
Non-covalent immobilisation of p-toluenesulfonic acid in a porous molecular crystal for size-specific acid-catalysed reactions†
Shohei
Tashiro
*,
Hirotaka
Yonezawa
,
Ryou
Kubota‡
,
Tsutomu
Umeki
and
Mitsuhiko
Shionoya
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shionoya@chem.s.u-tokyo.ac.jp
First published on 26th April 2016
Abstract
Non-covalent immobilisation of catalysts in nanoporous materials is a promising way to apply homogeneous catalysts to heterogeneous catalytic reactions. Herein we report a size-specific catalytic reaction with an acid catalyst, p-toluenesulfonic acid, immobilised in a porous molecular crystal, metal–macrocycle framework (MMF), composed of metallo-macrocycles. A tritylated substrate which is smaller than the pore dimension of MMF was deprotected by the heterogeneous catalyst, whereas the reaction with a larger substrate was completely suppressed due to the steric restriction.
Immobilisation of homogeneous catalysts on a solid surface is key to the development of reusable, separable and selective catalysts.1 Recent progress in porous solid materials such as mesoporous silica, zeolites, porous coordination polymers (PCPs) and covalent-organic frameworks (COFs) has allowed various immobilisation methods of catalytic moieties through pre- and post-synthetic modification.2 Among these methods, non-covalent immobilisation of the highly catalytic molecules is a simple and easy-to-use method because any chemical modifications of catalysts are not necessary. To date, several examples of such immobilisation have been reported.3 For instance, ionic porous supports can immobilise a counter-ion catalyst as an ion-pair through ion-exchange.4 Also, encapsulation of a catalyst in porous supports, whose window size is smaller than that of the catalyst, is known as a “ship-in-a-bottle” approach.4e,5 In addition, another method is simple adsorption of unmodified catalytic molecules on a pore surface through significant interactions. However, a drawback of this method is leaching of catalysts due to the weak adsorption, and this approach is therefore limited to some cases using porous supports with effective binding sites on their pore surface.6
We have recently developed a new porous molecular crystal, metal–macrocycle framework (MMF), self-assembled from four isomers of trinuclear PdII complexes of a macrocycle having tris(o-phenylenediamine) moieties.7 In this crystal, nanochannels with a pore size of 1.4 × 1.9 nm2 are arranged in parallel, and the pore surface provides five kinds of enantiomeric binding pockets along with several polar sites (NH and Cl) (Fig. 1a and b). Utilising this structural