
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired pH and magnetic responsive catechol-functionalized chitosan hydrogels with tunable elastic properties†
Ali
Ghadban
ab,
Anansa S.
Ahmed
a,
Yuan
Ping
ab,
Ricardo
Ramos
ab,
Najmul
Arfin
ab,
Bram
Cantaert
ab,
Raju V.
Ramanujan
a and
Ali
Miserez
*abc
aSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: ali.miserez@ntu.edu.sg; Fax: +65 6790-9081; Tel: +65 6592-7947
bCentre for Biomimetic Sensor Science, Nanyang Technological University, 50 Nanyang Drive, Singapore 637553, Singapore
cSchool of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore
First published on 30th October 2015
Abstract
We have developed pH- and magnetic-responsive hydrogels that are stabilized by both covalent bonding and catechol/Fe3+ ligands. The viscoelastic properties of the gels are regulated by the complexation valence and can be used to tune drug release profiles. The stable incorporation of magnetic nanoparticles further expands control over the mechanical response and drug release, in addition to providing magnetic stimuli-responsivity to the gels.
The field of hydrogels has rapidly expanded in recent years,1,2 with bioinspired and biopolymer-based hydrogels representing an increasingly large fraction of these efforts. A growing theme is the development of hydrogels that are multi-functional, stimuli-responsive, and with tailorable physico-chemical and mechanical properties.3,4 Towards obtaining gels with tunable properties, metal coordination bonding has recently emerged as a versatile strategy, notably by exploiting catechol moiety/transition metal complexation.5,6 Such ligands are particularly useful to stabilize and modulate gels viscoelastic properties owing to: (i) their pH dependence that triggers either mono-, bi- or tri-dentate complexes, and (ii) the ability to further tune their properties by simple modification of the transition metal ions used to coordinate the catecholic moieties.7 On the other hand, the conformational response of many hydrogels, which would be useful in many applications, remains slow. This limitation can be overcome by preparing magnetic hydrogels, composed of a discrete magnetic phase such as magnetic nanoparticles (MNPs) embedded within a polymeric matrix.8 Magnetically-controlled hydrogels provide unique functionalities in biomedical applications such as tissue engineering and drug delivery. For instance, they can be actuated in a controlled manner to prepare 3D tissue scaffolds during cell proliferation.9,10 Alternatively, the behavior of encapsulated cells can also be controlled in vitro by a remotely-activated mechanical stimulus, which is triggered by interactions between the MNPs and an external magnetic field.11 Using alternating magnetic fields, MNPs can also be remotely heated to induce: (i) conformational changes to the hydrogels, which can be exploited for the pulsatile release of drugs,12 or (ii) hydrophobic-to-hydrophilic phase transitions that can promote large dimensional changes.13 Finally, MNPs provide contrast in magnetic resonance imaging studies, enabling therapy and diagnostic (theranostic) related protocols.13,14 Thus, the introduction of magnetic responsiveness to the hydrogel increases its versatility to develop a truly multifunctional, multi-stimuli responsive material.7
Chitosan, the deacetylated version of the polysaccharide chitin, represents a convenient source of biodegradable biopolymers for a wide range of soft materials, including hydrogels.15 It is highly abundant, notably in the waste from the seafood industry; it is soluble in weak acidic solutions;16 and it can readily be functionalized to expand its range of functional properties for biomedical applications such as gene delivery and tissue repair.17,18 The merging of the two approaches, namely catechol functionalization of chitosan, is quickly emerging as a method of choice to combine the specific advantages of catechol complexation and the ease of processing of chitosan, as exemplified in recent studies.19–22 Herein, we explore the potential of catechol-functionalized chitosan to engineer pH-sensitive and magnetic-responsive hydrogels by taking advantage of metal–catechol coordination. Hydrocaffeic acid (HCA) bearing the catechol moiety was used to functionalize chitosan, allowing the modulation of the viscoelastic response of HCA–chitosan gels via pH triggering and variation of the network mesh size ξ, which in turn allowed the tuning of drug release profiles. In order to further expand the multi-functionality of these gels, we then incorporated iron oxide (γ-Fe2O3) MNPs in their formulation and established that this strategy adds an additional level of multi-functionality. First, it allows further modulation of the mechanical response; second it provides an additional control over drug release kinetics; and third, it confers magnetic responsivity on the gels, opening the door to their usage as stimuli-responsive, biocompatible hydrogels with tunability of drug release kinetics.
