
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual gold photoredox C(sp2)–C(sp2) cross couplings – development and mechanistic studies†
Vincent
Gauchot
and
Ai-Lan
Lee
 *
*
Institute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: A.Lee@hw.ac.uk
First published on 19th July 2016
Abstract
A dual visible light photoredox and gold-catalysed C(sp2)–C(sp2) cross coupling is described. The success of this mild, oxidant- and base-free cross coupling is highly dependent on the amount of water added. Mechanistic studies show two distinct pathways depending on the gold catalyst employed: transmetallation of the arylboronic acid with gold(I) occurs prior to oxidation of gold(I) to gold(III) using cationic gold(I) catalysts, whereas oxidation of gold(I) to gold(III) precedes transmetallation using neutral gold(I) catalysts.
Over the past two decades, homogenous gold catalysis has emerged as a powerful and versatile tool for C–C and C-heteroatom bond formation. By virtue of its excellent carbophilicity, gold(I) complexes have traditionally been used to activate unsaturated C–C bonds towards nucleophilic attack, whereby the gold species typically does not change oxidation states throughout the catalytic cycle.1 Nevertheless, in order to significantly expand the repertoire of gold-catalysed processes, there has been intense recent interest in developing Au(I)/Au(III) catalytic processes.2 However, unlike the readily accessible Pd(0)/Pd(II) cycle, the high redox potential of the Au(I)/Au(III) redox couple (E0 = 1.41 V)3 means that strong oxidative conditions are usually required to access Au(I)/Au(III) catalytic processes. While these seminal works4 have proven that Au(I)/Au(III) catalytic cycles are accessible and can be used as powerful cross-coupling strategies, the use of typically super-stoichiometric, strong external oxidants negates one of the attractive features of gold-catalysis: mild reaction conditions and functional group tolerance. In order to circumvent the use of strong oxidants, the groups of Glorius5 and Toste6,7 have recently demonstrated the elegant use of dual gold and photoredox8 catalytic systems:9 a photosensitiser and an aryl radical source combined with visible light irradiation to access the Au(I)/Au(III) catalytic cycles under mild conditions.10
While visible light-mediated gold-catalysed C(sp2)–C(sp) cross couplings to form aryl-alkynes have been very recently reported,5c,6b there are, to the best of our knowledge, no examples of C(sp2)–C(sp2) biaryl couplings using dual gold/photoredox catalysts.11,12 Since we recently disclosed that arylboronic acids 1 readily transmetallate with PPh3AuNTf2 to form the arylgold species I,13–15 which regenerates the gold(I) catalyst upon protodeauration, we hypothesised that it should be possible to trap the arylgold intermediate I using photoredox catalysis and diazonium salts 4, in order to achieve the first dual/photoredox catalysed C(sp2)–C(sp2) biaryl couplings (Scheme 1). Upon addition of an aryl radical onto the Au(I) catalyst and the subsequent SET, the resulting Au(III) species II should undergo reductive elimination16 to yield 3. However, a significant challenge lies in the propensity for I to protodeaurate in the presence of water, and our previous mechanistic studies showed that water is necessary for the transmetallation step to form I. Despite this conundrum, we herein disclose our successful attempts at overcoming this challenge to develop the first dual gold/photoredox catalysed C(sp2)–C(sp2) cross-couplings, along with our mechanistic studies.
