
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining cycloisomerization with trienamine catalysis: a regiochemically flexible enantio- and diastereoselective synthesis of hexahydroindoles†
V.
Chintalapudi
,
E. A.
Galvin
,
R. L.
Greenaway
and
E. A.
Anderson
*
Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: edward.anderson@chem.ox.ac.uk; Fax: +44 1865 285002; Tel: +44 1865 285000
First published on 11th November 2015
Abstract
The synthesis of polysubstituted hexahydroindoles through trienamine-organocatalyzed cycloadditions of pyrrolidinyl dienals, prepared by palladium-catalyzed cycloisomerization, is reported. The cycloadditions of this novel class of dienals proceed with excellent levels of enantio- and diastereoselectivity, with the regioselectivity of cycloaddition with respect to the tethering ring readily tuned through design of the cycloisomerization substrate. This work culminates in the first examples of double-stereodifferentiating trienamine catalysis, where catalyst stereocontrol dominates facial selectivity in the cycloaddition, affording azacyclic products that are specifically functionalized at every position.
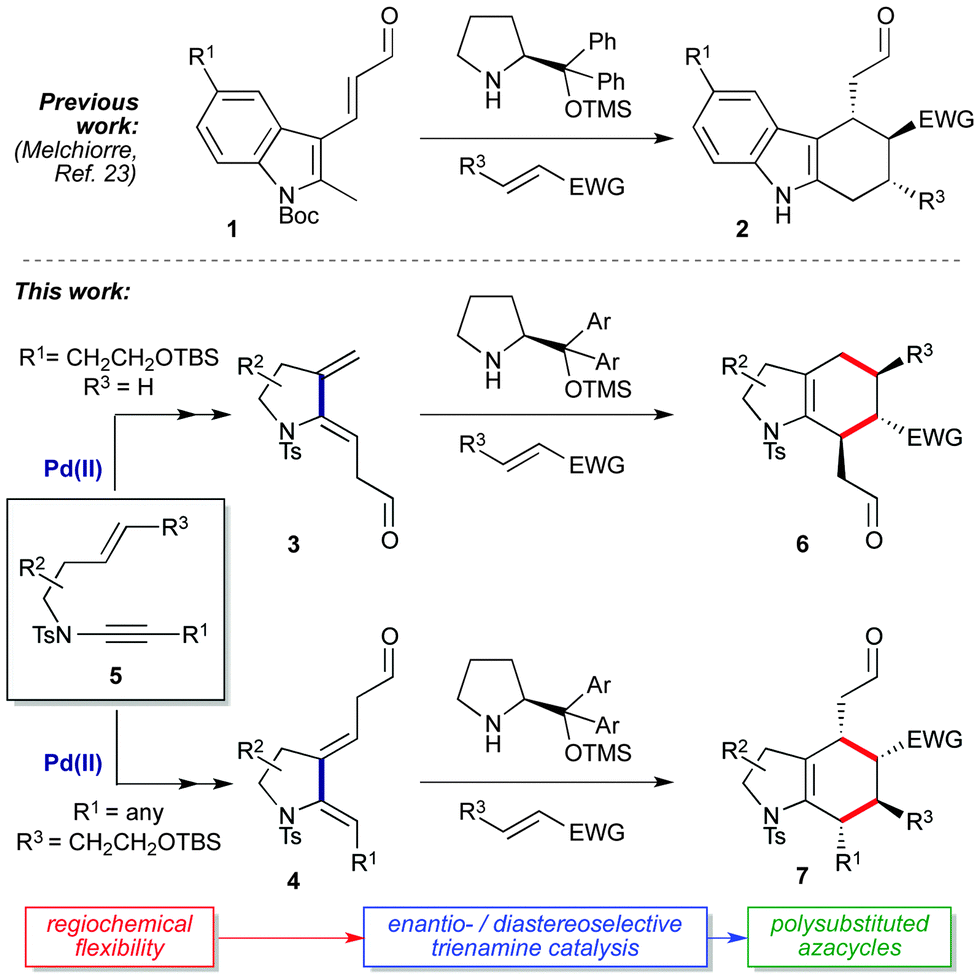
Trienamine organocatalysis represents a frontier of asymmetric synthesis,1–6 with trienamine-catalyzed cycloadditions of acyclic dienals7–16 and dienones17,18 enabling efficient syntheses of enantioenriched monocyclic cyclohexenes. In contrast, the use of exocyclic dienals in trienamine catalysis is comparatively rare, being limited mainly to elegant work on dearomatized aromatics such as indole-2,3-quinodimethane trienes (1 → 2Scheme 1).19–23 Furthermore, this chemistry has to date accessed only one of two possible cycloaddition regioisomers with respect to the tethering ring, presumably due to synthetic constraints in the positioning of the aldehyde. The wider extension of trienamine chemistry to cyclic substrates thus depends on the availability of suitable ring-tethered dienals, and methods that streamline dienal synthesis whilst simultaneously expanding reaction scope would offer a valuable entry to densely functionalized chiral (hetero)cyclic scaffolds.