First, we functionalized high molecular weight chitosan (Mw ≅ 94 KDa) with the HCA catecholic moiety, with a degree of substitution of ∼15% as measured by 1H NMR (Fig. S1, ESI†). The preparation of hydrogels in the absence of MNPs was then investigated. Upon addition of Fe3+ (HCA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe3+ = 3:1), increasing the pH of the HCA–chitosan solution to 6 and 12 led to the formation of bis- and tris-complexes, respectively. As a control, we also prepared covalently cross-linked hydrogels (hereafter referred to as “chemical hydrogels”) by increasing the pH of HCA–chitosan to 13 (Scheme 1). Fig. 1 shows the UV-Vis spectra of the chemical and physical (stabilized by coordination complexation) hydrogels prepared with and without Fe3+ (HCA:Fe3+ = 3:1) at different pHs. The presence of an absorbance peak at 280 nm for HCA–chitosan confirms the successful grafting of HCA to the chitosan backbone. Increasing the pH from acidic to basic, the color of HCA–chitosan solution changed from transparent to a yellowish/orange color with a simultaneous gel formation, an observation already reported in earlier studies.18 Additional peaks at ∼260 and ∼350 nm appeared, which can be attributed to covalent crosslinking resulting from either catechol–catechol or amino–quinone bond formation, respectively.18,22 Conducting the gelation in the presence of Fe3+ at pH 6 and 12, blue/purple and red-colored hydrogels were observed, with absorbance peaks at 550 and 495 nm associated with the formation of bis- and tris-complexes, respectively. The latter result is in agreement with recent reports on catechol-functionalized PEG and chitosan.5,22 The presence of chemical crosslinks is inevitable under such conditions due to the high molecular weight of chitosan (resulting in abundant free amino groups), the high degree of substitution (DS ∼ 15%), and the redox effect of Fe3+. This is also reflected by the appearance of peaks at ∼260 and 350 nm. It is worth mentioning that the mono-complex associated with a green color formation was observed when the pH of the HCA–chitosan solution was increased from ∼2 to 3–4 in the presence of Fe3+ (data not shown). The visco-elastic properties of the synthesized hydrogels were characterized two hours after gelation by rheology in the dynamic mode at 20 °C (Fig. 2). In the tested frequency range, HCA–chitosan (without Fe3+) at pH 13 showed the characteristics of a soft hydrogel, with a constant storage modulus G′ of ca. 500 Pa. The damping factor (tanδ = G′′/G′) was approximately 1 below 0.1 Hz, implying network properties at the boundary between fluid- and solid-like gel. However, above 0.1 Hz the gel was solid-like, with G′/G′′ > 1. The addition of Fe3+ greatly increased the storage modulus of the gels, which increased from 6 to ca. 20 KPa (at 1 Hz) as the pH of the HCA–chitosan solution was increased from 6 to 12. At pH 3 in the presence of Fe3+, G′ values were considerably lower (<1 Pa, Fig. S3, ESI†). This increase in stiffness can unambiguously be attributed to catechol/Fe3+ complexation since no other bonds are introduced into the network. As previously reported,23,24 increasing the pH modifies the nature of the coordination complexes between the catechol side-chains and Fe3+, with mono-complexes present below pH 3, bis-complexes at pH 6 and tris-complexes being the dominant species at pH 12. Using the classical relationship between storage modulus and mesh size (G′ ∼ kBT/ξ3), we determined that ξ decreased from ca. 22 nm at pH 13 in the absence of Fe3+ down to 10 nm (pH 6, Fe3+) and 5 nm (pH 12, Fe3+), suggesting a direct correlation between the complexation state and the mesh size (Fig. 2B).