feature, we have also demonstrated site-selective arrangements of many types of guest molecules on the pore surface through non-covalent interactions such as hydrogen bonding, CH–π interaction and van der Waals interaction.7c In particular, two typical acid catalysts, p-toluenesulfonic acid and benzenesulfonic acid, were site-selectively adsorbed to the bottom corners of the pore surface through non-covalent interactions as confirmed by single-crystal X-ray diffraction (XRD) analyses (Fig. 1c).7b This finding then prompted us to use the MMF crystals as novel porous solid supports for non-covalent immobilisation of unmodified acid catalysts in the pores. Herein we report heterogeneous catalytic reactions in MMF crystals with p-toluenesulfonic acid (1) non-covalently bound to the pore surfaces. As a first step, we immobilised 1 on the inner surface of MMF crystals. The resulting crystals enabled an acid-catalytic deprotection reaction of the trityl (Trt) group in a pore/guest size-specific manner. The immobilised acid catalyst was not leached away from the MMF channel in halogenated solvents, while it can be easily rinsed off using CH3CN due to the non-covalent immobilisation. This feature is in sharp contrast to previously-reported crystals with covalently immobilised sulfonic acid in their pore surfaces.8
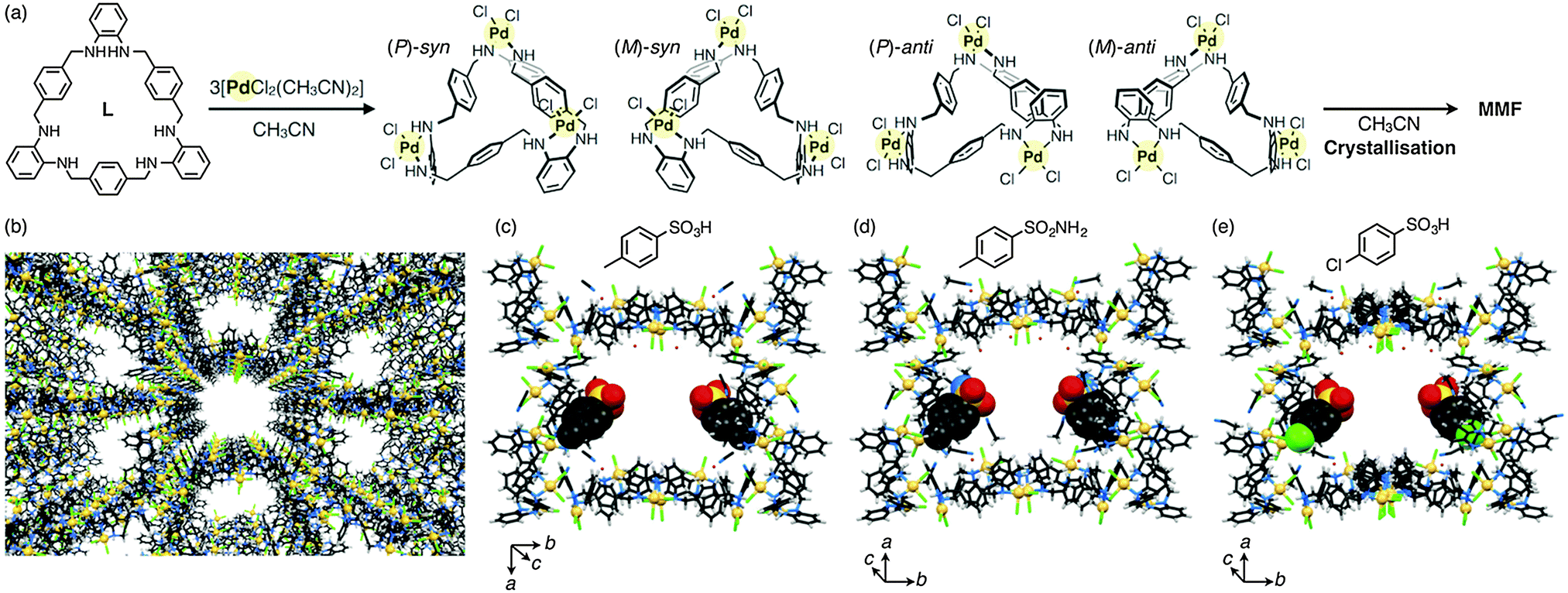 | ||
| Fig. 1 (a) Crystallisation of MMF by complexation between L and [PdCl2(CH3CN)2] in CH3CN via the formation of four stereoisomers of [Pd3LCl6], (P)-syn, (M)-syn, (P)-anti and (M)-anti forms. (b) Crystal packing structure of MMF. Unit-space structures of MMF (a half of the unit cell) incorporating (c) p-toluenesulfonic acid (1),7b (d) p-toluenesulfonamide and (e) 4-chlorobenzenesulfonic acid. Their occupancies in the crystal structures were 50%, 40% and 50%, respectively. MMF: stick model, each guest: CPK model. C: black, H: white, N: blue, O: red, S: yellow, Cl: green and Pd: lemon yellow. | ||
First, we examined guest inclusion ability of a variety of acid molecules such as p-toluenesulfonic acid monohydrate (1·H2O),7bp-toluenesulfonamide and 4-chlorobenzenesulfonic acid hydrate. MMF crystals were soaked in a CH3CN solution containing each guest (0.5 M) for several days at 20 °C, and then analysed by single-crystal XRD measurements. As expected for their 1,4-disubstituted benzene framework, every acid molecule was adsorbed on the bottom corners of the pores (Fig. 1c–e). This result also indicates the excellent adsorption capability of MMF for sulfonic acid derivatives. We then utilised p-toluenesulfonic acid (1) hydrate as a heterogeneous acid catalyst in MMF.