 | ||
| Scheme 1 Trienamine-organocatalyzed cycloadditions of exocyclic dienals. | ||
Here we report a flexible and atom-efficient synthesis of a new class of azacyclic dienals (3 and 4) from enynamides (5) via palladium-catalyzed cycloisomerization,24–26 where the relative positioning of the aldehyde on the dienal framework is dictated by simple variation of the enynamide starting material. The enantio- and diastereoselective cycloadditions of these dienals give a range of regioisomeric hexahydroindole cycloadducts (6, 7), including spirooxindoles and azlactams which are of significant interest as medicinal chemistry scaffolds.27–29 These investigations culminate with the first examples of double stereodifferentiation in trienamine catalysis, which afford fully-functionalized hexahydroindole frameworks with precise control over the stereochemistry of all ring substituents, and thus expand the field of trienamine catalysis to access products of unprecedented complexity.
Our work began with the preparation of the regioisomeric ring-constrained dienals 3a and 4a (Scheme 2) by high-yielding palladium-catalyzed cycloisomerization24 of enynamides 5a and 5b,30 followed by desilylation and oxidation. In the case of 5b, either the partially- (4a) or fully-conjugated (8a) dienal could be accessed, depending on the oxidation conditions. As the success of trienamine catalysis can depend crucially on the degree of conjugation of