:Fe3+ = 3:1), increasing the pH of the HCA–chitosan solution to 6 and 12 led to the formation of bis- and tris-complexes, respectively. As a control, we also prepared covalently cross-linked hydrogels (hereafter referred to as “chemical hydrogels”) by increasing the pH of HCA–chitosan to 13 (Scheme 1). Fig. 1 shows the UV-Vis spectra of the chemical and physical (stabilized by coordination complexation) hydrogels prepared with and without Fe3+ (HCA:Fe3+ = 3:1) at different pHs. The presence of an absorbance peak at 280 nm for HCA–chitosan confirms the successful grafting of HCA to the chitosan backbone. Increasing the pH from acidic to basic, the color of HCA–chitosan solution changed from transparent to a yellowish/orange color with a simultaneous gel formation, an observation already reported in earlier studies.18 Additional peaks at ∼260 and ∼350 nm appeared, which can be attributed to covalent crosslinking resulting from either catechol–catechol or amino–quinone bond formation, respectively.18,22 Conducting the gelation in the presence of Fe3+ at pH 6 and 12, blue/purple and red-colored hydrogels were observed, with absorbance peaks at 550 and 495 nm associated with the formation of bis- and tris-complexes, respectively. The latter result is in agreement with recent reports on catechol-functionalized PEG and chitosan.5,22 The presence of chemical crosslinks is inevitable under such conditions due to the high molecular weight of chitosan (resulting in abundant free amino groups), the high degree of substitution (DS ∼ 15%), and the redox effect of Fe3+. This is also reflected by the appearance of peaks at ∼260 and 350 nm. It is worth mentioning that the mono-complex associated with a green color formation was observed when the pH of the HCA–chitosan solution was increased from ∼2 to 3–4 in the presence of Fe3+ (data not shown). The visco-elastic properties of the synthesized hydrogels were characterized two hours after gelation by rheology in the dynamic mode at 20 °C (Fig. 2). In the tested frequency range, HCA–chitosan (without Fe3+) at pH 13 showed the characteristics of a soft hydrogel, with a constant storage modulus G′ of ca. 500 Pa. The damping factor (tanδ = G′′/G′) was approximately 1 below 0.1 Hz, implying network properties at the boundary between fluid- and solid-like gel. However, above 0.1 Hz the gel was solid-like, with G′/G′′ > 1. The addition of Fe3+ greatly increased the storage modulus of the gels, which increased from 6 to ca. 20 KPa (at 1 Hz) as the pH of the HCA–chitosan solution was increased from 6 to 12. At pH 3 in the presence of Fe3+, G′ values were considerably lower (<1 Pa, Fig. S3, ESI†). This increase in stiffness can unambiguously be attributed to catechol/Fe3+ complexation since no other bonds are introduced into the network. As previously reported,23,24 