily oxidized to the corresponding sulfonamide 5. Finally, β-lactam 6 was obtained in quantitative yield by treatment with ozone. This last result can be considered particularly relevant, given the interest in the spirocyclic oxindolo-β-lactam skeleton, which, combining two privileged heterocycles such as indoles and β-lactams, is a motif strictly related to natural products and synthetic compounds with significant biological activities.17
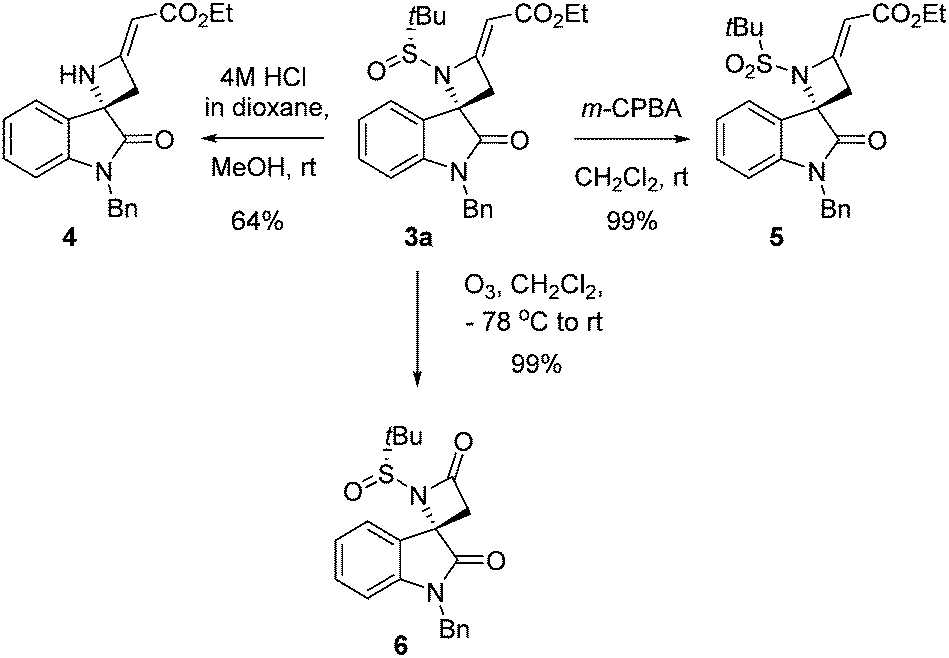 | ||
| Scheme 1 Further transformations of azetidine 3a. | ||
In conclusion, an efficient asymmetric approach for the preparation of unprecedented, chiral spirooxindole-fused 4-methyleneazetidines has been developed, based on a diastereoselective, DABCO-catalysed formal [2+2] annulation reaction. The method provides easy access to a range of highly functionalized compounds, which are obtained as single diastereoisomers, under mild conditions. By taking advantage of the functional groups transformations, a rapid asymmetric construction of the relevant spirocyclic oxindolo-β-lactam has also been achieved.
Further work is currently underway, aimed at establishing spirooxindole-fused 4-methyleneazetidines as possible lead compounds for drug discovery programs.
Notes and references
- (a) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988–4035 CrossRef CAS PubMed; (b) Y. Dejaegher, N. M. Kuz'menok, A. M. Zvonok and N. De Kimpe, Chem. Rev., 2002, 102, 29–60 CrossRef CAS PubMed.
- For example, see: (a) E. Rubenstein, H. Zhou, K. M. Krasinska, A. Chien and C. H. Becker, Phytochemistry, 2006, 67, 898–903 CrossRef CAS PubMed; (b) Y. Aoyagi, Phytochemistry, 2006, 67, 618–621 CrossRef CAS PubMed.
