
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Screening of a virtual mirror-image library of natural products†
Taro
Noguchi
a,
Shinya
Oishi
*a,
Kaori
Honda
b,
Yasumitsu
Kondoh
b,
Tamio
Saito
b,
Hiroaki
Ohno
a,
Hiroyuki
Osada
b and
Nobutaka
Fujii
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: soishi@pharm.kyoto-u.ac.jp; nfujii@pharm.kyoto-u.ac.jp; Fax: +81 75-753-4570
bRIKEN CSRS, Wako, Saitama 351-0198, Japan
First published on 11th May 2016
Abstract
We established a facile access to an unexplored mirror-image library of chiral natural product derivatives using D-protein technology. In this process, two chemical syntheses of mirror-image substances including a target protein and hit compound(s) allow the lead discovery from a virtual mirror-image library without the synthesis of numerous mirror-image compounds.
Natural products and their derivatives have been valuable resources for drug discovery.1 A number of natural products and their derivatives are used in clinical practice as anticancer, antibacterial and immunosuppressive agents; these compounds were originally produced by plants, fungi, bacteria, or others as secondary metabolites for defensive and reproductive purposes.2 The desirable biological activities are attributed to the unique, complex, and sp3-carbon-rich scaffolds. While many natural products are produced as a single enantiomer when bearing a chiral structure(s), in some limited cases, two enantiomeric forms of chiral natural products are produced by a single or various species.3 The two mirror-image isomers with identical chemical and physicochemical properties often exhibit different functions and/or biological activities.4 The underlying chiral interactions between natural products and the target biomolecules are fundamental to the remarkable stereospecificity of these substances.
Recent reports have described attempts to expand the diversity of natural-product-like scaffolds.5 Using complex natural product structures as starting materials, a variety of natural-product-like scaffolds were prepared via a ring-distortion strategy using chemical modifications.6 Similarly, the component sesquiterpenes in the extract of Curcuma zedoaria were converted into a variety of derivatives with unprecedented scaffolds by treatment with oxidizing agents.7 Manipulation of the biosynthetic pathway by modification of culture conditions increases the chemical diversity of metabolite products.8 Epigenetic remodeling via activation of silent gene cluster(s) also leads to the production of unprecedented natural products from fungi.9 However, the stereochemical outcomes of the newly obtained scaffolds from these approaches are highly dependent on those of the parent substrates. To improve the access to mirror-image natural products and their derivatives with potentially desirable bioactivities as valuable resources in drug discovery, a complementary approach needs to be developed. Herein, we report a novel strategy to expand the structural diversity of chiral natural products and their derivatives by assessment of mirror-image forms.
We designed a new screening strategy for a virtual mirror-image library of chiral natural products with favorable drug-like properties, taking advantage of the mirror-image protein technology. The mirror-image proteins have been used for understanding a variety of biological phenomena.10 Mirror-image phage display has also led to the identification of antiviral and anticancer D-peptides, which have resistance against peptidase-mediated degradation.11 By using D-proteins, the interactions with compounds are diastereomeric to those afforded by the same compounds with native proteins (L-proteins).12 In this sense, chiral compounds could effectively bind in entirely new ways. We anticipated that screening performed using mirror-image proteins could potentially double the chiral natural product resources available for drug discovery (Fig. 1). This process involves the following: (1) initially, the D-protein form of the target protein molecule is prepared by chemical protein synthesis; (2) using the synthetic D-protein, chiral natural products are screened to identify hit compound(s); (3) the mirror-image structure(s) of the hit compound(s) is synthesized and (4) the biological activities for the target L-protein could subsequently be assessed.