increasing the pH modifies the nature of the coordination complexes between the catechol side-chains and Fe3+, with mono-complexes present below pH 3, bis-complexes at pH 6 and tris-complexes being the dominant species at pH 12. Using the classical relationship between storage modulus and mesh size (G′ ∼ kBT/ξ3), we determined that ξ decreased from ca. 22 nm at pH 13 in the absence of Fe3+ down to 10 nm (pH 6, Fe3+) and 5 nm (pH 12, Fe3+), suggesting a direct correlation between the complexation state and the mesh size (Fig. 2B).
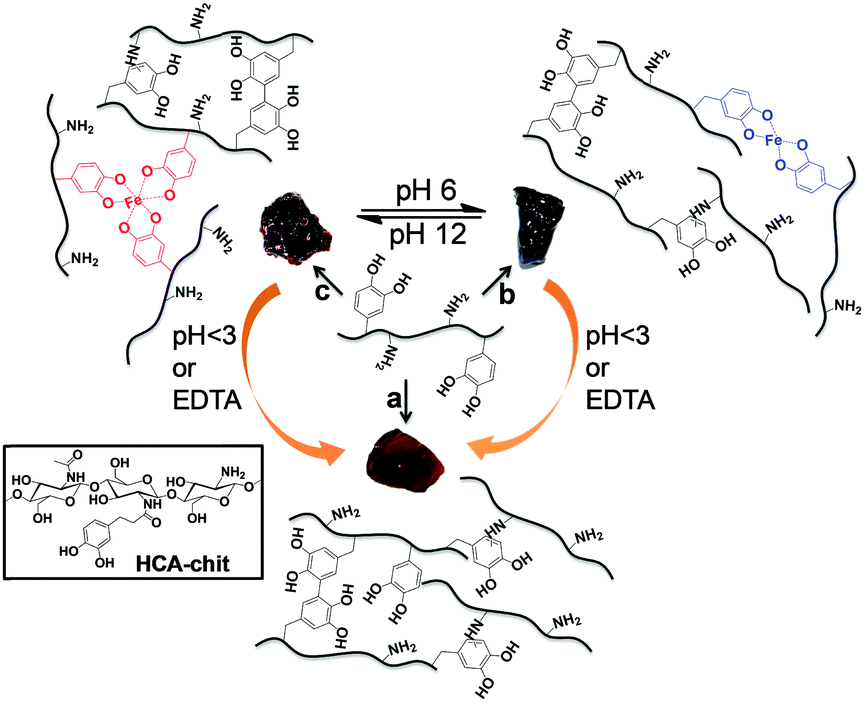 | ||
| Scheme 1 Schematic of pH-tunable HCA–chitosan hydrogels stabilized by covalent bonding (“chemical gels”) and catechol/Fe3+ complexation (“physical gels”). Gelation conditions used: (a) NaOH pH 13; (b) FeCl3 (Fe3+:HCA = 1:3), pH 6; (c) FeCl3 (Fe3+:HCA = 1:3), pH 12. Note the yellow-orange color of the covalently cross-linked hydrogel, and the blue-purple and red colors of the metal coordinated cross-linked hydrogels at pH 6 and 12, respectively. | ||
 | ||
| Fig. 1 UV-Vis monitoring of hydrogel formation for HCA–chitosan at different pH in the absence (A) and presence (B) of Fe3+ (HCA:Fe3+ = 3:1). | ||
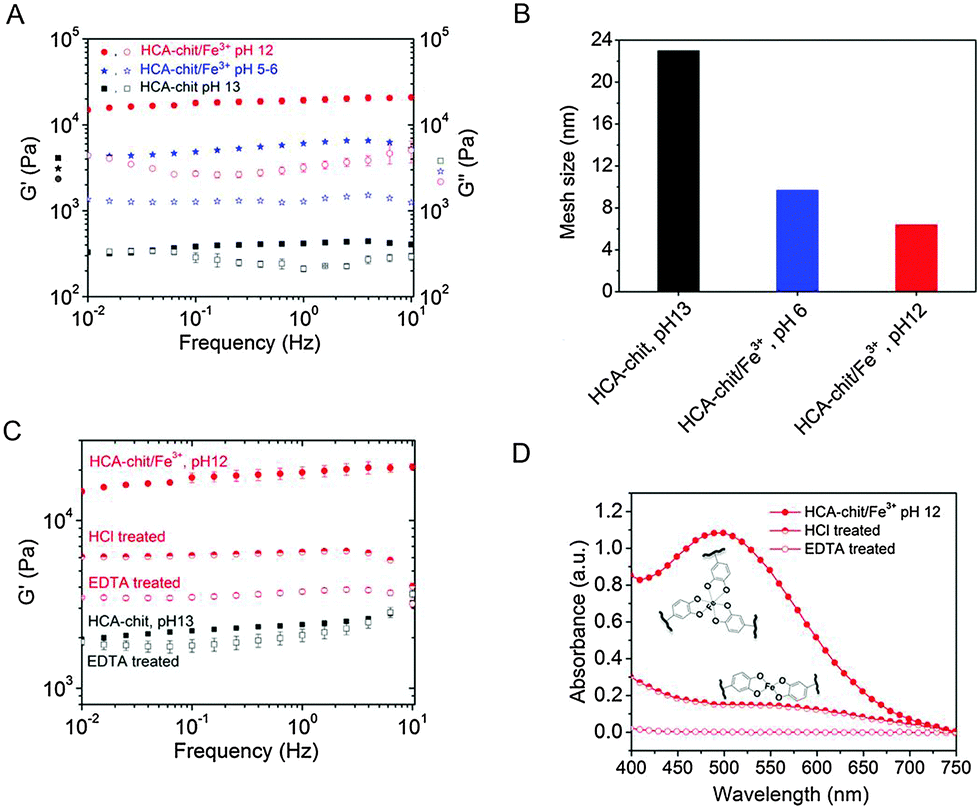 | ||
| Fig. 2 Rheological properties of synthesized hydrogels (A) in the absence and presence of Fe3+ at various pHs. (B) Estimated mesh size ξ for the different gels. (C) Rheological properties and (D) UV-Vis spectra of physicals gels after treatment with HCl or EDTA. | ||
To assess the reversibility of the gelation process, the tris-complex hydrogel was treated with either HCl or EDTA overnight. The final pH of HCl treated hydrogel solution was found to be 5.5. As a control, the chemical hydrogel was also treated with EDTA. The EDTA-treated chemical hydrogels exhibited a storage modulus comparable to that of the initial hydrogel, as expected, given the absence of metallic cations in the initial gel (Fig. 2C). On the other hand, the tris-complex treated gel featured a drop in stiffness, with G′ decreasing from ∼20 KPa to ∼6.5 and ∼3.7 KPa (at 1 Hz) after HCl and EDTA treatments, respectively. The UV-Vis analysis supports this finding: a peak at 570 nm appeared after HCl treatment corresponding to the bis-complex at this lower pH. Treating the gel with EDTA showed no evidence for any absorbance peak above 450 nm, confirming the removal of all Fe3+ from the gel (Fig. 2D). The higher storage modulus of this hydrogel compared to that of the chemical hydrogel may be attributed to the formation of chemical crosslinks concomitantly taking place during metal chelation, leading to a gel with a higher covalent cross-link density. Collectively, these results indicate that the viscoelastic response of HCA–chitosan gels can be reversibly modulated by chelation or re-incorporation of metal ions.