the carbonyl substrate,16–18 it is notable that this cycloisomerization/oxidation approach permits such regiocontrol in dienal synthesis; in the event this indeed proved important. We were pleased to find that 3a underwent smooth cycloaddition with oxindole 9a, promoted by the Jørgensen–Hayashi catalyst 10, giving cycloadduct 11a in high yield and enantioselectivity after just 2 h at room temperature (84%, 98% ee, 3.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Reaction of 4a was similarly successful, giving the regioisomeric product 12a (78%, 94% ee, 6:1 dr). In contrast, no reaction was observed between 8a and 9a, even under heating, emphasizing the importance of the deconjugated nature of dienal 4a.16–18
:1 dr). Reaction of 4a was similarly successful, giving the regioisomeric product 12a (78%, 94% ee, 6:1 dr). In contrast, no reaction was observed between 8a and 9a, even under heating, emphasizing the importance of the deconjugated nature of dienal 4a.16–18
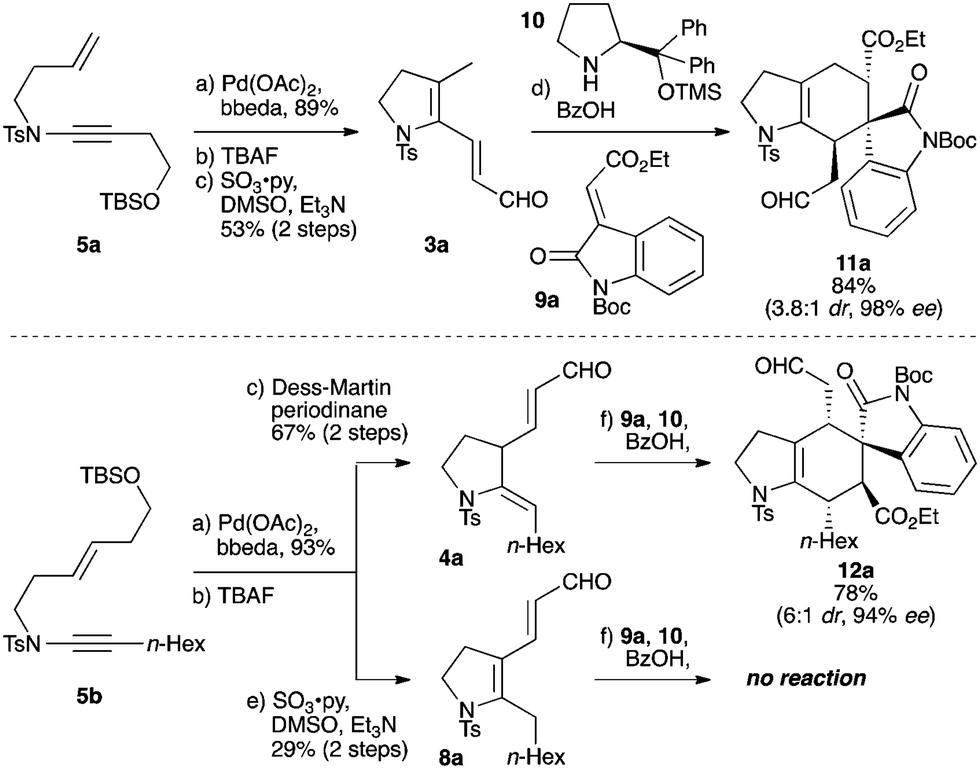 | ||
| Scheme 2 Palladium-catalyzed synthesis of regioisomeric dienals 3a, 4a and 8a, and initial study of trienamine-organocatalyzed cycloadditions. Reagents and conditions: (a) Pd(OAc)2 (5 mol%), bis-benzylidene ethylenediamine (bbeda) (5 mol%), toluene, 60 °C, 30 min; (b) TBAF, THF, rt, 2 h; (c) Dess–Martin periodinane, CH2Cl2, rt, 1.5 h; (d) 10 (20 mol%), BzOH (20 mol%), 9a (1.0 equiv.), 3a (1.5 equiv.), toluene, rt, 2 h; (e) SO3·py, Et3N, DMSO, CH2Cl2, 0 °C, 2 h; (f) 10 (20 mol%), BzOH (20 mol%), 9a (1.0 equiv.), 4a (1.5 equiv.), toluene, rt, 6 h. | ||