p-Toluenesulfonic acid monohydrate (1·H2O) was easily adsorbed in MMF pores by crystal soaking in a CH3CN solution of 1·H2O (0.8 M) for several hours at 20 °C. The MMF crystal was stable in the solution during the soaking experiment. The number of adsorbed molecules of 1 in a unit space, a half of the crystallographic unit cell, was estimated to be 4–5 by 1H NMR analysis of a DMSO–DCl solution of digested crystals (Fig. 2). Further time-course NMR digestion analysis also indicated that the amount of adsorbed 1 was saturated in 1 h of soaking. In contrast, only 1.0 molecule of 1 per unit space was crystallographically observed (50% occupancies at both corners) by XRD analysis.7b This discrepancy suggests that the remaining 3–4 molecules of 1 are highly disordered probably in a fully solvated manner in the unit space (Fig. 2). Indeed, the mobile molecules of 1 are easily leached from the crystals as detected by 1H NMR analysis of its CDCl3 supernatant.
 | ||
| Fig. 2 Schematic representation for the preparation of 1@MMF through crystal soaking in a CH3CN solution of 1·H2O and washing with CH2Cl2 and CHCl3. | ||
To wash out only the mobile molecules of 1, several washing conditions were tested with three types of solvents, CH3CN, n-hexane and CH2Cl2. In the case of CH3CN which is a good solvent for 1·H2O (solubility ∼0.8 M), the number of molecules of 1 adsorbed in the unit space was significantly decreased to 0.6 after washing three times because most molecules of 1 were leached from the MMF crystals. In contrast, a poor solvent for 1·H2O, n-hexane (solubility ≪1 mM), did not wash out any molecules of 1. Finally, we found that MMF crystals firmly immobilising 1 were successfully prepared by washing with CH2Cl2, a moderately good solvent for 1·H2O (solubility ∼1 mM). The number of molecules of 1 adsorbed in a unit space was roughly estimated to be 1.4–2.0 (1.7 ± 0.3 in average) after washing with CH2Cl2 three to four times, and further washing decreased the number of immobilised molecules of 1 little. We then prepared acid-immobilised crystals, 1@MMF, including 1.5 molecules of 1 in average in a unit space by repeated CH2Cl2 washing and additional washing with CHCl3 (Fig. 2). As expected, the resulting 1@MMF crystals did not show any acid leaching of 1 in CDCl3 as proven by 1H NMR analysis.