We previously reported HCA–chitosan films reinforced with MNPs, in which the mechanical properties of the films were enhanced through load transfer from the functionalized-chitosan to MNPs via coordination interactions between catechol and Fe2O3.20 To confer dual pH and magnetic responsiveness, MNP particles were added to the hydrogels. To this end, HCA–chit were mixed with Fe2O3 MNPs (2.5 and 5 wt%) and gelled by increasing the pH in the absence or presence of Fe3+ (HCA:Fe3+ = 3:1), to yield both chemical and physical hybrid hydrogels. The chemical hydrogels featured a light-brown visual appearance consistent with the color of maghemite Fe2O3, whereas the physically cross-linked hydrogels maintained a dark red color, suggesting a faster kinetics of complexation for Fe3+/catechol than for Fe2O3/catechol. The addition of 2.5 and 5 wt% of MNPs to the physical gels resulted in a linear increase in the storage modulus with MNP content of up to 26 kPa at 1 Hz (Fig. 3A and Fig. S4A, ESI†). However, we note that the average mesh size ξ estimated from G′ ∼ kBT/ξ3 decreased only moderately upon addition of MNPs (Fig. 3B), suggesting that catechols favour interactions with Fe3+ and that MNPs primarily act as fillers in the physical gels but do not specifically interact with MNPs. In contrast, the relative increase of G′ with MNP content for chemical gels was more distinct – though lower in absolute values than for the physical gels – and is reflected by the more pronounced relative decrease in mesh size upon addition of MNPs (Fig. 3B). In the absence of Fe3+, HCA moieties in the chemical gels are free to coordinate the surface of Fe2O3, thus allowing load transfer from the soft chitosan–HCA matrix to the stiff MNPs.20 The increase in G′ may also be assessed in terms of decreased swelling upon addition of MNPs. The storage modulus of swollen networks is well-established to exhibit an inverse power law dependence on the swelling ratio Q (G′ ∼ Q−m),25 where m is in the range of 2.2 to 3 depending on the solvent. To verify this dependency, we plotted G′ vs. Q (Fig. S4B, ESI†) and obtained G′ ∼ Q−2.9 for chemical/MNPs gels, which is in good agreement with the power law predicted for theta solvents and similar to that measured for various PEG hybrid hydrogels.26,27 In contrast, in physical/MNPs hydrogels a weaker dependence of G′ on swelling was observed with G ∼ Q−1.2, which is consistent with a network where the catecholic moieties are saturated with Fe3+ coordination bonds and thus less available for water uptake. In summary, these data reveal a distinct dependency of G′ on the MNP content, which can be attributed to substantial differences in the swelling behaviour in the presence or absence of catechol/Fe3+ coordination bonds.