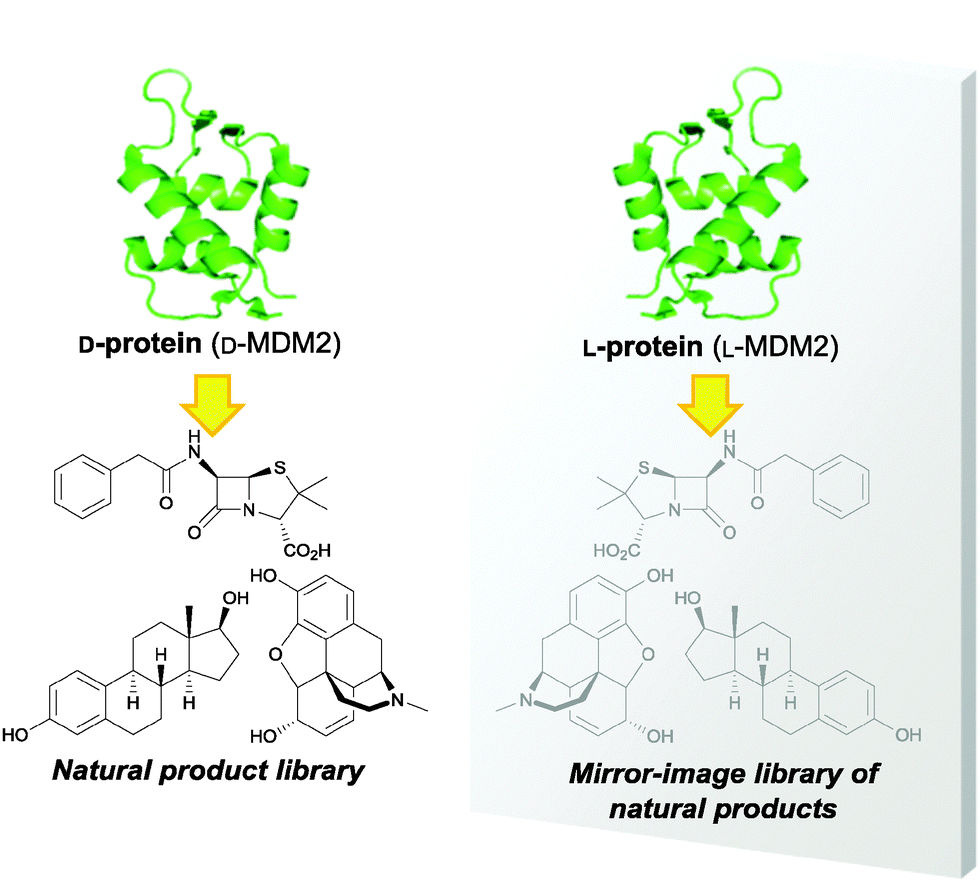 | ||
| Fig. 1 Concept of the screening strategy for a virtual mirror-image library of natural products. Screening of chiral natural products using D-protein corresponds to that of mirror-image natural products using L-protein in a mirror. | ||
MDM2 is a negative regulator of tumor-suppressor protein p53.13 We chose the p53-binding domain of MDM2 (MDM225–109) as the target, because a number of chiral inhibitors against MDM2–p53 interaction have been identified as potential anticancer agents.14 For example, nutlin-3a potently binds to MDM2, while nutlin-3b (enantiomer of nutlin-3a) shows significantly less activity.15 Initially, we synthesized D-MDM225–109 by standard Fmoc-based solid-phase peptide synthesis according to the protocol used in our previous study.16 The tetramethylrhodamine (TMR)-labeled protein (D-MDM2TMR) was also prepared by conjugation of TMR-(PEG)3-azide onto the N-terminal alkyne tag of D-MDM225–109. The folded L-MDM225–109 and D-MDM225–109 had symmetric circular dichroism spectra, supporting the presence of mirror-image α-helices (Fig. S3, ESI†).11f To verify the biological activity of the synthetic MDM2 proteins, the binding affinities toward p53 peptides (biotinyl-aminocaproyl-GSGSSQETFSDLWKLLPEN-NH2) were evaluated by surface plasmon resonance (SPR) analysis (Fig. S4, ESI†). The high-affinity bindings of the L-MDM2–L-p53 peptide and the D-MDM2–D-p53 peptide were observed [KD (L-MDM2–L-p53): 0.75 ± 0.04 μM; KD (D-MDM2–D-p53): 0.51 ± 0.02 μM], while the binding activities of the L-MDM2–D-p53 peptide and the D-MDM2–L-p53 peptide were practically nil, suggesting high enantiomeric recognition of MDM2 toward p53.
Chemical array screening of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 293 compounds including natural products and their derivatives from RIKEN NPDepo was carried out using L-MDM2TMR and D-MDM2TMR.16,17 Among 43 initial hit compounds (Table S1, ESI†), four compounds showed inhibitory activity against L-MDM2–L-p53 and/or D-MDM2–D-p53 interactions in the competitive binding inhibition assay by fluorescence polarization (FP) (Fig. 2 and Fig. S5, ESI†). Of these, a chiral α-tocopherol derivative NP843 (1a) was identified to be a selective inhibitor against D-MDM2–D-p53 interaction (Fig. 3).