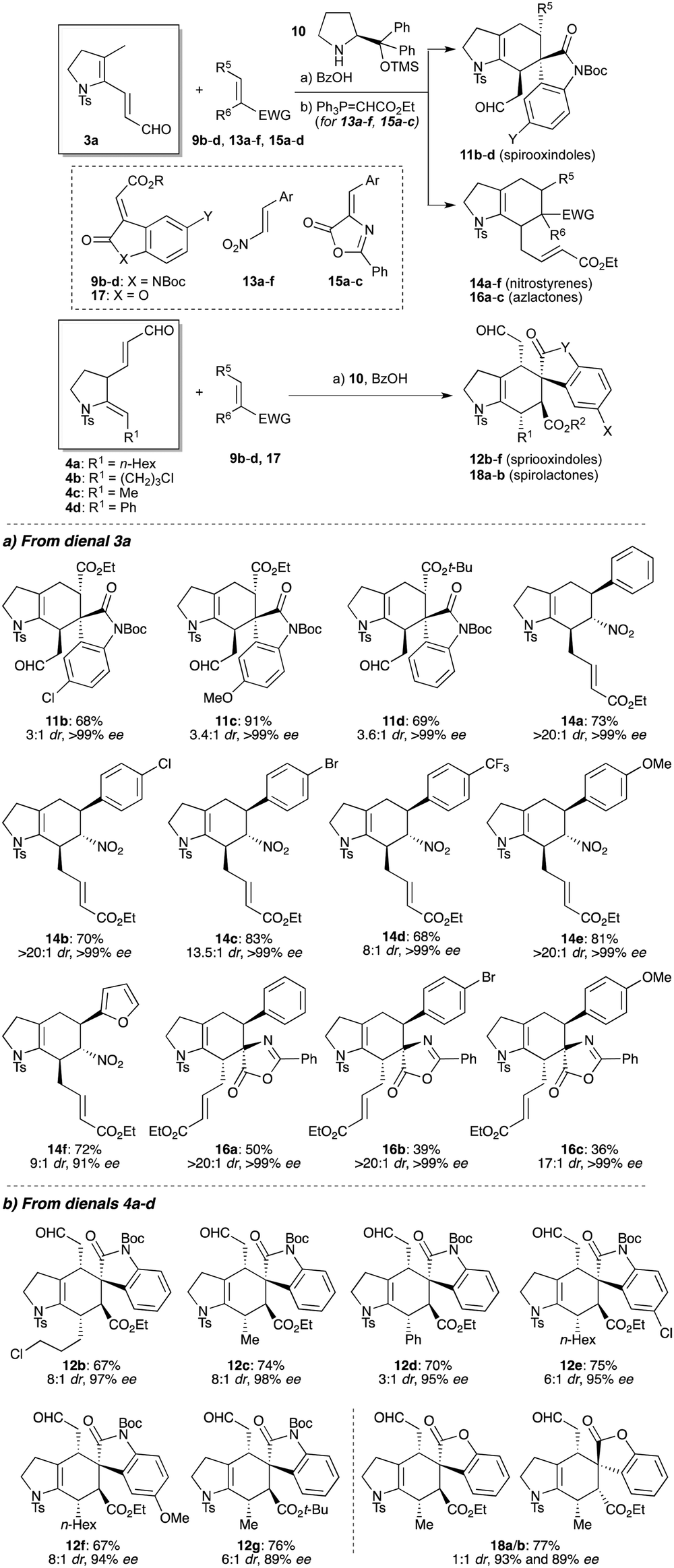
With conditions to effect organocatalyzed Diels–Alder reactions established, we next evaluated the scope of the enantioselective cycloadditions (Table 1). The reactions of 3a with various oxindoles were first examined, which afforded spirooxindoles 11b–d with high enantioselectivity. A selection of electron-deficient and electron-rich nitroalkenes were next tested, which pleasingly also underwent high yielding cycloadditions, giving cycloadducts 14a–f with excellent enantio- and diastereoselectivities.31 Azlactone cycloadditions provide a useful route to masked quaternary amino acids;32 for dienal 3a, these reactions were again found to be exceptionally diastereoselective and enantioselective (16a–d, >99% ee, >20:1 dr), albeit moderate yielding. Collectively, these reactions show a marked increase in rate compared to related acyclic1,2,16 or aromatic substrates,19 which may be due to the intrinsic s-cis constraint of the reacting diene in the trienamine intermediate, an effect that appears to override any electron-withdrawing effects from the sulfonamide group. It is also notable that formation of the presumed trienamine intermediate from 3a is regioselective for the formation of an exocyclic, rather than endocyclic double bond.