 | ||
| Fig. 3 Rheological, magnetic, and drug-release properties of MNPs-loaded hydrogels. (A) Frequency sweep experiments of magnetic chemical and physical hydrogels. (B) Estimated mesh size ξ for the different gels. (C) VSM of chemical and physical magnetic hydrogels. (D) Time-dependent release of dextran-FITC (MW: 20000) from different hydrogel at pH 12. The data represent the mean ± SD (n = 3). | ||
Vibrating Sample Magnetometer (VSM) measurements were conducted in order to confirm the magnetism of both chemical and physical hydrogels. Fig. 3C illustrates the magnetization versus magnetic field (M–H loop) for the chemical and physical hydrogels at various weight fractions of MNPs. All hydrogels displayed a low hysteresis behavior similar to native Fe2O3 (Fig. S2D, ESI†). The magnetic property values of the hydrogels are fully consistent with the weight fraction of MNPs in the gels. No change was observed in the magnetic properties even after maintaining the gels in water for 10 days (data not shown). This indicates that the MNPs remained entrapped within the gel without further oxidation. For the same weight fraction of MNPs, physical gels exhibited slightly higher values (∼0.18 vs. 0.16 and 0.09 vs. 0.07 emu g−1 at 5 and 2.5 wt%, respectively), which can be attributed to the smaller swelling ratio of physical gels compared to their chemical analogues (Fig. S5, ESI†). A video illustrating the magnetically-induced shape morphing of the magnetic hydrogels is provided in the ESI.†
In addition to tuning the mechanical properties, MNP hydrogels can also generate additional stress and/or strain in DC magnetic fields, while an AC field could generate a temperature change, enabling thermal triggering. The quick magnetic responsivity could thus be used as implantable magnetic gels containing drugs or cells, which could be delivered on-demand by external stimuli.28 To specifically test whether our gels could entrap and release drugs in a controlled manner, both low- and high-molecular weight drugs were incorporated in the gel prior to gelation, and we reasoned that changes in the coordination state would allow the tuning of the release profile. Both physical and chemical gels were able to efficiently entrap rhodamine B, a low-molecular-weight dye that is often used as a probe molecule for drug release studies, after gel formation (Fig. S6, ESI†). We further used fluorescein isothiocyanate (FITC)-dextran as a macromolecular model drug to evaluate the release kinetics of the drug-loaded gels. As shown in Fig. 3D, whereas the chemical gel released FITC-dextran rapidly, the physical gel exhibited moderate release rate under the same conditions. The difference in release kinetics between both gels is attributed to the mesh size differences (Fig. 2B), which is believed to play an important role in gel diffusivity.29 Specifically, physical gels form a denser network reinforced by tris-catechol/Fe3+ complexes as compared to the chemical gel, which is stabilized by a lower cross-link density of covalent bonds. With the incorporation of MNPs into the physical gels, marginal FITC-dextran release (<15%) was observed within 120 h, indicating that the release profile could be further modulated. Since swelling is known to directly influence drug release kinetics,30 we attribute the slower release of FITC-dextran in MNP-loaded physical gels to their reduced swelling degree. We also noticed that the release kinetics of the physical gels (with or without MNP incorporation) is pH-dependent. At pH 6, both physical gels exhibited a faster release rate than that at pH 12 (Fig. S7, ESI†). Collectively, these results indicate that the release of cargos from physical hydrogels can be manipulated by pH and Fe2O3 nanoparticle incorporation via variation of mesh size and swelling degree. Expanding this multi-functionality with a modulated mechanical response triggered by pH-dependence metal/catechol complexation open further opportunities, for instance to adjust the visco-elastic properties of the magnetic hydrogels to specific micro-environments or tissues. Magnetic hydrogels also constitute an interesting approach to design tissue scaffolds for bone and muscle tissue engineering, where the application of stress is required for cell differentiation.31 Finally, magnetic fields could be exploited to tailor the mechanical properties of the scaffold to suit optimal growth conditions, while the pH responsiveness could be used to trigger the release of growth factors at the appropriate times and/or spatial locations. Although the cytotoxicity of these stimuli-responsive gels remains to be assessed, recent studies have indicated a low cytotoxicity of metal coordination reinforced gels using vanadium ions as ligand centers,32 suggesting a promising alternative to Fe3+ cross-linked gels should the latter prove cytotoxic.
This research was funded by the Singapore National Research Foundation (NRF) through a NRF Fellowship awarded to A.M.
Notes and references
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed
.