293 compounds including natural products and their derivatives from RIKEN NPDepo was carried out using L-MDM2TMR and D-MDM2TMR.16,17 Among 43 initial hit compounds (Table S1, ESI†), four compounds showed inhibitory activity against L-MDM2–L-p53 and/or D-MDM2–D-p53 interactions in the competitive binding inhibition assay by fluorescence polarization (FP) (Fig. 2 and Fig. S5, ESI†). Of these, a chiral α-tocopherol derivative NP843 (1a) was identified to be a selective inhibitor against D-MDM2–D-p53 interaction (Fig. 3).
 | ||
| Fig. 2 Two-step screening process using D-MDM2. Initially, chemical array screening was carried out using TMR-labeled MDM2 proteins (L-MDM2TMR or D-MDM2TMR) for 22293 compounds. Among 43 hit compounds with selective binding affinity toward L-MDM2 or D-MDM2 (Table S1, ESI†), four compounds were identified as L-MDM2–L-p53 and/or D-MDM2–D-p53 interaction inhibitors by a competitive binding experiment in the FP assay. Abbreviation: FAM = carboxyfluorescein. | ||
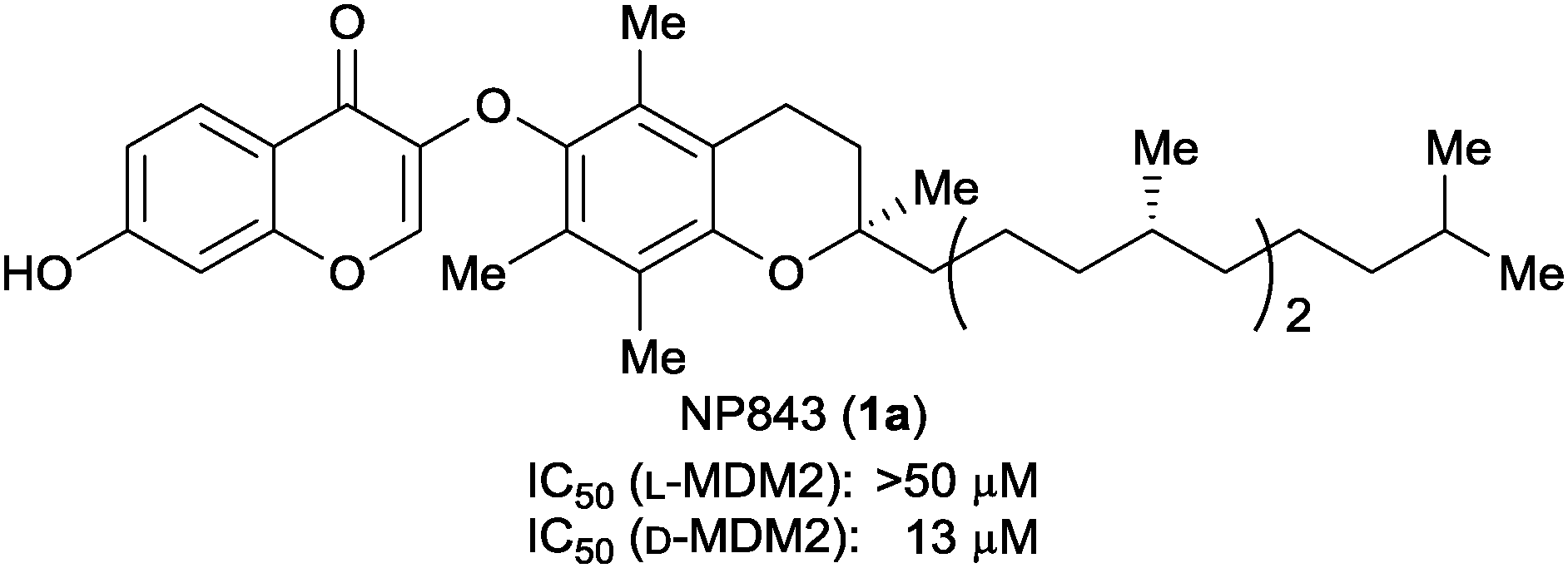 | ||
| Fig. 3 Structure and biological activity of NP843. IC50 values were measured by the FP assay using a 0.5 nM fluorescein-labeled p53-like peptide (P4 peptide)18 and 10 nM MDM2 protein. | ||
The next step was to verify the bioactivity of the enantiomeric form of 1a (ent-NP843, 1b) toward L-MDM2–L-p53 interaction. Compound 1b was prepared by chiral pool synthesis from three commercially available optically pure components (Scheme 1). Initially, (R)-Roche ester-derived tosylate 2 was reacted with Grignard reagent 3, prepared from (S)-citronellyl bromide, in the presence of a copper catalyst. The subsequent deprotection of the trityl-group gave alcohol 4, after treatment with 1 N