To determine the binding mode of 1 in the MMF pore, we used single-crystal XRD analysis of 1@MMF prepared in a CH2Cl2 washing protocol as above. We successfully observed X-ray diffraction of this crystal, though the diffraction intensity was not sufficient probably due to the decreasing crystallinity with crystal shrinkage through washing. The preliminary result with low-resolution data (1.2 Å) indicated that the framework structure was intact. The analysis of the electron map on the pore surface suggests the presence of CH2Cl2 and water molecules and not 1. If so, how does 1 stay in a pore of 1@MMF? It may be assumed that an ionic pair of p-toluenesulfonate (p-TsO−) and H3O+ is formed in CH2Cl2 in which H3O+ binds to the pore surface through hydrogen bonding and its concomitant counter anion, p-TsO−, with severely disordered structures. To confirm this we added Et3N to a suspension of 1@MMF in CDCl3 to neutralise the acid species. 1H NMR analysis of this supernatant indicated the leaching of p-TsO(Et3NH) salts. This result may suggest that H3O+ serves as an effective anchor when 1·H2O is immobilised on the pore surface in non-polar solvents to form an ionic pair, p-TsO− and H3O+ (Fig. 2). Such a binding mode is reminiscent of previously reported examples of non-covalent immobilisation approaches of transition metal catalysts on polar supports with triflate or heteropoly acids as anchors.6c,9
The acid catalyst 1@MMF including 1.5 molecules of 1 in a unit space was next applied to a typical deprotection reaction of an acid-labile Trt group. We chose BnOTrt (2) as a substrate because the kinetic diameter of 2 was calculated to be approximately 1.1 nm,10 which is smaller than the pore size of MMF (1.4 × 1.9 nm2) (Fig. 3). The crystals of 1@MMF (6 mol%: the mole number was calculated based on the total amount of 1 included in MMF) were soaked in a mixed solution of CDCl3 and H2O containing 2 ([2] = 2.2 mM, [H2O] = 23 mM) at 20 °C, which was monitored by 1H NMR analyses. While almost no reactions took place for 2 days, the conversion gradually proceeded to approach ca. 70% in 3 weeks (Fig. 3). As the reaction speed was a little dependent on the size or crystallinity of 1@MMF crystals, the reproducibility of this catalytic reaction was confirmed by three independent reaction batches. In contrast, since acid-free MMF did not exhibit any reactivity (0% after 3 weeks), we could confirm that 1@MMF evidently served as an acid catalyst. Not surprisingly, homogeneous catalyst 1·H2O (4 mol%) rapidly promoted this reaction (93% after 1.5 h) in bulk solution. The extremely low reactivity in the early stage of the reaction with the 1@MMF catalyst is presumably due to the poor accessibility and slow diffusion of 2 into the pore of 1@MMF. Indeed, when guest-free MMF crystals were soaked in a CDCl3 solution of 2 ([2] = 500 mM), guest 2 was slowly included in one day and the number of included molecules of 2 was estimated to be 0.2 per unit space as proven by 1H NMR digestion experiments.
 | ||
| Fig. 3 (top) Channel dimension of MMF and kinetic diameters of 2 and 4. (bottom) Deprotection reaction of 2 or 4 in the presence of 6 mol% of 1@MMF. Circles and crosses show the conversion rates of 2 and 4, respectively. The inset shows the results of control experiments with homogeneous catalyst 1·H2O (4–5 mol%) in CDCl3 at 20 °C. | ||
Because acid 1 was non-covalently immobilised in MMF crystals, the catalytic activity of 1@MMF was easily lost by desorption of 1 through CH3CN washing. The number of molecules of 1 in a unit space of MMF decreased from 1.5 to 0.7 by repeated CH3CN washing. We confirmed that the MMF crystals containing 0.7 molecules of 1 in a unit space did not initiate the deprotection of 2 at all (0% conversion after 4 weeks). This behaviour is due to the non-covalent immobilisation of catalyst 1 in the unique solid support MMF that can control molecular binding behaviours depending on the kind of solvent.
Next, to confirm the character of 1@MMF as a heterogeneous catalyst, the crystals of 1@MMF were filtered off after the reaction, and its filtrate with additional 2 was allowed to stand at 20 °C. As expected, no reaction occurred in the filtrate even after 5 weeks, which strongly suggests that acid leaching from 1@MMF was negligible and that 1@MMF indeed serves as a heterogeneous catalyst. On the other hand, the reusability of 1@MMF collected after the first reaction, which did not contain 2 and 3 in the channel as confirmed by NMR digestion analysis, was moderate because the secondary use of 1@MMF resulted in no reaction with 2 at 20 °C.11 One possible reason for the loss of the catalytic activity of the reused 1@MMF is that the entrances of the pores may be collapsed and blocked as the crystallinity was significantly decreased.