- C. J. Kearney and D. J. Mooney, Nat. Mater., 2013, 12, 1004–1017 CrossRef CAS PubMed
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed
- M. S. Menyo, C. J. Hawker and J. H. Waite, Soft Matter, 2013, 9, 10314–10323 RSC
- N. Holten Andersen, A. Jaishankar, M. Harrington, D. Fullenkamp, G. DiMarco, L. He, G. McKinley, P. Messersmith and K. Y. C. Lee, J. Mater. Chem. B, 2014, 2, 2467–2472 RSC
- G. Bayramoglu, B. Altintas and M. Y. Arica, Appl. Microbiol. Biotechnol., 2013, 97, 1149–1159 CrossRef CAS PubMed
- B. S. Kim, J. Nikolovski, J. Bonadio and D. J. Mooney, Nat. Biotechnol., 1999, 17, 979–983 CrossRef CAS PubMed
- J. R. Mauney, S. Sjostorm, J. Blumberg, R. Horan, J. P. O'Leary, G. Vunjak-Novakovic, V. Volloch and D. L. Kaplan, Calcif. Tissue Int., 2004, 74, 458 CrossRef CAS PubMed
- H. Fujita, K. Shimizu, Y. Yamamoto, A. Ito, M. Kamihira and E. Nagamori, J. Tissue Eng. Regener. Med., 2010, 4, 437–443 CAS
- T. Hoare, J. Santamaria, G. F. Goya, S. Irusta, D. Lin, S. Lau, R. Padera, R. Langer and D. S. Kohane, Nano Lett., 2009, 9, 3651–3657 CrossRef CAS PubMed
- S. Purushotham and R. V. Ramanujan, Acta Mater., 2010, 6, 502–510 CAS
- S. Purushotham, P. E. J. Chang, H. Rumpel, I. H. C. Kee, R. T. H. Ng, P. K. H. Chow, C. K. Tan and R. V. Ramanujan, Nanotechnology, 2009, 20, 305101 CrossRef CAS PubMed
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS
- M. Rinaudo, G. Pavlov and J. Desbrieres, Polymer, 1999, 40, 7029–7032 CrossRef CAS
- Y. Ping, C. D. Liu, G. P. Tang, J. S. Li, J. Li, W. T. Yang and F. J. Xu, Adv. Funct. Mater., 2010, 20, 3106–3116 CrossRef CAS
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS PubMed
- K. Kim, J. H. Ryu, D. Y. Lee and H. Lee, Biomater. Sci., 2013, 1, 783–790 RSC
- O. Zvarec, S. Purushotham, A. Masic, R. V. Ramanujan and A. Miserez, Langmuir, 2013, 29, 10899–10906 CrossRef CAS PubMed
- J. H. Ryu, S. Jo, M.-Y. Koh and H. Lee, Adv. Funct. Mater., 2014, 24, 7709–7716 CrossRef CAS
- P. S. Yavvari and A. Srivastava, J. Mater. Chem. B, 2015, 3, 899–910 RSC
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed
- B. P. Lee and S. Konst, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed
- K. Urayama, T. Kawamura and S. Kohjiya, J. Chem. Phys., 1996, 105, 4833–4840 CrossRef CAS
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed
- A. T. Metters, C. N. Bowman and K. S. Anseth, AIChE J., 2006, 47, 1432–1437 CrossRef
- X. Zhao, J. Kim, C. A. Cezar, N. Huebsch, K. Lee, K. Bouhadir and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2010, 108, 67–72 CrossRef PubMed
- S. Lee, X. Tong and F. Yang, Acta Biomater., 2014, 10, 4167 CrossRef CAS PubMed
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 CrossRef CAS PubMed
- Y. Li, G. Huang, X. Zhang, B. Li, Y. Chen, T. Lu, T. J. Lu and F. Xu, Adv. Funct. Mater., 2013, 23, 660–672 CrossRef CAS
- J. P. Park, I. T. Song, J. Lee, J. H. Ryu, Y. Lee and H. Lee, Chem. Mater., 2015, 27, 105–111 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials, methods, analytical techniques, synthesis of hydrogels, 1H NMR of HCA–chit, TEM, DLS, VSM, XRD of MNPs, supplementary rheology data, swelling ratio of hydrogels, videos on hydrogels and drug release study. See DOI: 10.1039/c5cc08617e |
| This journal is © The Royal Society of Chemistry 2016 |