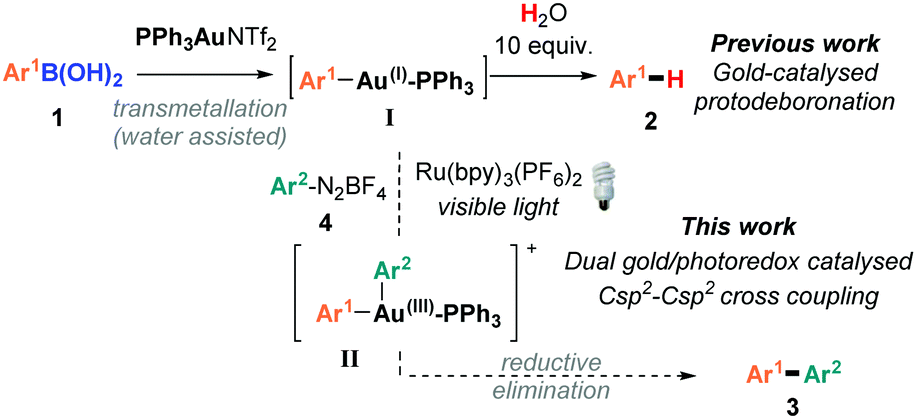 | ||
| Scheme 1 Dual gold/photoredox coupling between arylboronic acids and aryldiazonium salts. | ||
We initiated our studies using 1a and 4a, with PPh3AuNTf2 as the gold(I) catalyst17 and Ru(bpy)3(PF6)2 as the photocatalyst (Table 1). As expected, a significant amount of the unwanted protodeboronated product 2a was initially observed alongside 3aa. A solvent screen identified MeCN as the most promising solvent (see the ESI†), but the most significant change to improve the 3aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a ratio proved to be the impact of water (Table 1).
:2a ratio proved to be the impact of water (Table 1).
|
|
|||
|---|---|---|---|
| Entrya | H2Ob (equiv.) |
3aa:2ac |
Yield 3aac (%) |
| a 0.1 mmol scale in degassed MeCN (1 mL). b With respect to 1a. c Determined by 1H NMR analysis using dimethylsulfone as internal standard. d Isolated yield. e 4a (2 equiv.), PPh3AuNTf2 (5 mol%), Ru(bpy)3(PF6)2 (2.5 mol%). | |||
| 1 | — | 0.7:1 |
16 |
| 2 | 1 | 1.7:1 |
41 |
| 3 | 5 | 0.6:1 |
18 |
| 4 | 10 | 2.7:1 |
42 |
| 5 | 50 | 4.2:1 |
70 |
| 6 | 60 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
71 (70) |
| 7 | 75 | 3.5:1 |
66 |
| 8 | 60 |
>20:1
|
83 , |
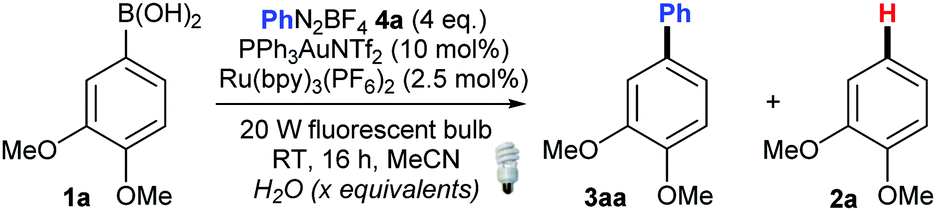
As shown in Table 1, addition of 1 equiv. of water instantly increases the amount of coupling product (1.7:1 3aa:2a, entries 1 and 2). However, this trend is non-linear: 5 equiv. of water produces a poor result (entry 3). Interestingly, when 10 or more equiv. of water are added, the reaction mixture becomes homogeneous, thus allowing for better light irradiation through the reaction mixture. This translates into a significant increase in yield observed in favour of the coupling product 3aa (entries 4–7). Surprisingly, this increase is once again non-linear. Our best results in this screen were obtained with 60 equiv. of water, yielding 70% 3aa (entry 6). However, going above 60 equiv. leads to a significant drop in the product ratio (entry 7, see the ESI† for full study). We believe that this double-edged effect of water, as well as the non-linearity of the effect, can be attributed to three factors. Firstly, water is involved in aiding the transmetallation step (1 → I, Scheme 1),13 which explains its beneficial effect. Working against this is the fact that water also promotes the undesired competing protodeboronation (I → 2, Scheme 1). Thirdly, water affects the homogeneity of the reaction mixture, and therefore the ability of light to efficiently penetrate the mixture to promote photoredox coupling. Therefore, a delicate balance needs to be struck between all three factors. Finally, further optimisation (see the ESI†) improved the yield to 83% with >20:1 3aa:2a (entry 8). Control reactions show that only a trace amount of 3aa is produced in the absence of the gold catalyst, the Ru catalyst, or light, confirming that the reaction is dual gold/photoredox catalysed (see the ESI†).
With these optimised conditions in hand, we commenced our substrate scope studies by investigating a range of arylboronic acids (Table 2). Since the system is base-free and mild, we expected the reaction to be tolerant to a range of different functional groups. Electron-rich aryl boronic acids are ideal coupling partners for this system, yielding the desired products with good to excellent yields (70–92%, 3aa–3ha). The substitution pattern seems to have little effect, with electron-rich ortho-, meta- and para-substituted aryls all providing good yields (3aa–3ha). Furthermore, steric hindrance seems to be well tolerated as exemplified by the formation of 3ha in 77% yield. In terms of functional group tolerance, we were pleased to see that phenols (3da), amides (3eb), esters (3ma) and carboxylic acids (3na) are compatible. However, the acid-sensitive THP group is cleaved during the reaction, producing the deprotected product 3ia′. The performance of electron-poor arylboronic acids is slightly more variable. While the presence of mildly electron-withdrawing p-bromo- and p-chloro substituents provide coupling products 3ka and 3la, respectively, in high yields (85–88%), the yields are more moderate with p-ester (3ka, 55%) and carboxylic acid (3la, 61%) substituted arylboronic acids. Noticeably lower yields are achieved with strongly electron-withdrawing substituents: nitro- (3pa, 47%) and CF3- (3qa, 34%), as well as with iodoboronic acid (3oa, 34%). Alkeneboronic acids are tolerated, albeit in a modest yield (39%, 3ja).
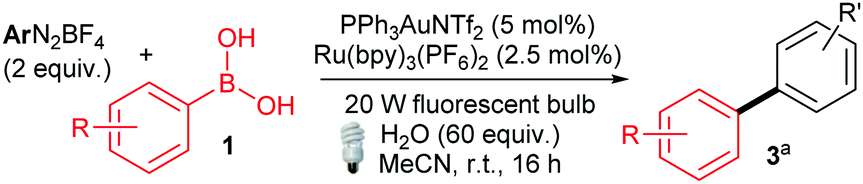
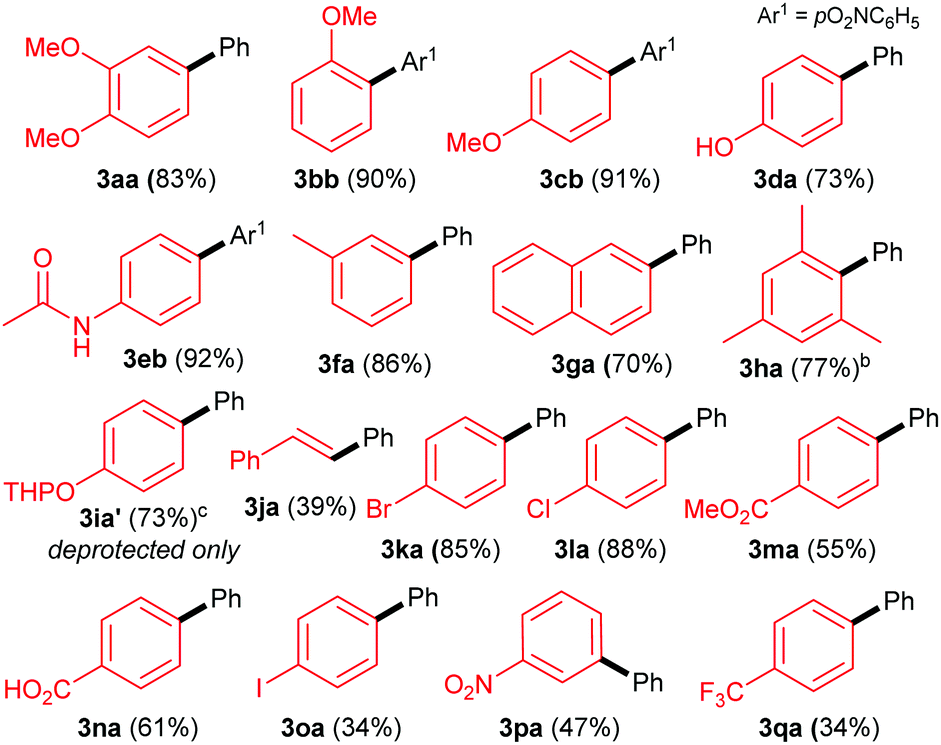
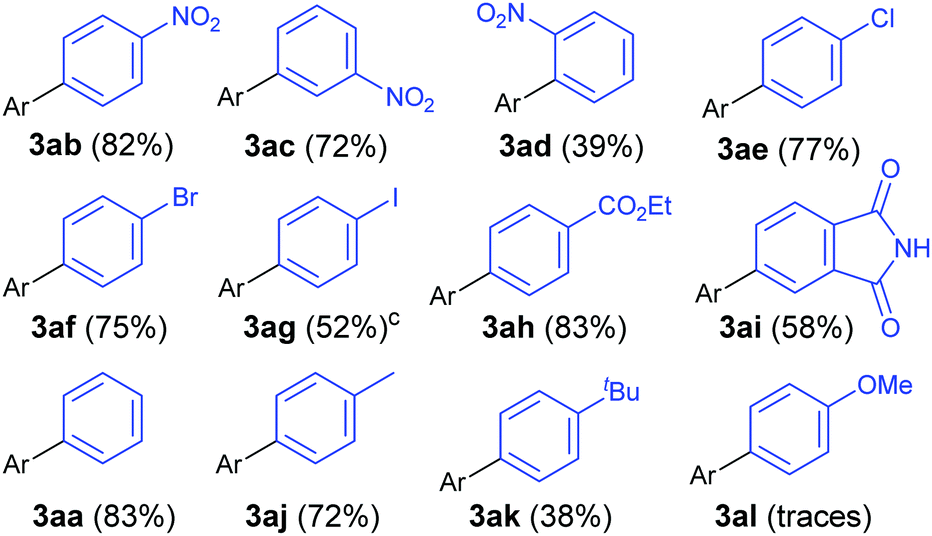
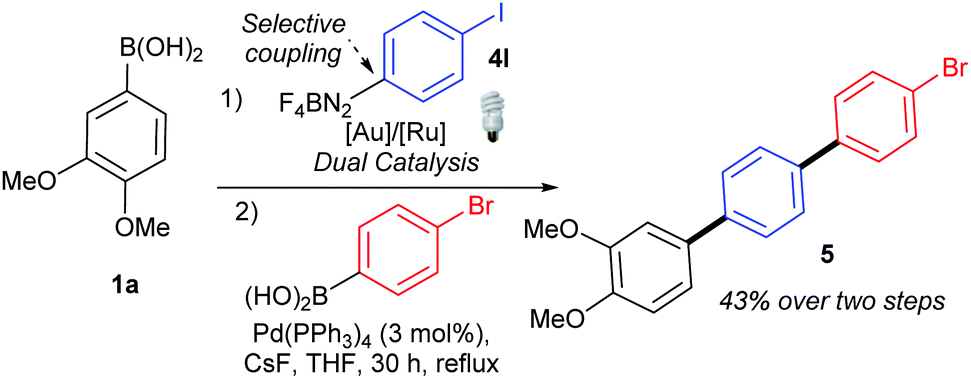
Gratifyingly, the opposite trend is observed in the aryldiazonium substrate scope (Table 3), which means that good yields can generally be achieved by the judicious choice of coupling partners. While electron-rich arylboronic acid coupling partners generally perform better than very electron-poor ones, electron-withdrawing substituents on the aryldiazonium coupling partners now provide the best yields (3ab–3ai, up to 83%). Para- (3ab) and meta-substituted (3ac) substrates are tolerated well, but a drop in yield is observed for more hindered ortho-substituted substrates (3ad). Predictably,18 electron-rich aryldiazonium salts react more sluggishly, with the yields decreasing as the substituent gets progressively more electron-donating (72% 3aj, 38% 3ak, traces 3al). Halogen substituted 4-chloro-, 4-bromo- and 4-iodophenyldiazonium salts once again perform well, producing the corresponding biaryls 3ae, 3af and 3ag in 77%, 75% and 52%. These results are of significance because the tolerance to C-halogen bonds19 renders the dual gold/photoredox coupling orthogonal to Pd(0)-catalysed reactions. The dual catalytic system can therefore be applicable to sequential cross-coupling reactions as exemplified by the reaction in Scheme 2. The cross coupling of 1a and 4l occurs without competitive cleavage of the C–I bond, which is then available for further Pd(0) cross-coupling. Subjecting the crude mixture of 3ag to Suzuki coupling20 subsequently produces triaryl 5 in 43% over the two steps.

 | ||
| Scheme 2 Sequential cross-coupling. | ||
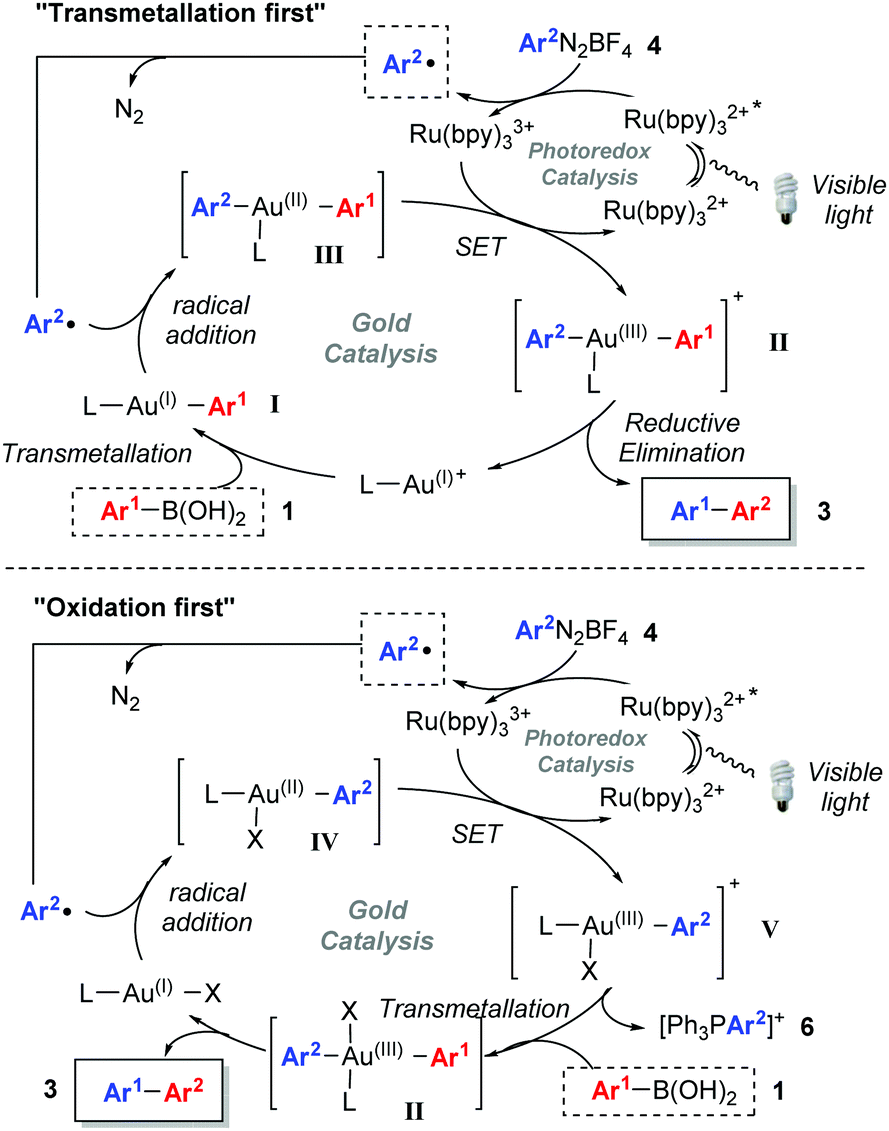
Based on a combination of literature reports,5b,c,6c,21 we postulated that the reaction can proceed through two different mechanisms (Scheme 3). In the “transmetallation first” pathway, LAu(I)+ undergoes transmetallation with 1 to form I prior to radical addition and oxidation to Au(III) (II). The alternative involves oxidation of the Au(I) species to Au(III) V before transmetallation (“oxidation first” pathway). Mechanistic studies21 on Glorius’ dual gold/photoredox-catalysed oxyarylation of alkenes5b as well as Toste's elegant studies on arylative ring expansions6c both point towards the oxidation first pathway. Nevertheless, as the reaction developed here involves a transmetallation rather than alkene activation with Au(I), we decided to carry out mechanistic studies.
 | ||
| Scheme 3 Postulated mechanistic pathways. | ||
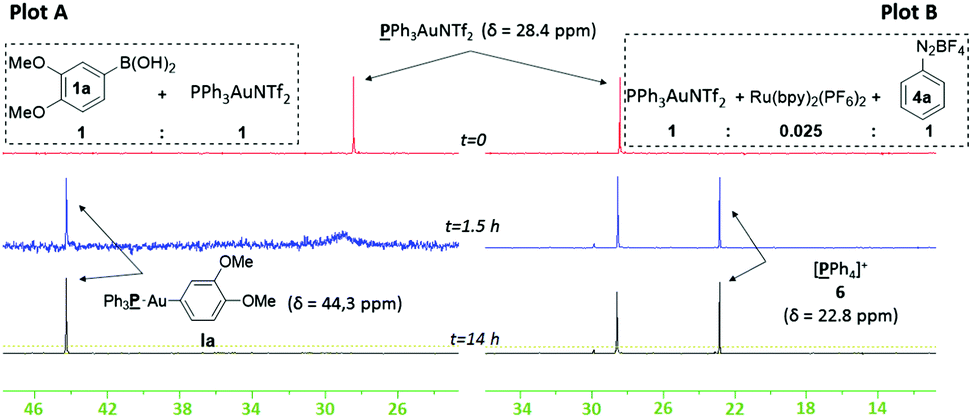
We chose to study both cationic PPh3AuNTf2 as well as neutral PPh3AuCl catalysts using 31P NMR analysis, as the latter successfully produces an appreciable amount of coupled product 3aa (37% conversion) even though previous reports have paradoxically shown that it is unable to undergo transmetallation with phenylboronic acid.22 Indeed, stirring stoichiometric amounts of PPh3AuCl and 1a in CD3CN and D2O revealed no transmetallation product (see the ESI†). In stark contrast, the same procedure using PPh3AuNTf2 gives the transmetallated Au(I) species Ia (Fig. 1, Plot A).23 Next, stoichiometric amounts of PPh3AuNTf2 and 4a with a catalytic amount of Ru(bpy)3(PF6)2 were irradiated under visible-light to produce a new signal at 22.1 ppm, corresponding to 6 (Fig. 1, Plot B).21,246 has been shown to arise from reductive elimination of gold(III) species V (Scheme 3),6c therefore, the detection of 6 can be used to imply that intermediate V exists in solution. In the absence of light, only the original PPh3AuNTf2 signal at 28.4 ppm was present.