NaOH for hydrolysis of the formyl ester derived from 4. C1 elongation of 4 was then performed via tosylation and cyanylation followed by reduction to give an alcohol 6, which was converted into phosphonium salt 8. (R)-Trolox-derived aldehyde 9 was employed as a chiral chromane component for the Wittig reaction. The subsequent two-step catalytic reduction including PtO2-mediated hydrogenation of olefins and subsequent Pd/C-mediated hydrogenolysis of the benzyl group provided the L-α-tocopherol (10b) in 84% yield. Alkylation of 10b with bromide 11 followed by deprotection gave ketone 12b. Construction of the left-part of the chromone substructure by treatment of 12b with N,N-dimethylformamide dimethyl acetal (DMF-DMA) afforded the mirror-image compound 1b.
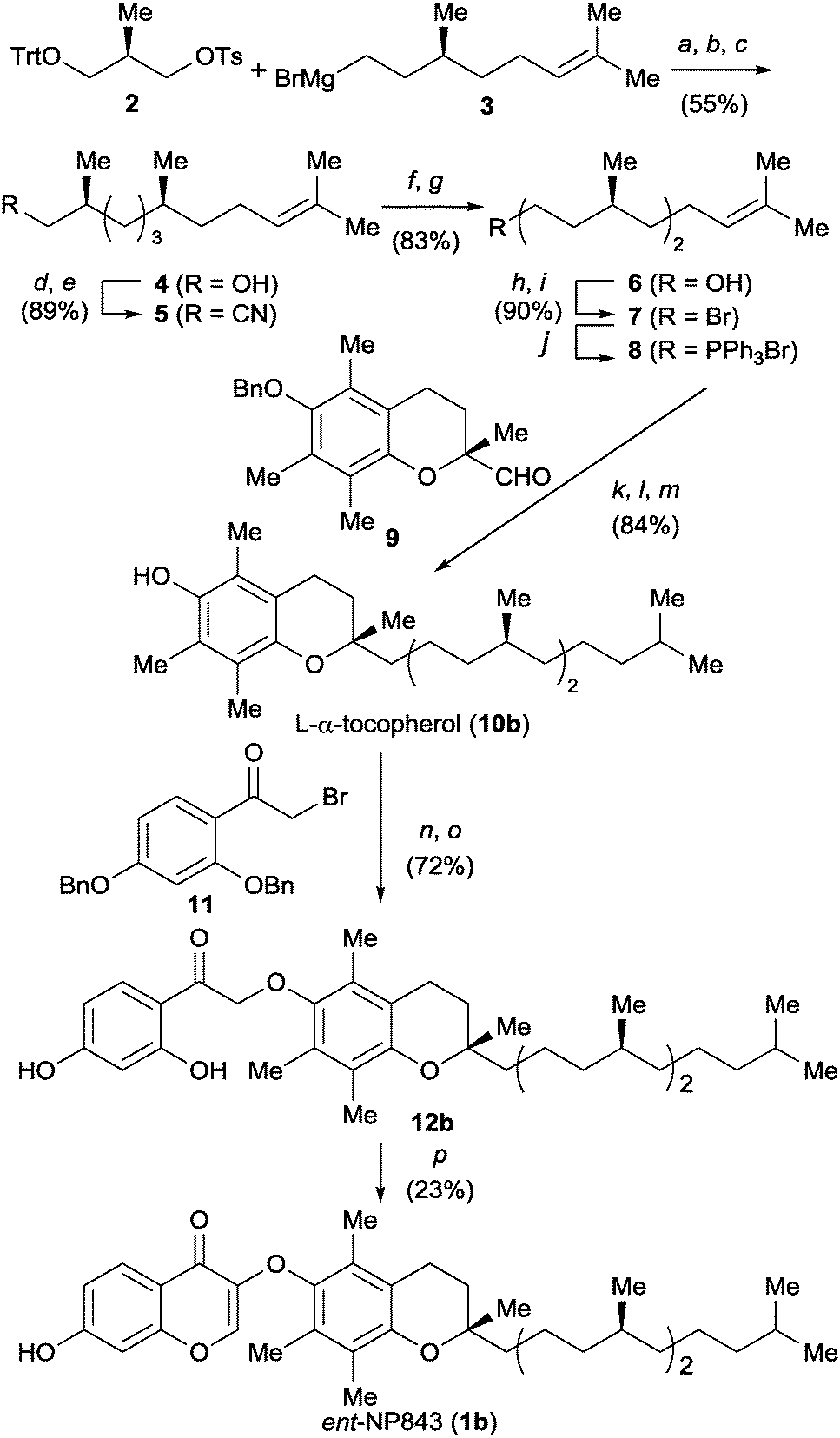 | ||
| Scheme 1 Synthetic scheme for ent-NP843. Reagents and conditions: (a) Li2CuCl4, THF, 0 °C; (b) HCO2H, Et2O, rt; (c) NaOH, MeOH/H2O, rt; (d) TsCl, pyridine, 0 °C; (e) NaCN, DMSO, rt; (f) DIBAL-H, THF/CH2Cl2, −78 °C, then H3O+; (g) DIBAL-H, THF/CH2Cl2, −78 °C; (h) TsCl, pyridine, 0 °C; (i) LiBr, acetone, reflux; (j) PPh3, neat, 100 °C; (k) LiHMDS, 9, THF, −40 °C to rt; (l) H2, PtO2, TBME, rt; (m) H2, Pd/C, MeOH, rt; (n) 11, K2CO3, DMF, rt; (o) H2, Pd/C, EtOAc, rt; (p) DMF-DMA, THF, reflux. | ||
The inhibitory activity of mirror-image compound 1b against the interaction between MDM2 and the p53 peptide was evaluated by an FP assay (Table 1 and Fig. S6, ESI†). Compound 1b inhibited L-MDM2–L-p53 interaction [IC50 (1b) = 7.6 ± 1.9 μM for L-MDM2–L-p53], while no inhibition was observed against D-MDM2–D-p53 interaction at 30 μM of 1b. This corresponded with the inhibitory activities of the original hit 1a against D-MDM2–D-p53 and L-MDM2–L-p53 interactions, respectively [IC50 (1a) = 6.5 ± 0.5 μM for D-MDM2–D-p53].19 These data identified the mirror-image α-tocopherol derivative 1b (ent-NP843) as a selective inhibitor of the L-MDM2–L-p53 interaction.20