|
A similar exploration of reactivity was now conducted with the regioisomeric aldehyde system (4), which provided an opportunity to vary the enamide substituent (R1 in 4a–d, Table 1); these cycloadditions afford products that are substituted at all positions of the cyclohexene ring. We first tested oxindole dienophile cycloadditions, where variation of the enamide sidechain (12b–d) and oxindole (12e–g) led to high yields and excellent enantioselectivities. Notably, the diastereoselectivities of these ‘regiocomplementary’ cycloadditions were enhanced compared to those of dienal 3a, and showed some influence from the enamide substituent R1. Although cycloadditions with nitrostyrenes and azlactones proved ineffective, use of the more reactive lactone 17 successfully delivered cycloadducts 18a/b in high enantioselectivity, but without endo/exo preference.33
The cycloisomerization approach to these dienals provides the opportunity to functionalize the tethering pyrrolidine scaffold. This raised a question in trienamine catalysis that has not been addressed in previous studies: what levels of catalyst stereocontrol could be achieved in a double stereo-differentiating setting, where a chiral, single enantiomer substrate is reacted under the influence of enantiomeric catalysts?34–41 To investigate this, we prepared ynamides 5c and 5d (Scheme 3) as single enantiomers.30 These were subjected to palladium-catalyzed cycloisomerization, followed by desilylation and oxidation, to give dienals 3b and 4e respectively. Reaction of monosubstituted dienal 3b with nitrostyrene, under the influence of catalyst 10, afforded cycloadduct 19a in short reaction time (2 h at rt) with high diastereoselectivity (58%, 8:1 dr). This matched combination of substrate and catalyst reflects a preference of both components for approach of the dienophile to the top face of the molecule (as drawn). The corresponding mismatched reaction of 3b with nitrostyrene and catalyst ent-10 proceeded at a reduced reaction rate (7 h at rt), but pleasingly with a complete reversal of facial selectivity. To our surprise, an increased level of diastereoselectivity was observed (19b, 53%, 20:1 dr), interestingly in favour of the endo isomer with respect to the nitro group30 – an unprecedented observation in trienamine-organocatalyzed nitrostyrene Diels–Alder reactions.42 This suggests that although the catalyst completely controls the facial selectivity of cycloaddition, the substrate has a significant, and in this case dominant influence over endo/exo selectivity, such that steric interactions between the phenyl groups are minimized, irrespective of electronic2,19 or other stereocontrolling effects (see TS in Scheme 3).43
We next examined the reactions of 3b with oxindole 9a, which generated spirooxindoles 11e and 11f. In the matched case (giving 11e), we were delighted to find that previously observed levels of diastereoselectivity for oxindole cycloadditions (see Scheme 2) were increased (63%, 7:1 dr), illustrating a reinforcing influence of the conformation of the substrate on reaction diastereoselectivity. The mismatched combination gave a moderate yield of the cycloadducts 11f, arising from exclusive addition to the opposite face, but with poor selectivity. Finally, we addressed the challenge of the stereochemical influence of the doubly-substituted backbone in dienal 4e. To our delight, the reactions of 4e with 9a, which generate hexahydroindole spirooxindoles featuring a stereogenic centre at every position on the indole skeleton, proceeded with excellent yield and stereoselectivity in both the matched (12h, 60%, 20:1 dr) and mismatched (12i, 45%, 7:1 dr) settings; both reactions again illustrate strong substrate conformational effects that enhance diastereoselectivity.30
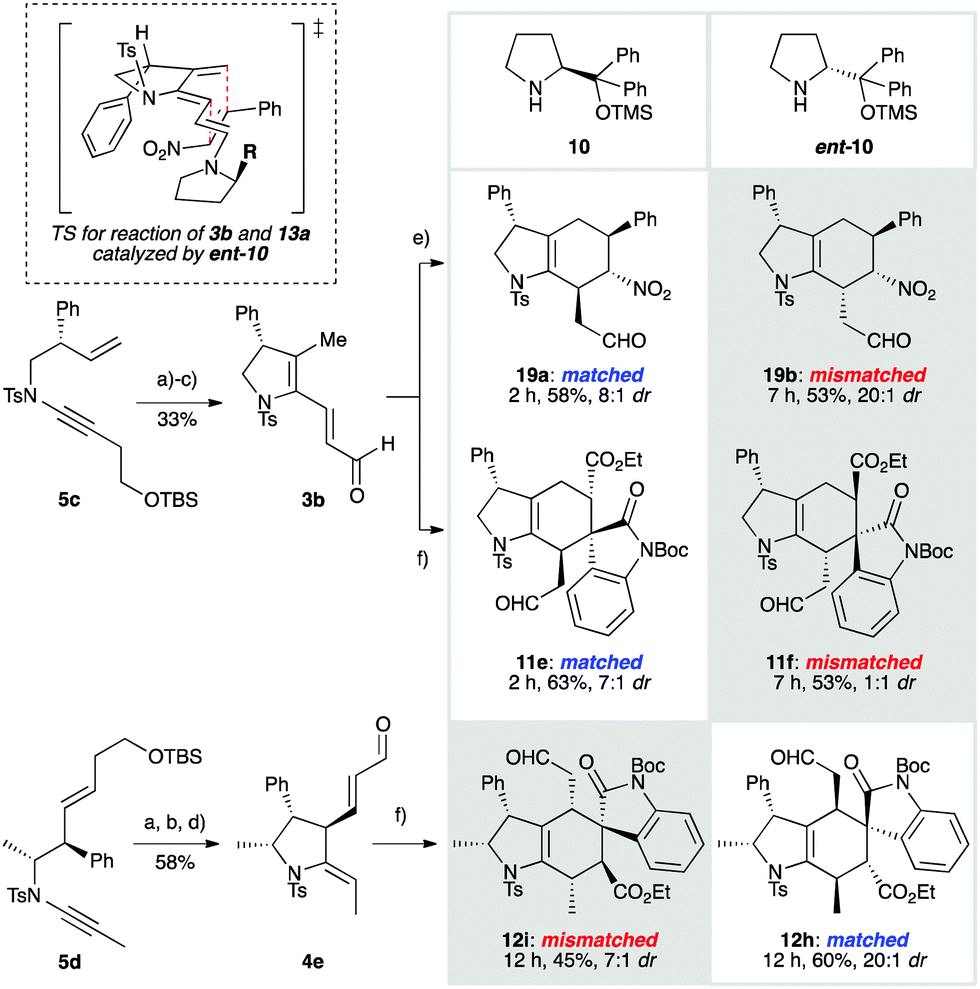 | ||
| Scheme 3 Double stereodifferentiating cycloadditions of dienals 3b and 4e. Diastereomer ratios reflect endo/exo selectivity; in all cases only a single face of the diene reacts. Cycloadditions were conducted using the dienophile (3.0 equiv.), aldehyde (1.0 equiv.), catalyst (10 or ent-10, 20 mol%) and BzOH (20 mol%) in toluene at rt under Ar (18 h). Other reagents and conditions: (a) Pd(OAc)2 (5 mol%), bbeda (5 mol%), toluene, 60 °C, 30 min, 85% from 5c, 95% from 5d; (b) TBAF, THF, rt, 2 h, 97% from 5c, 93% from 5d; (c) SO3·py, Et3N, DMSO, CH2Cl2, 0 °C, 2 h, 40%; (d) Dess–Martin periodinane, CH2Cl2, rt, 1.5 h, 66%; (e) reaction with 13a; (f) reaction with 9a. See the ESI† for structural assignment. | ||
In conclusion, palladium-catalyzed cycloisomerization provides a powerful and efficient entry to regioisomeric azacycle-tethered exocyclic dienals, which undergo enantio- and diastereoselective trienamine-organocatalyzed cycloadditions. These represent the first examples of exocyclic dienes to engage in such chemistry that arise from non-aromatic precursors. In addition to exploring the relative reactivity of these regioisomeric substrates across a range of dienophiles, we show that double stereodifferentiating cycloadditions proceed under high levels of catalyst stereocontrol for both regioisomers, thus permitting the tuneable synthesis of fully-functionalized hexahydroindole frameworks, including complex spirooxindoles.
We thank the EPSRC (EP/K005391/1) for financial support. VC thanks the European Union for an EU-Namaste Fellowship. RLG thanks Syngenta for an Industrial CASE studentship.