In light of the pore window size of MMF (1.4 × 1.9 nm2), the size effect on acid-catalysed deprotection of trityl groups was examined using a large sized tritylated PdII porphyrin derivative 4 with a 1.7 nm kinetic diameter.10 The acid lability of 4 to a catalytic amount of 1·H2O in CDCl3 was comparable to that of 2, and a PdII ion was not practically dissociated from the porphyrin moiety of 4 under the acidic conditions. Then the reactivities of 2 and 4 to 1@MMF were experimentally compared separately under the identical conditions in the presence of 6 mol% 1@MMF in CDCl3 ([2] = 2.2 mM, [4] = 2.1 mM, [H2O] = 23 mM). As a result, the conversion of 2 was almost completed after three weeks, but the larger substrate 4 did not react at all (Fig. 3). The size specificity observed here strongly supported the fact that the trityl group of 2 was removed from the MMF pore. Although the reaction rate somewhat became altered depending on the size and shape of 1@MMF, the difference of the reaction rate between 2 and 4 was significant and reproducible.
In summary, we have established that porous MMF crystals non-covalently immobilising p-toluenesulfonic acid serve as size-specific heterogeneous catalysts for the deprotection reaction of a trityl group. This study successfully demonstrated non-covalent heterogenisation of an unmodified catalyst in the porous solid support. Also, it should be noted that MMF crystals with several binding sites on the pore surface have great potential to immobilise not only a single catalytic molecule but also multiple catalysts in a site-selective manner,7a,c leading to concerted catalytic reactions as observed in multi-functional enzymes.
This research was supported by JSPS KAKENHI Grant Number 23655117 (Challenging Exploratory Research) for M. S. and 15H05478 (Young Scientists(A)) for S. T.
References
- (a) in Catalysts for Fine Chemical Synthesis: Microporous and Mesoporous Solid Catalysts, ed. E. G. Derouane, Wiley Ltd, Chichester, England, 2006 Search PubMed; (b) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- (a) D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed; (b) S. Kitagawa, S. Noro and T. Nakamura, Chem. Commun., 2006, 701–707 RSC; (c) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC; (d) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (e) D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS; (f) K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC; (g) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (h) J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- (a) J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS PubMed; (b) L. Zhang, S. Luo and J.-P. Cheng, Catal. Sci. Technol., 2011, 1, 507–516 RSC; (c) M. Gruttadauria, F. Giacalone and R. Noto, Green Chem., 2013, 15, 2608–2618 RSC.
- (a) C. Simons, U. Hanefeld, I. W. C. E. Arends, A. J. Minnaard, T. Maschmeyer and R. A. Sheldon, Chem. Commun., 2004, 2830–2831 RSC; (b) A. Crosman and W. F. Hoelderich, J. Catal., 2005, 232, 43–50 CrossRef CAS; (c) P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515–529 CrossRef CAS PubMed; (d) D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed; (e) S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178 CrossRef CAS PubMed.
- (a) N. Herron, Inorg. Chem., 1986, 25, 4714–4717 CrossRef CAS; (b) D. E. De Vos, J. L. Meinershagen and T. Bein, Angew. Chem., Int. Ed. Engl., 1996, 35, 2211–2213 CrossRef CAS; (c) A. Corma and H. Garcia, Eur. J. Inorg. Chem., 2004, 1143–1164 CrossRef CAS; (d) R. W. Larsen, L. Wojtas, J. Perman, R. L. Musselman, M. J. Zaworotko and C. M. Vetromile, J. Am. Chem. Soc., 2011, 133, 10356–10359 CrossRef CAS PubMed; (e) M. Shakeri, R. J. M. K. Gebbink, P. E. de Jongh and K. P. de Jong, Angew. Chem., Int. Ed., 2013, 52, 10854–10857 CrossRef CAS PubMed; (f) B. Nepal and S. Das, Angew. Chem., Int. Ed., 2013, 52, 7224–7227 CrossRef CAS PubMed; (g) B. Li, Y. Zhang, D. Ma, T. Ma, Z. Shi and S. Ma, J. Am. Chem. Soc., 2014, 136, 1202–1205 CrossRef CAS PubMed.