Further structure–activity study of compound 1b and its derivatives revealed the structural requirements for inhibition of L-MDM2–L-p53 interaction (Table 1 and Fig. S6, ESI†). We designed two diastereomers of compound 1b: epimer 1c at the tetrasubstituted carbon on the chromane scaffold, and diastereomer 1d with two epimeric methyl groups on the right-hand alkyl chain (mirror-image of 1c) (Fig. 4). Compound 1d, having a tetrasubstituted carbon with (S)-configuration on the chromane skeleton (identical to that of compound 1b), reproduced the bioactivity of 1b against L-MDM2–L-p53 interaction [IC50 (1d) = 6.9 ± 1.9 μM for L-MDM2–L-p53], while compound 1c with (R)-configuration (identical to that of compound 1a) showed no inhibition. However, for D-MDM2–D-p53 inhibition, compound 1c was potent [IC50 (1c) = 7.8 ± 2.0 μM for D-MDM2–D-p53]. These results indicate that the stereochemistry of the tetrasubstituted carbon on the chromane skeleton is dominant in the chiral recognition by L-MDM2 protein when compared with the stereocenters on the extended alkyl chain. To demonstrate the significance of the aliphatic chain length of 1b, we also designed derivatives 13a,b and 14a,b with shorter isoprene units (Fig. 4). Derivatives 13a,b and 14a,b showed no inhibitory activity against L-MDM2–L-p53 interaction (nor against D-MDM2–D-p53 interaction). These results suggest that the chain length of three isoprene units is indispensable for the biological activity of compound 1b.
 | ||
| Fig. 4 Structures of NP843 derivatives. The methyl group in blue indicates the identical stereochemical configuration to that in ent-NP843 (1b). | ||
In this study, isolated natural products and derivatives, which were immobilized on microarray slides, were used as screening resources. This approach would be further applicable to the mirror-image screening of mixture samples from natural resources including crude plant extracts and fermentation broth extracts, which could provide an unprecedented opportunity to explore diastereomeric interactions between the mirror-image structures of chiral ingredients and native proteins. Once the hit extract(s) is identified, structural determination of the active ingredient(s) and synthesis of its enantiomer(s) would be needed. In this way, a novel hit compound(s) with “unnatural” chirality would be obtained with a high probability. It is our expectation that future analysis of screening data sets against a number of target molecules may give some insights into the value of mirror-image natural resources for drug discovery. In this strategy, two chemical syntheses of mirror-image substances (target protein and initial hit compound), and chemical array technology using synthetic D-protein facilitated access to a variety of virtual mirror-image natural products. For further application to other molecular targets, rapid and efficient preparation of mirror-image biomolecules and natural product hit(s) is essential because these cannot be generated via protein expressions or conventional cultivation. Recent progress in chemical protein synthesis has increased the availability of synthetic proteins, even with highly functional modification(s).21 Although this study demonstrates mirror-image screening for protein–protein interaction inhibitors, the targets could be extended to enzyme inhibitors and receptor ligands once the counterpart mirror-image biomolecule(s) are available.12 A number of recent advances in efficient synthetic strategies to access unique and complex frameworks in natural products should also contribute to the concise preparation of mirror-image structure(s) of natural product hit compounds. Thus, investigation of the bioactivities and functions of virtual mirror-image compounds is achievable only by using advanced synthetic organic chemistry technologies.
In summary, we have established a novel screening process for a virtual mirror-image library of chiral natural products and derivatives. Chemically synthesized mirror-image MDM2 was applied for chemical array screening of a library of natural product derivatives, identifying novel tocopherol derivative NP843 (1a) as a D-MDM2–D-p53 interaction inhibitor. Chemical synthesis of the enantiomeric compound (1b) enabled the validation of the inhibitory activity of 1b against L-MDM2–L-p53 interaction. The selective recognition by MDM2 was attributed to the stereochemistry of the tetrasubstituted carbon on the chromane skeleton of 1b. The aliphatic side chain of three isoprene units was also needed for inhibition. In contrast to conventional screenings, this process could identify hit compounds from unavailable mirror-image chiral natural products, thus providing unprecedented lead compounds for drug discovery. To our knowledge, this is the first application of mirror-image protein technology in the screening of chiral small molecules including natural products.
This work was supported by Grants-in-Aid for Scientific Research from JSPS, Japan; the Targeted Protein Research Program; and the Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT, Japan. T. N. is grateful for Research Fellowships for Young Scientists from JSPS, Japan.
Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed.
- M. S. Bulter, A. A. B. Robertson and M. A. Cooper, Nat. Prod. Rep., 2014, 31, 1612 RSC.
- J. M. Finefield, D. H. Sherman, M. Kreitman and R. M. Williams, Angew. Chem., Int. Ed., 2012, 51, 4802 CrossRef CAS PubMed.
- (a) G. Haniotakis, W. Francke, K. Mori, H. Redlich and V. Schurig, J. Chem. Ecol., 1986, 12, 1559 CrossRef CAS PubMed; (b) H. Wolosker, E. Dumin, L. Balan and V. N. Foltyn, FEBS J., 2008, 275, 3514 CrossRef CAS PubMed.
- As an alternative approach to compound collection with natural-product-like scaffolds, there have been a number of reports on natural product-inspired synthesis. For reviews, see: (a) K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2009, 48, 3224 CrossRef CAS PubMed; (b) S. Rizzo and H. Waldmann, Chem. Rev., 2014, 114, 4621 CrossRef CAS PubMed.
- R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195 CrossRef PubMed.
- H. Kikuchi, K. Sakurai and Y. Oshima, Org. Lett., 2014, 16, 1916 CrossRef CAS PubMed.