- For example, see: (a) J. T. Lowe, M. D. Lee IV, L. B. Akella, E. Davoine, E. J. Donckele, L. Durak, J. R. Duvall, B. Gerard, E. B. Holson, A. Joliton, S. Kesavan, B. C. Lemercier, H. Liu, J. C. Marié, C. A. Mulrooney, G. Muncipinto, M. Welzel-O'Shea, L. M. Panko, A. Rowley, B. C. Suh, M. Thomas, F. F. Wagner, J. Wei, M. A. Foley and L. A. Marcaurelle, J. Org. Chem., 2012, 77, 7187–7211 CrossRef CAS PubMed; (b) G. B. Evans, R. H. Furneaux, B. Greatrex, A. S. Murkin, V. L. Schramm and P. C. Tyler, J. Med. Chem., 2008, 51, 948–956 CrossRef CAS PubMed.
- (a) W. Van Brabandt, M. Vanwalleghem, M. D'hooghe and N. De Kimpe, J. Org. Chem., 2006, 71, 7083–7086 CrossRef CAS PubMed; (b) F. von Nussbaum, M. Brands, B. Hinzen, S. Weigand and D. Habich, Angew. Chem., Int. Ed., 2006, 45, 5072–5129 CAS; (c) G. S. Singh, Mini-Rev. Med. Chem., 2004, 4, 69–92 CrossRef CAS PubMed; (d) G. S. Singh, Mini-Rev. Med. Chem., 2004, 4, 93–109 CrossRef CAS PubMed; (e) J. D. Buynak, Curr. Med. Chem., 2004, 11, 1951–1964 CrossRef CAS PubMed.
- (a) M. Hatano, T. Horibe and K. Ishihara, J. Am. Chem. Soc., 2010, 132, 56–57 CrossRef CAS PubMed; (b) A. Bhalla, P. Venugopalan and S. S. Bari, Eur. J. Org. Chem., 2006, 4943–4950 CrossRef CAS.
- (a) R. Jain, K. Sharma and D. Kumar, J. Heterocycl. Chem., 2013, 50, 315–319 CrossRef CAS; (b) B. V. Subba Reddy, G. Karthik, T. Rajasekaran, A. Antony and B. Sridhar, Tetrahedron Lett., 2012, 53, 2396–2401 CrossRef CAS; (c) Y. Cheng, B. Wang and L. Q. Cheng, J. Org. Chem., 2006, 71, 4418–4427 CrossRef CAS PubMed.
- (a) S. T. M. Orr, R. Beveridge, S. K. Bhattacharya, K. O. Cameron, S. Coffey, D. Fernando, D. Hepworth, M. V. Jackson, V. Khot, R. Kosa, K. Lapham, P. M. Loria, K. F. McClure, J. Patel, C. Rose, J. Saenz, I. A. Stock, G. Storer, M. von Volkenburg, D. Vrieze, G. Wang, J. Xiao and Y. Zhang, ACS Med. Chem. Lett., 2015, 6, 156–161 CrossRef CAS PubMed; (b) S. Karlsson, R. Bergman, C. Löfberg, P. R. Moore, F. Pontén, J. Tholander and H. Sörensen, Org. Process Res. Dev., 2015, 19, 2067–2074 CrossRef CAS; (c) R. Xu, M. Czarniecki, J. de Man, J. Pan, L. Qiang, Y. Root, S. Ying, J. Su, X. Sun, Y. Zhang, T. Yu, Y. Zhang, T. Hu and S. H. Chen, Tetrahedron Lett., 2011, 52, 3266–3270 CrossRef CAS; (d) M. J. Meyers, I. Muizebelt, J. van Wiltenburg, D. L. Brown and A. Thorarensen, Org. Lett., 2009, 11, 3523–3525 CrossRef CAS PubMed; (e) G. Deguest, L. Bischoff, C. Fruit and F. Marsais, Org. Lett., 2007, 9, 1165–1167 CrossRef CAS PubMed.
- M. Lüthy, M. C. Wheldon, C. Haji-Cheteh, M. Atobe, P. S. Bond, P. O'Brien, R. E. Hubbard and I. J. S. Fairlamb, Bioorg. Med. Chem., 2015, 23, 2680–2694 CrossRef PubMed.
- (a) M. Stucchi, G. Lesma, F. Meneghetti, G. Rainoldi, A. Sacchetti and A. Silvani, J. Org. Chem., 2016, 81, 1877–1884 CrossRef CAS PubMed; (b) G. Lesma, F. Meneghetti, A. Sacchetti, M. Stucchi and A. Silvani, Belstein J. Org. Chem., 2014, 10, 1383–1389 CrossRef PubMed; (c) A. Sacchetti, A. Silvani, F. G. Gatti, G. Lesma, T. Pilati and B. Trucchi, Org. Biomol. Chem., 2011, 9, 5515–5522 RSC; (d) G. Lesma, N. Landoni, A. Sacchetti and A. Silvani, Tetrahedron, 2010, 66, 4474–4478 CrossRef CAS; (e) G. Lesma, N. Landoni, T. Pilati, A. Sacchetti and A. Silvani, J. Org. Chem., 2009, 74, 4537–4541 CrossRef CAS PubMed.
- For reviews on bioactive spirooxindoles, see: (a) N. Ye, H. Chen, E. A. Wold, P.-Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef CAS; (b) P. Saraswat, G. Jeyabalan, Z. H. Ansari, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016 DOI:10.1080/00397911.2016.1211704; (c) B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed . For example, see: ; (d) S. Wang, Y. Jiang, S. Wu, G. Dong, Z. Miao, W. Zhang and C. Sheng, Org. Lett., 2016, 18, 1028–1031 CrossRef CAS PubMed; (e) A. Bertamino, M. Soprano, S. Musella, M. R. Rusciano, M. Sala, E. Vernieri, V. Di Sarno, A. Limatola, A. Carotenuto, S. Cosconati, P. Grieco, E. Novellino, M. Illario, P. Campiglia and I. Gomez-Monterrey, J. Med. Chem., 2013, 56, 5407–5421 CrossRef CAS PubMed.
- (a) G.-L. Zhao and M. Shi, J. Org. Chem., 2005, 70, 9975–9984 CrossRef CAS PubMed; (b) G.-L. Zhao, J. W. Huang and M. Shi, Org. Lett., 2003, 5, 4737–4739 CrossRef CAS PubMed.
- L. J. Yang, S. Li, S. Wang, J. Nie and J. A. Ma, J. Org. Chem., 2014, 79, 3547–3558 CrossRef CAS PubMed.
- X. Y. Chen, R. C. Lin and S. Ye, Chem. Commun., 2012, 48, 1317–1319 RSC.
- (a) S. Takizawa, F. Arteaga, Y. Yoshida, M. Suzuki and H. Sasai, Org. Lett., 2013, 15, 4142–4145 CrossRef CAS PubMed; (b) J. B. Denis, G. Masson, P. Retailleau and J. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5356–5360 CrossRef CAS PubMed.
- B. S. Santos, A. L. Cardoso, A. Matos Beja, M. Ramos Silva, J. A. Paixão, F. Palacios and T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2010, 3249–3256 CrossRef CAS.
- (a) V. U. Bhaskara Rao, A. P. Jadhav, D. Garad and R. P. Singh, Org. Lett., 2014, 16, 648–651 CrossRef PubMed; (b) D. Chen and M. H. Xu, Chem. Commun., 2013, 49, 1327–1329 RSC.
- (a) H. M. Zhang, Z. H. Gao and S. Ye, Org. Lett., 2014, 16, 3079–3081 CrossRef CAS PubMed; (b) S. Sato, M. Shibuya, N. Kanoh and Y. Iwabuchi, Chem. Commun., 2009, 6264–6266 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, crystallographic data in CIF and NMR spectra. CCDC 1487568. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc05838h |
| This journal is © The Royal Society of Chemistry 2016 |