Notes and references
- For seminal work, see: Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen and K. A. Jørgensen, J. Am. Chem. Soc., 2011, 133, 5053–5061 CrossRef CAS PubMed.
- Z.-J. Jia, Q. Zhou, Q.-Q. Zhou, P.-Q. Chen and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 8638–8641 CrossRef CAS PubMed.
- For reviews, see: I. D. Jurberg, I. Chatterjee, R. Tannert and P. Melchiorre, Chem. Commun., 2013, 49, 4869–4883 RSC.
- I. Kumar, P. Ramaraju and N. A. Mir, Org. Biomol. Chem., 2013, 11, 709–716 CAS.
- J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491–1500 CrossRef CAS PubMed.
- K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS PubMed.
- For recent examples, see: H. Jiang, B. Gschwend, Ł. Albrecht, S. G. Hansen and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 9032–9036 CrossRef CAS PubMed.
- Ł. Albrecht, F. Cruz Acosta, A. Fraile, A. Albrecht, J. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 9088–9092 CrossRef PubMed.
- C. Ma, Z.-J. Jia, J.-X. Liu, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 948–951 CrossRef CAS PubMed.
- K. Zhu, H. Huang, W. Wu, Y. Wei and J. Ye, Chem. Commun., 2013, 49, 2157–2159 RSC.
- Z.-J. Jia, K. Jiang, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Chem. Commun., 2013, 49, 5892–5894 RSC.
- J.-X. Liu, Q.-Q. Zhou, J.-G. Deng and Y.-C. Chen, Org. Biomol. Chem., 2013, 11, 8175–8178 CAS.
- C. Ma, J. Gu, B. Teng, Q.-Q. Zhou, R. Li and Y.-C. Chen, Org. Lett., 2013, 15, 6206–6209 CrossRef CAS PubMed.
- X. Li, M.-H. Lin, Y. Han, F. Wang and J.-P. Cheng, Org. Lett., 2014, 16, 114–117 CrossRef CAS PubMed , and references therein.
- X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 14173–14176 CrossRef CAS PubMed.
- L. Prieto, G. Talavera, U. Uria, E. Reyes, J. L. Vicario and L. Carrillo, Chem. – Eur. J., 2014, 20, 2145–2148 CrossRef CAS PubMed.
- X.-F. Xiong, Q. Zhou, J. Gu, L. Dong, T.-Y. Liu and Y.-C. Chen, Angew. Chem., Int. Ed., 2012, 51, 4401–4404 CrossRef CAS PubMed.
- P.-Q. Chen, Y.-C. Xiao, C.-Z. Yue and Y.-C. Chen, Org. Chem. Front., 2014, 1, 490–493 RSC.
- Y. Liu, M. Nappi, E. Arceo, S. Vera and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 15212–15218 CrossRef CAS PubMed.
- Y. Liu, M. Nappi, E. C. Escudero-Adán and P. Melchiorre, Org. Lett., 2012, 14, 1310–1313 CrossRef CAS PubMed.
- H. Jiang, C. Rodríguez-Escrich, T. K. Johansen, R. L. Davis and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 10271–10274 CrossRef CAS PubMed.
- For an elegant example of a cross-conjugated cyclohexadiene-containing trieneamine which proceeds via a stepwise mechanism, see: K. S. Halskov, T. K. Johansen, R. L. Davis, M. Steurer, F. Jensen and K. A. Jørgensen, J. Am. Chem. Soc., 2012, 134, 12943–12946 CrossRef CAS PubMed.
- A. Dieckmann, M. Breugst and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 3237–3242 CrossRef CAS PubMed.
- P. R. Walker, C. D. Campbell, A. Suleman, G. Carr and E. A. Anderson, Angew. Chem., Int. Ed., 2013, 52, 9139–9143 CrossRef CAS PubMed.
- For seminal work on enynes, see: B. M. Trost, G. J. Tanoury, M. Lautens, C. Chan and D. T. Macpherson, J. Am. Chem. Soc., 1994, 116, 4255–4267 CrossRef CAS.
- V. Michelet, P. Y. Toullec and J. P. Genet, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- A. P. Antonchick, C. Gerding-Reimers, M. Catarinella, M. Schürmann, H. Preut, S. Ziegler, D. Rauh and H. Waldmann, Nat. Chem., 2010, 2, 735–740 CrossRef CAS PubMed.
- M. M. M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef CAS.
- See the ESI† for details of substrate synthesis and structural assignment.
- To facilitate isolation and analysis, the aldehyde product was directly derivatized via a Wittig olefination to the corresponding (E)-enoate.
- R. A. Mosey, J. S. Fisk and J. J. Tepe, Tetrahedron: Asymmetry, 2008, 19, 2755–2762 CrossRef CAS.
- Given the greater diastereoselectivity (typically >4:1) observed in previous cycloadditions between 17 and linear dienals (see ref. 33), steric effects from the substrate may be responsible for this reduced selectivity.
- To the best of our knowledge, no examples of double stereodifferentiating dienamine or trienamine catalysis have been reported. For examples of double stereodifferentiation in enamine organocatalysis, see: J. Marjanovic, V. Divjakovic, R. Matovic, Z. Ferjancic and R. N. Saicic, Eur. J. Org. Chem., 2013, 5555–5560 CrossRef CAS.
- F. Calderón, E. G. Doyagüez and A. Fernández-Mayoralas, J. Org. Chem., 2006, 71, 6258–6261 CrossRef PubMed.
- F. Calderón, E. G. Doyagüez, P. H.-Y. Cheong, A. Fernández-Mayoralas and K. N. Houk, J. Org. Chem., 2008, 73, 7916–7920 CrossRef PubMed.
- B. Alcaide, P. Almendros, A. Luna and M. R. Torres, J. Org. Chem., 2006, 71, 4818–4822 CrossRef CAS PubMed.
- N. Palyam and M. Majewski, J. Org. Chem., 2009, 74, 4390–4392 CrossRef CAS PubMed.
- J. Carpenter, A. B. Northrup, d. Chung, J. J. M. Wiener, S.-G. Kim and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2008, 47, 3568–3572 CrossRef CAS PubMed.
- D. Enders and C. Grondal, Angew. Chem., Int. Ed., 2005, 44, 1210–1212 CrossRef CAS PubMed.
- For an example of enhanced diastereoselectivity in enantioselective organocatalysis through catalyst design, see: L. Caruana, F. Kniep, T. K. Johansen, P. H. Poulsen and K. A. Jørgensen, J. Am. Chem. Soc., 2014, 136, 15929–15932 CrossRef CAS PubMed.
- For these highly challenging mismatched cycloadditions, we found it essential to degas the reaction solution to avoid aldehyde decomposition, a measure which allowed us to successfully employ an excess of dienophile and only one equivalent of aldehyde, which contrasts with previous stoichiometries employed in trienamine catalysis.
- A question arises as to whether the terms ‘matched’ and ‘mismatched’ should refer to the rate of reaction (where 19a would be the ‘matched’ product), or its selectivity (where 19b would be the ‘matched’ product). For the purposes of this work, we use the former definition; as the reactions of 3b to generate 11e or 11f proceed with the same trend in facial selectivity and rate as those to give 19a and 19b, we feel the superior (inverse) diastereoselectivity arising in the case of 13c is a result solely of steric interactions imparted by the substrate. In other words, we propose that this reaction is ‘mismatched’ with respect to the dienophile, and an overturn in endo/exo selectivity for 19b is a consequence of the enforced approach of this dienophile to the more hindered face of the molecule.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for novel compounds, stereochemical assignment of cycloadducts, copies of 1H and 13C NMR spectra and HPLC traces. See DOI: 10.1039/c5cc08886k |
| This journal is © The Royal Society of Chemistry 2016 |