- (a) J. A. Takahashi, A. P. C. Teles, A. de A. P. Bracarense and D. C. Gomes, Phytochem. Rev., 2013, 12, 773 CrossRef CAS; (b) A. Marmann, A. H. Aly, W. Lin, B. Wang and P. Proksch, Mar. Drugs, 2014, 12, 1043 CrossRef PubMed.
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Org. Biomol. Chem., 2008, 6, 1895 CAS.
- (a) B. L. Pentelute, Z. P. Gates, V. Tereshko, J. L. Dashnau, J. M. Vanderkooi, A. A. Kossiakoff and S. B. H. Kent, J. Am. Chem. Soc., 2008, 130, 9695 CrossRef CAS PubMed; (b) B. L. Pentelute, Z. P. Gates, J. L. Dashnau, J. M. Vanderkooi and S. B. H. Kent, J. Am. Chem. Soc., 2008, 130, 9702 CrossRef CAS PubMed; (c) M. T. Weinstock, M. T. Jacobsen and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11679 CrossRef CAS PubMed.
- (a) T. N. M. Schumacher, L. M. Mayr, D. L. Minor Jr, M. A. Milhollen, M. W. Burgess and P. S. Kim, Science, 1996, 271, 1854 CAS; (b) D. M. Eckert, V. N. Malashkevich, L. H. Hong, P. A. Carr and P. S. Kim, Cell, 1999, 99, 103 CrossRef CAS PubMed; (c) K. Wiesehan, K. Buder, R. P. Linke, S. Patt, M. Stoldt, E. Unger, B. Schmitt, E. Bucci and D. Willbold, ChemBioChem, 2003, 4, 748 CrossRef CAS PubMed; (d) B. D. Welch, A. P. VanDemark, A. Heroux, C. P. Hill and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16828 CrossRef CAS PubMed; (e) T. van Groen, K. Wiesehan, S. A. Funke, I. Kadish, L. Nagel-Steger and D. Willbold, ChemMedChem, 2008, 3, 1848 CrossRef CAS PubMed; (f) M. Liu, M. Pazgier, C. Li, W. Yuan, C. Li and W. Lu, Angew. Chem., Int. Ed., 2010, 49, 3649 CrossRef CAS PubMed; (g) M. Liu, C. Li, M. Pazgier, C. Li, Y. Mao, Y. Lv, B. Gu, G. Wei, W. Yuan, C. Zhan, W. Y. Lu and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14321 CrossRef CAS PubMed.
- A similar concept was introduced in the previous patent publication, see: S. B. H. Kent, S. C. F. Milton and R. C. deL. Milton, WO 9325667, 1993.
- F. Toledo and G. M. Wahl, Nat. Rev. Cancer, 2006, 6, 909 CrossRef CAS PubMed.
- S. Shangary and S. Wang, Annu. Rev. Pharmacol. Toxicol., 2009, 49, 223 CrossRef CAS PubMed.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844 CrossRef CAS PubMed.
- T. Noguchi, S. Oishi, K. Honda, Y. Kondoh, T. Saito, T. Kubo, M. Kaneda, H. Ohno, H. Osada and N. Fujii, Bioorg. Med. Chem. Lett., 2013, 23, 3802 CrossRef CAS PubMed.
- N. Kanoh, S. Kumashiro, S. Simizu, Y. Kondoh, S. Hatakeyama, H. Tashiro and H. Osada, Angew. Chem., Int. Ed., 2003, 42, 5584 CrossRef CAS PubMed.
- A. Czarna, G. M. Popowicz, A. Pecak, S. Wolf, G. Dubin and T. A. Holak, Cell Cycle, 2009, 8, 1176 CrossRef CAS PubMed.
- The similar inhibitory activities of 1b were observed by SPR analysis and ELISA using synthetic and recombinant L-MDM2 proteins, respectively (Fig. S6, ESI†).
- Unfortunately, no antiproliferative activities of ent-NP843 (1b) against SJSA-1 (MDM2 overexpression) and H1299 (no p53 expression) cells were observed at 30 μM (Fig. S7, ESI†).
- (a) P. Wang, S. Dong, J. H. Shieh, E. Peguero, R. Hendrickson, M. A. S. Moore and S. J. Danishefsky, Science, 2013, 342, 1357 CrossRef CAS PubMed; (b) M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc03114e |
| This journal is © The Royal Society of Chemistry 2016 |