
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid protein immobilization for thin film continuous flow biocatalysis†
Joshua
Britton
ab,
Colin L.
Raston
*b and
Gregory A.
Weiss
*a
aDepartments of Chemistry, Molecular Biology and Biochemistry, University of California, Irvine, California 92697-2025, USA. E-mail: gweiss@uci.edu
bCentre for NanoScale Science and Technology, School of Chemical and Physical Sciences, Flinders University, Bedford Park, Adelaide, South Australia 5001, Australia. E-mail: colin.raston@flinders.edu.au
First published on 19th July 2016
Abstract
A versatile enzyme immobilization strategy for thin film continuous flow processing is reported. Here, non-covalent and glutaraldehyde bioconjugation are used to immobilize enzymes on the surfaces of borosilicate reactors. This approach requires only ng of protein per reactor tube, with the stock protein solution readily recycled to sequentially coat >10 reactors. Confining reagents to thin films during immobilization reduced the amount of protein, piranha-cleaning solution, and other reagents by ∼96%. Through this technique, there was no loss of catalytic activity over 10 h processing. The results reported here combines the benefits of thin film flow processing with the mild conditions of biocatalysis.
Nature builds diverse and complex natural products through assembly line biosynthesis. Polyketide synthases for example, are multi-domain proteins that perform iterative processes to synthesize a large range of secondary metabolites.1,2 Continuous flow has emerged as an analogous, in vitro process, for synthesizing compounds through multistep processes.
In the laboratory, enzymes can perform a wide range of transformations including reductions,3,4 oxidations,5,6 cyclization,7,8 aziridinations9 and nitration reactions.10 Improving the performance of these enzymes typically relies on directed evolution11,12 and computational design.13,14 These widely used techniques can improve reaction rates and enzyme promiscuity to accept non-natural substrates. Although such approaches increase the utility and adoption of biocatalyzed transformations, scaling up enzyme-catalyzed reactions can be challenging.
Translating reactions into continuous flow can increase reaction yields and safety,15,16 aid multistep transformations,17,18 and decrease human effort and waste.19,20 Furthermore, enzymes in synthetic pathways can improve sustainability metrics by avoiding hazardous solvents and toxic metals. Combining the benefits of continuous flow and biocatalysis offers numerous advantages such as processing with immobilized enzymes and rapid scale-up. Continuous flow biocatalysis has thus increasingly become a focus of many laboratories, as shown in a few examples.21–24
Immobilizing enzymes can increase their industrial viability by creating reusable biocatalysts with potentially improved reactivity, purity, specificity, selectivity, thermal stability and pH tolerance.25–30 Given this importance, many immobilization strategies have been described, including attachment to magnetic nanoparticles and nanomaterials,31,32 supports through antibody-specific epitopes and crosslinking,33 and also entrapment within a polymer network.34 Glutaraldehyde crosslinking was chosen here due to its simplicity, commercial availability, and success in previous immobilization studies.35,36
Recently, our laboratories have focused on utilizing thin films to mediate protein folding,37 biocatalysis38 and molecular assembly line processes.39 This involves processing in a vortex fluidic device (VFD) which confines reagents to a ≈250 μM thin film. Here, micromixing, shear stress and mechanical vibrations40,41 can operate upon reagents to increase reaction yields and efficiencies. Processing in a single VFD with a 20 mm external diameter reactor can achieve flow rates up to 20 mL min−1. Larger scale processing is possible by applying multiple VFDs. In pursuing new multistep transformations, we have recently embarked on exploring thin film continuous flow biocatalysis. Future experiments, using enzymes alone, or in conjunction with organic reagents will require immobilization of minute quantities of protein for efficient continuous flow reactors.
Unlike other continuous flow systems, the VFD reactor is made from borosilicate glass. This material can simplify bioconjugation, as explored systematically here. APTES (3-aminopropyl triethoxysilane) was coupled to the reactor surface to create a layer of nucleophilic amines (Fig. 1a). This APTES modified reactor was then used for rapid covalent and non-covalent immobilization. Non-covalent immobilization can be achieved though surface-exposed functionalities on the protein interacting with the APTES layer through salt bridges and hydrogen bonds (Fig. 1b). In contrast, covalent immobilizations used surface-exposed lysine sidechains (also thiols, phenols and imidazoles36) on the protein to form imine and amine bonds with a glutaraldehyde-modified APTES linker (Fig. 1c and d). The structure of the glutaraldehyde linker and resultant cross-link has been simplified in Fig. 1; in aqueous solution, for example, many different forms of glutaraldehyde can exist.35,36
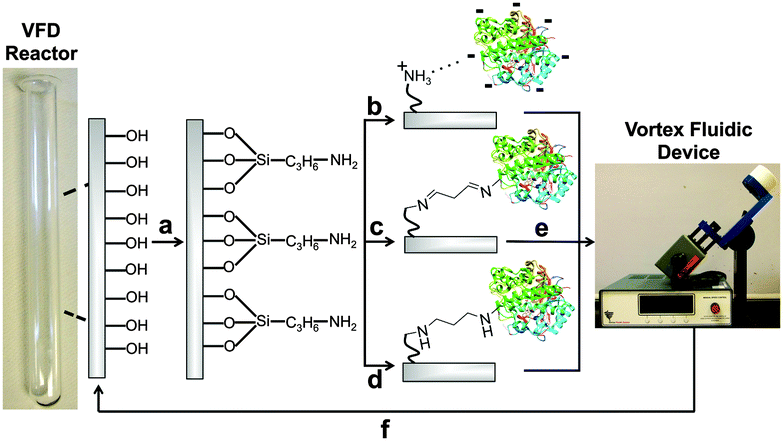 | ||
| Fig. 1 Enzyme immobilization onto the surface of the VFD reactor. (a) First, the surface of the sample tube is coated with APTES (3-aminopropyl triethoxysilane) to generate a high concentration of surface-bound amines; a simplified depiction of this surface coating is shown here. (b) β-Glucosidase is added directly to the APTES-coated sample tube for non-covalent immobilization. (c) After derivatization of the APTES layer with glutaraldehyde, β-glucosidase is attached in this simplified structure of the linker and cross-link. (d) The imine-glutaraldehyde is reduced with NaBH3CN. (e) The immobilization efficiency was tested by VFD processing in the presence of the β-glucosidase substrate, 4-nitrophenyl β-D-glucopyranoside. (f) The sample tube can be regenerated through rapid treatment with a thin film of piranha solution. Some of the reactions in this manuscript were performed in continuous flow, further information on the reactor setup has been previously reported.39 | ||
Coating the reactor with APTES required optimizing a three-step process. First, treatment with piranha solution exposes high concentrations of silinols on the reactor surface. Although the reactor can be filled with piranha solution (50 mL), confining 3 mL to a thin film for one min offers the same cleaning efficiency, whilst reducing the volume of this highly hazardous fluid by 94%. After washing and drying, the reactor surface is then derivatized with a dilute APTES solution (79.5 mM, 60 μL in 3 mL MeOH). Again, confining reagents to a thin film reduced the quantities of MeOH and APTES required by 94%. Lastly, the APTES-modified surface is heated to 160 °C to drive the condensation reaction to completion (Fig. 1a).
Non-covalent immobilization is sometimes preferred to covalent immobilization as introducing random covalent bonds can distort enzymes' structures.28 For testing a large number of non-covalent immobilization variables, a colorimetric enzyme-substrate assay was used, β-glucosidase and 4-nitrophenyl β-D-glucopyranoside, respectively. This assay offers high throughput conditions (5 min per reaction), an effective quench solution, and stability to vortexing conditions (Fig. 2d).38
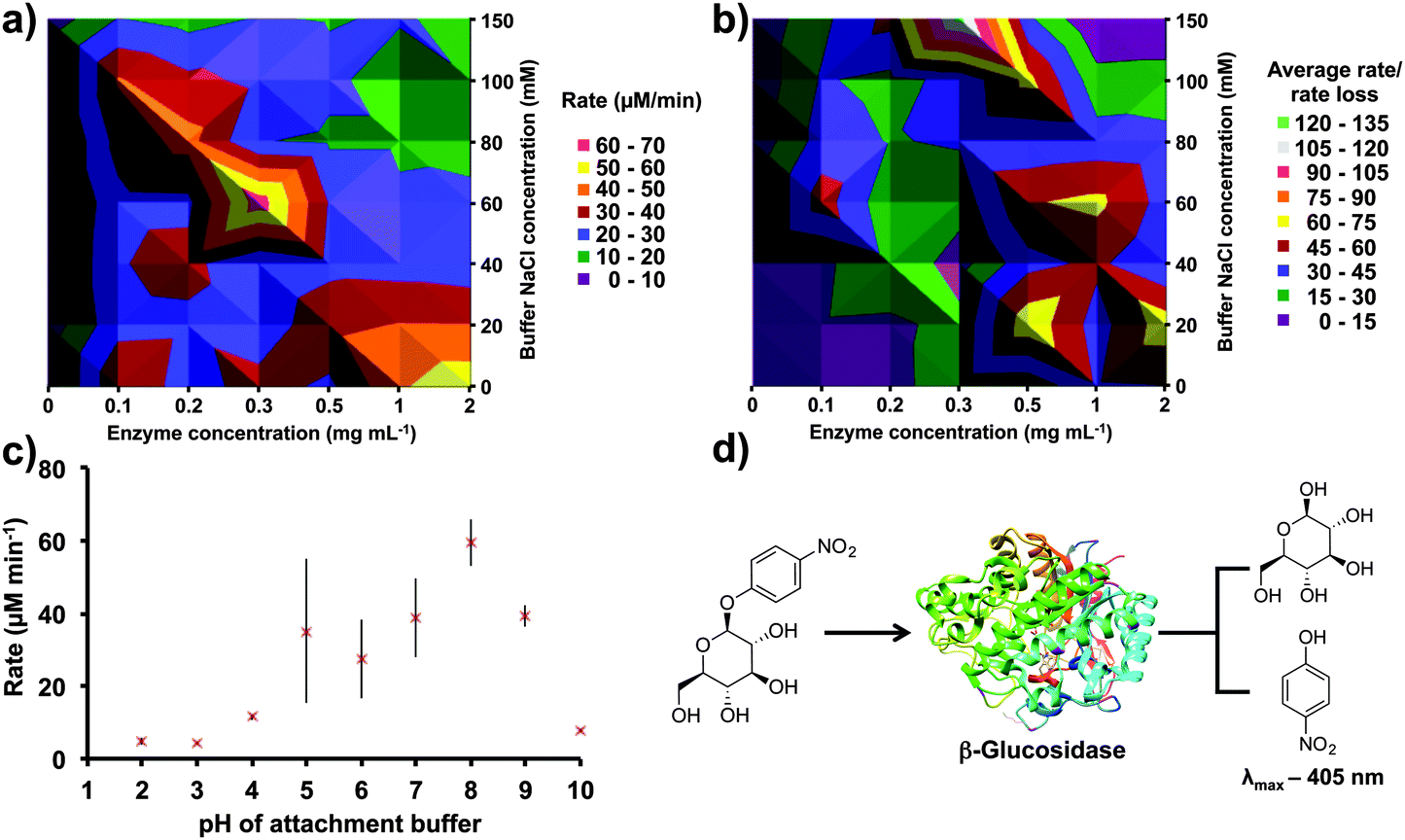 | ||
| Fig. 2 Non-covalent immobilization using β-glucosidase and 4-nitrophenyl β-D-glucopyranoside for optimization. (a) The enzyme and buffer salt concentration were varied during the immobilization step. The contour plot reveals that a 0.3 mg mL−1 enzyme concentration in a 60 mM NaCl PBS buffer is optimal for high substrate conversion. (b) Decreased catalytic activity due to enzyme leaching is depicted in this contour plot. Thus, higher salt concentrations are revealed as beneficial for immobilization longevity. (c) Varying the pH of the attachment buffer established optimal immobilization in PBS at pH 8.0. The deviation from the trend at pH 5.0 is due to the isoelectric point of β-glucosidase. (d) For all optimization experiments, a β-glucosidase and 4-nitrophenyl β-D-glucopyranoside (10 mM, 1.50 mL) system was used. β-Glucosidase hydrolyses the substrate, releasing p-nitrophenol (λmax 405 nm) and β-D-glucopyranoside. Each assay was performed in the VFD for five min, and each reactor was assayed six times. Two separate reactors were used per data point, and the error is a standard deviation around the mean (n = 12). | ||
β-Glucosidase and buffer salt concentrations play an integral role in non-covalent immobilization efficiency and activity. Varying both of these variables simultaneously generated a contour plot (Fig. 2a). A β-glucosidase concentration of 0.3 mg mL−1 was optimal, with variation either side of this concentration decreasing immobilization efficiency. Furthermore, confining the protein solution to a thin film for immobilization reduced the volume of protein solution used from 50 to 3 mL, i.e. a reduction of 94% (15 mg to 0.9 mg). Additionally, 60 mM NaCl in PBS was found to be optimal, but taking into account rate loss over time revealed that 150 mM NaCl in PBS is superior, with no decrease in substrate transformation rate over 30 min (Fig. 2b). The higher salt concentration during adsorption could increase the strength of the enzyme–APTES interaction.42
Next we next examined the conditions required for covalent immobilization. Covalent immobilization can increase enzymes' stability greatly through the addition of short spacers off the reactor surface.27 Reacting glutaraldehyde with the APTES-coated reactor, followed by the sequential addition of β-glucosidase solution afforded an imine linker for immobilization (Fig. 1c). Furthermore, this imine can be reduced to the amine with NaBH3CN solution (Fig. 1d).43,44 Notably, lysine residues in the active site are typically uninvolved in catalysis, and this immobilization strategy is therefore unlikely to perturb enzyme function.36 Once again, these steps were performed in the thin film, resulting in a 96% reduction in quantity of buffer and reagents required.
Switching to covalent immobilization increased the yields of conjugated enzyme with a concomitant increase in the rates of substrate conversion. Although a slight increase in enzyme immobilization efficiency and reaction rate results from switching to covalent immobilization, the reduction of imine to amine provides a dramatic improvement. This reduction prevents hydrolysis of the imine, thus increasing the concentration of protein on the surface of the reactor tube (Fig. 3a). To test the stability of these immobilization strategies, each immobilized enzyme was subjected to a continuous flow reaction at 1.0 mL min−1; all immobilizations demonstrated excellent stability, with no loss of activity after 10 h of processing recorded with the amine linker (Fig. 3a).
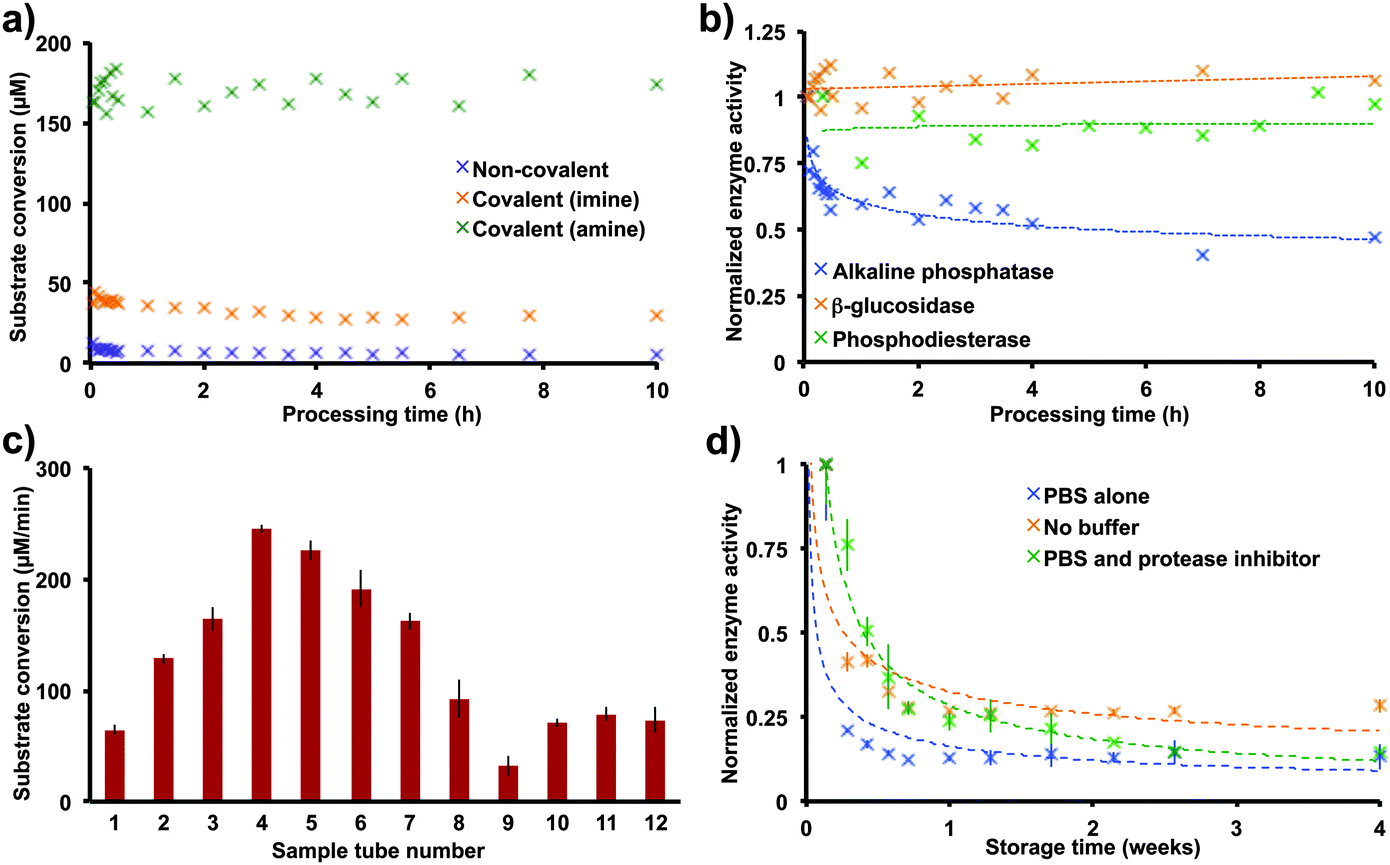 | ||
| Fig. 3 The conditions optimized here are general for a range of proteins. The protein solutions used in the immobilization step can be recycled to coat more than ten sample tubes, with the coated sample tubes still maintaining catalytic activity for weeks. (a) Switching from non-covalent to covalent attachments increased substrate conversion levels dramatically. (b) Applying the symmetrical amine-glutaraldehyde cross linker optimized for β-glucosidase to alkaline phosphatase and phosphodiesterase establishes the generality of the method, with all three proteins having good stability over 10 h of processing. (c) The β-glucosidase solution (3 mL, 0.3 mg mL−1) used in the immobilization step can be recycled to coat more than ten sample tubes, with the first sample tube having the same substrate transformation rate as the last. We anticipate that this solution could coat tens of sample tubes given the small amount of protein used in each immobilization. (d) Storing the enzyme-immobilized tubes devoid of buffer allowed >20% catalytic activity after one month. The rates displayed above are the average rates as described in Fig. 2 (n = 12). The data in (a) and (b) were from continuous flow experiments with a flow rate of 1.0 mL min−1. | ||
Our second requirement for this immobilization strategy was to make it general. Given that proteins have a hydrophilic surface, most enzymes have a surface-exposed lysine residue for immobilization. As small quantities of protein are used in this immobilization strategy (0.9 mg), we were able to explore phosphodiesterase, a poorly overexpressing recombinant protein. Immobilizing phosphodiesterase and a commercially available alkaline phosphatase via amine-glutaraldehyde immobilization (Fig. 1c) resulted in stable levels of substrate conversion for 10 h in continuous flow (1 mL min−1, Fig. 3b).
The final criterion for this immobilization method was to increase immobilization efficiency. This process already uses a low quantity of protein, but, to address efficiency further, it would be useful to know how much protein is on the surface of the reactor. Two complementary experiments revealed that 15.4 to 69.8 ng of β-glucosidase are present on the surface of the reactor after covalent immobilization (Fig. S1 and S2, ESI†). This surprising result opened up the possibility to recycle the protein stock solution (0.3 mg mL−1). Indeed, recycling the stock solution of β-glucosidase allowed the coating of 12 reactor tubes with no observable decrease in substrate conversion between the first tube and the last (Fig. 3c). We were unable to identify why recycling the enzyme solution increased substrate conversion levels for sample tubes 2–8. This trend subsides with additional sample tubes, and we believe that it is an experimental artifact.
Lastly, sample tube storage was investigated, which was deemed important given that sample tubes are often transported to other laboratories. Surprisingly, a dry sample tube bearing surface bound β-glucosidase provided reasonable substrate conversion after one month of storage (4 °C storage, 19 μM min−1 conversion, Fig. 3d). Presumably the decrease in substrate conversion results from a combination of protein leaching and unfolding.
In conclusion, a rapid and general technique for protein immobilization onto a thin film continuous flow reactor has been developed. Importantly, using thin films for reagent confinement reduced the volume of protein solution, piranha solution, APTES, MeOH, glutaraldehyde, NaBH3CN and a range of buffers by an average of 95%. The ability to use a small amount of protein (900 μg) to coat >10 sample tubes provides a general strategy to increase the efficiency of enzyme-mediated transformations in continuous flow. Incorporating biocatalysts into multistep processes offers the potential to create complex molecules using nature's machinery. The findings reported here will facilitate biocatalysts by allowing low expressing proteins to be used in complex substrate transformations such as natural products and pharmaceutical ingredients.
JB was supported in part by a Taihi Hong Memorial award. GW gratefully acknowledges the National Institute of General Medical Sciences of the NIH (1RO1-GM100700-01). CR acknowledges the Australian Research Council and the Government of South Australia for their financial support during this project.
Notes and references
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
- H. Sato, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2015, 54, 4488–4492 CrossRef CAS PubMed.
- N. G. Turrini, M. Hall and K. Faber, Adv. Synth. Catal., 2015, 357, 1861–1871 CrossRef CAS.
- A. Ilie, R. Agudo, G.-D. Roiban and M. T. Reetz, Tetrahedron, 2015, 71, 470–475 CrossRef CAS.
- S. Sirajuddin and A. C. Rosenzweig, Biochemistry, 2015, 54, 2283–2294 CrossRef CAS PubMed.
- G. K. T. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, J. Am. Chem. Soc., 2015, 137, 15398–15401 CrossRef CAS PubMed.
- E. Oueis, M. Jaspars, N. J. Westwood and J. H. Naismith, Angew. Chem., Int. Ed., 2016, 128, 5936–5939 CrossRef PubMed.
- C. C. Farwell, R. K. Zhang, J. A. McIntosh, T. K. Hyster and F. H. Arnold, ACS Cent. Sci., 2015, 1, 89–93 CrossRef CAS PubMed.
- S. C. Dodani, G. Kiss, J. K. B. Cahn, Y. Su, V. S. Pande and F. H. Arnold, Nat. Chem., 2016, 8, 419–425 CrossRef CAS PubMed.
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS PubMed.
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS PubMed.
- C. E. Tinberg, S. D. Khare, J. Dou, L. Doyle, J. W. Nelson, A. Schena, W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard and D. Baker, Nature, 2013, 501, 212–216 CrossRef CAS PubMed.
- J. B. Siegel, A. L. Smith, S. Poust, A. J. Wargacki, A. Bar-Even, C. Louw, B. W. Shen, C. B. Eiben, H. M. Tran, E. Noor, J. L. Gallaher, J. Bale, Y. Yoshikuni, M. H. Gelb, J. D. Keasling, B. L. Stoddard, M. E. Lidstrom and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3704–3709 CAS.
- A. Herath, R. Dahl and N. D. P. Cosford, Org. Lett., 2010, 12, 412–415 CrossRef CAS PubMed.
- D. Obermayer, T. N. Glasnov and C. O. Kappe, J. Org. Chem., 2011, 76, 6657–6669 CrossRef CAS PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- A. R. Bogdan, S. L. Poe, D. C. Kubis, S. J. Broadwater and D. T. McQuade, Angew. Chem., Int. Ed., 2009, 48, 8547–8550 CrossRef CAS PubMed.
- S. Newton, C. F. Carter, C. M. Pearson, L. de C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915–4920 CrossRef CAS PubMed.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- L. H. Andrade, W. Kroutil and T. F. Jamison, Org. Lett., 2014, 16, 6092–6095 CrossRef CAS PubMed.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- R. Porcar, V. Sans, N. Ríos-Lombardía, V. Gotor-Fernández, V. Gotor, M. I. Burguete, E. García-Verdugo and S. V. Luis, ACS Catal., 2012, 2, 1976–1983 CrossRef CAS.
- D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- U. Guzik, K. Hupert-Kocurek and D. Wojcieszyńska, Molecules, 2014, 19, 8995 CrossRef PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250–6261 RSC.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- H. Vaghari, H. Jafarizadeh-Malmiri, M. Mohammadlou, A. Berenjian, N. Anarjan, N. Jafari and S. Nasiri, Biotechnol. Lett., 2016, 38, 223–233 CrossRef CAS PubMed.
- K. Min and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2014, 19, 553–567 CrossRef CAS.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33, 435–456 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790–802 CAS.
- T. Z. Yuan, C. F. G. Ormonde, S. T. Kudlacek, S. Kunche, J. N. Smith, W. A. Brown, K. M. Pugliese, T. J. Olsen, M. Iftikhar, C. L. Raston and G. A. Weiss, ChemBioChem, 2015, 16, 393–396 CrossRef CAS PubMed.
- J. Britton, L. M. Meneghini, C. L. Raston and G. A. Weiss, Angew. Chem., Int. Ed., 2016 Search PubMed , in press.
- J. Britton, J. M. Chalker and C. L. Raston, Chem. – Eur. J., 2015, 21, 10660–10665 CrossRef CAS PubMed.
- J. Britton, S. B. Dalziel and C. L. Raston, RSC Adv., 2015, 5, 1655–1660 RSC.
- J. Britton, S. B. Dalziel and C. L. Raston, Green Chem., 2016, 18, 2193–2200 RSC.
- B. C. C. Pessela, M. Fuentes, C. Mateo, R. Munilla, A. V. Carrascosa, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2006, 39, 909–915 CrossRef CAS.
- A. W. Miller and J. F. Robyt, Biotechnol. Bioeng., 1983, 25, 2795–2800 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: With all experimental procedures and additional graphical material. See DOI: 10.1039/c6cc04210d |
| This journal is © The Royal Society of Chemistry 2016 |