 | ||
| Fig. 1 31P NMR studies in CD3CN:D2O. | ||
In light of these preliminary results, two separate gold/photoredox coupling experiments were carried out with stoichiometric amounts of PPh3AuCl or PPh3AuNTf2 respectively (Fig. 2). As expected, no traces of the transmetallated product Ia were observed using PPh3AuCl as catalyst (Plot C). Instead, the upfield signal appearing at 22.8 ppm matches that of 6 observed during control reaction B (Fig. 1, Plot B), implying that intermediate V is formed during the reaction.25 Concurrently, 1H NMR analysis confirmed the formation of coupling product 3aa after 14 h. These results suggest that the “oxidation first” catalytic cycle is the most plausible mechanism when neutral PPh3AuCl is used as catalyst. In stark contrast, a single signal matching the chemical shift of organogold species Ia appeared over time and no signal corresponding to 6 (and by implication, no formation of V) was observed using PPh3AuNTf2 as catalyst (Plot D). Concurrently, 1H NMR analysis clearly showed the formation of coupling product 3aa over the same time period. These results suggest that the “transmetallation first” catalytic cycle is the most plausible mechanism when cationic PPh3AuNTf2 is used as catalyst.
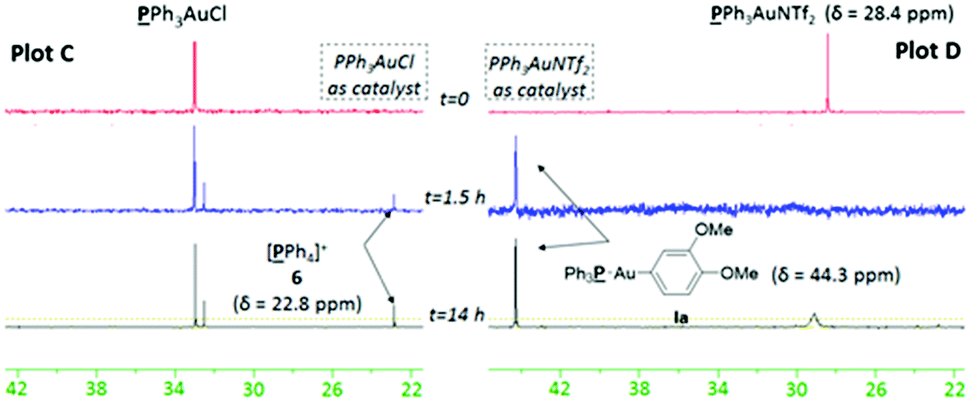 | ||
| Fig. 2 31P NMR monitoring of PPh3AuCl-catalysed reaction (plot C) and PPh3AuNTf2-catalysed reaction (plot D). | ||
In conclusion, we have developed the first application of dual gold/photoredox catalysis for C(sp2)–C(sp2) coupling. The successful cross-coupling was found to significantly depend on the amount of water added. While studies have shown that oxidation of Au(I) to Au(III) species V is the first step in several dual gold/photoredox catalysed reactions, our mechanistic studies reveal that the mechanistic sequence is in fact highly catalyst dependent. Cationic PPh3AuNTf2 actually undergoes transmetallation with arylboronic acid prior to oxidation of Au(I) to Au(III), whereas neutral PPh3AuCl undergoes the expected “oxidation first” pathway.
We thank the Leverhulme Trust (RPG-2014-345) for funding. Mass spectrometry data were acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University.
References
- Selected reviews: (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (b) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (c) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (d) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (e) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (f) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16 RSC; (g) D. Pflasterer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331 RSC.
- For review, see: M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1 CrossRef CAS.
- For selected examples of gold-catalysed oxidative coupling of arenes, see: (a) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (b) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636 CrossRef CAS PubMed; (c) Q. Wu, C. Du, Y. Huang, X. Liu, Z. Long, F. Song and J. You, Chem. Sci., 2015, 6, 288 RSC; (d) A. Kar, N. Mangu, H. M. Kaiser, M. Beller and M. K. Tse, Chem. Commun., 2008, 386 RSC.
- (a) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794 CrossRef CAS; (b) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (c) A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo and F. Glorius, Chem. Sci., 2016, 7, 89 RSC.
- (a) Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194 RSC; (b) S. Kim, J. Rojas-Martin and F. D. Toste, Chem. Sci., 2016, 7, 85 RSC; (c) X.-z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed.
- See also: (a) S. Cai, K. Yang and D. Z. Wang, Org. Lett., 2014, 16, 2606 CrossRef CAS PubMed; (b) D. V. Patil, H. Yun and S. Shin, Adv. Synth. Catal., 2015, 357, 2622 CrossRef CAS.
- For selected recent reviews on visible light photoredox catalysis, see: (a) T. P. Yoon, M. A. Ischay and J. N. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (c) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (d) D. M. Schultz and T. P. Yoon, Science, 2014, 343 Search PubMed; (e) E. Meggers, Chem. Commun., 2015, 51, 3290 RSC; (f) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2013, 4, 355 CrossRef; (g) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC.
- For review on cooperative photoredox catalysis, see: X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026 RSC.
- For an elegant alternative; photoredox using only Au catalysts with blue LEDs by Hashmi and Barriault: (a) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808 CrossRef CAS PubMed; (b) G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342 CrossRef CAS PubMed; (c) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435 RSC; (d) J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046 CrossRef CAS PubMed.
- Although it is known with ligand- and base-assisted redox gold-catalysis (mechanistically distinct from photoredox): (a) R. Cai, M. Lu, E. Y. Aguilera, Y. Xi, N. G. Akhmedov, J. L. Petersen, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2015, 54, 8772 CrossRef CAS PubMed . See also Pd-catalysis review: ; (b) H. Bonin, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2011, 353, 3063 CrossRef CAS.
- During the preparation of this manuscript, Barriault disclosed a photoredox gold catalysed C(sp3)–C(sp2) coupling to alkylated heterocycles: T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4757 RSC.
- G. Barker, S. Webster, D. G. Johnson, R. Curley, M. Andrews, P. C. Young, S. A. Macgregor and A.-L. Lee, J. Org. Chem., 2015, 80, 9807 CrossRef CAS PubMed.
- Stoichiometric: D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2006, 45, 8188 CrossRef CAS PubMed.
- See also: M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211 CrossRef CAS PubMed.
- Reductive elimination steps involving Au(III) can be extremely fast, see: W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159 CrossRef CAS PubMed.
- N. Mezailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef CAS PubMed.
- This is the same trend observed previously with aryldiazonium salts, see ref. 5.
- (a) M. Livendahl, C. Goehry, F. Maseras and A. M. Echavarren, Chem. Commun., 2014, 50, 1533 RSC; (b) M. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed; (c) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. De Buck Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133 CrossRef CAS.
- M. Schiek, K. Al-Shamery and A. Lutzen, Synthesis, 2007, 613 CAS.
- Q. Zhang, Z.-Q. Zhang, Y. Fu and H.-Z. Yu, ACS Catal., 2016, 6, 798 CrossRef CAS.
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293 CrossRef CAS PubMed.
- D. Weber, T. D. Jones, L. L. Adduci and M. R. Gagne, Angew. Chem., Int. Ed., 2012, 51, 2452 CrossRef CAS PubMed.
- D. Marcoux and A. B. Charette, J. Org. Chem., 2008, 73, 590 CrossRef CAS PubMed.
- Neutral Au(III)-boron transmetallation: M. Hofer, E. Gomez-Bengoa and C. Nevado, Organometallics, 2014, 33, 1328 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full optimisation and control studies, mechanistic studies, characterisation data and copies of NMR spectra of new compounds. See DOI: 10.1039/c6cc05078f |
| This journal is © The Royal Society of Chemistry 2016 |