- (a) X.-G. Zhou, X.-Q. Yu, J.-S. Huang, S.-G. Li, L.-S. Li and C.-M. Che, Chem. Commun., 1999, 1789–1790 RSC; (b) S. Krijnen, B. L. Mojet, H. C. L. Abbenhuis, J. H. C. Van Hooff and R. A. Van Santen, Phys. Chem. Chem. Phys., 1999, 1, 361–365 RSC; (c) F. M. de Rege, D. K. Morita, K. C. Ott, W. Tumas and R. D. Broene, Chem. Commun., 2000, 1797–1798 RSC; (d) H. M. L. Davies, A. M. Walji and T. Nagashima, J. Am. Chem. Soc., 2004, 126, 4271–4280 CrossRef CAS PubMed; (e) M. Tada, T. Taniike, L. M. Kantam and Y. Iwasawa, Chem. Commun., 2004, 2542–2543 RSC; (f) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed; (g) H. Tabe, S. Abe, T. Hikage, S. Kitagawa and T. Ueno, Chem. – Asian J., 2014, 9, 1373–1378 CrossRef CAS PubMed.
- (a) S. Tashiro, R. Kubota and M. Shionoya, J. Am. Chem. Soc., 2012, 134, 2461–2464 CrossRef CAS PubMed; (b) R. Kubota, S. Tashiro, T. Umeki and M. Shionoya, Supramol. Chem., 2012, 24, 867–877 CrossRef CAS; (c) S. Tashiro, T. Umeki, R. Kubota and M. Shionoya, Angew. Chem., Int. Ed., 2014, 53, 8310–8315 CrossRef CAS PubMed; (d) R. Kubota, S. Tashiro, M. Shiro and M. Shionoya, Nat. Chem., 2014, 6, 913–918 CrossRef CAS PubMed; (e) S. Tashiro and M. Shionoya, Bull. Chem. Soc. Jpn., 2014, 87, 643–654 CrossRef CAS; (f) R. Kubota, S. Tashiro and M. Shionoya, Chem. Sci., 2016, 7, 2217–2221 RSC.
- (a) J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705–710 CrossRef CAS PubMed; (b) G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef CAS PubMed; (c) P. Ramaswamy, R. Matsuda, W. Kosaka, G. Akiyama, H. J. Jeon and S. Kitagawa, Chem. Commun., 2014, 50, 1144–1146 RSC; (d) B. Li, K. Leng, Y. Zhang, J. J. Dynes, J. Wang, Y. Hu, D. Ma, Z. Shi, L. Zhu, D. Zhang, Y. Sun, M. Chrzanowski and S. Ma, J. Am. Chem. Soc., 2015, 137, 4243–4248 CrossRef CAS PubMed; (e) W. J. Phang, H. Jo, W. R. Lee, J. H. Song, K. Yoo, B. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2015, 54, 5142–5146 CrossRef CAS PubMed.
- R. Augustine, S. Tanielyan, S. Anderson and H. Yang, Chem. Commun., 1999, 1257–1258 RSC.
- T. C. Keller, S. Isabettini, D. Verboekend, E. G. Rodrigues and J. Pérez-Ramírez, Chem. Sci., 2014, 5, 677–684 RSC.
- Heating the mixture at 50 °C for 3–6 weeks promoted the deprotection of 2 to some extent. After heating, acid leaching was not detected as the supernatant of the sample did not show any reactivity for the deprotection of 2, and 1 was still included in the reused 1@MMF (1.2–1.5 molecules per a unit space) as proven by NMR digestion analysis.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic methods, analytic data, crystallographic data, crystal structure of 1@MMF, reaction procedures and reference experiments. CCDC 1468109 (MMF including 4-chlorobenzenesulfonic acid), 1468110 (1@MMF) and 1468503 (MMF including p-toluenesulfonamide). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc02621d |
| ‡ Current address: Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Kyoto 615-8510, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |