
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA one-pot amidation of primary nitroalkanes†
Kenneth E.
Schwieter
and
Jeffrey N.
Johnston
*
Department of Chemistry and Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: jeffrey.n.johnston@vanderbilt.edu
First published on 27th October 2015
Abstract
It has been over a half-century since Kornblum demonstrated the conversion of a primary nitroalkane to a carboxylic acid; addition of an amine results in carboxylic acid formation as well. We describe the formation of amides from terminal nitroalkanes in a two-step, one-pot reaction involving tandem halogenation/umpolung amide synthesis (UmAS).
Innovations in amide synthesis1 have taken the form of mechanistic complementarity, as well as the expansion of alternative starting materials, each offering innovative new access points leading to amide. Oxidative capture of hemiaminals,2 electrophilic capture of hydroxylamines,3 and electrophilic amination of nitronates4 are examples of orthogonal mechanistic pathways to amides; oxidative amidation can draw from aldehyde, alcohol, and alkyl halide feedstock. These methods provide alternatives to carboxylic acid derivatives (acyl halides and other electrophilic derivatives), arguably the most common access point with which to engage an amine for amide synthesis. In contrast to their primary alcohol brethren, primary nitroalkanes have not yet, to the best of our knowledge,5 served as precursors to amides (Scheme 1, eqn (1)).
 | ||
| Scheme 1 Amides from primary nitroalkanes (the Goal, eqn (1)) and related work (Kornblum's conversion of aliphatic nitroalkanes to carboxylic acids using mild conditions, eqn (2)); examination of the nitrolic acid as a precursor to acid and amide (eqn (3)–(5)). | ||
The conversion of secondary nitroalkanes to ketones,6,7 and primary nitroalkanes to carboxylic acids8 was first reported by Kornblum, beginning in 1956 (Scheme 1, eqn (2)) and further developed by Mioskowski.9 The procedure is strikingly mild, requiring a combination of nitrite ester and sodium nitrite, the latter acting as a base; a nitrolic acid intermediate was invoked, as these were known to readily convert to carboxylic acid.10,11 These transformations of nitronates exhibit umpolung reactivity relative to their nitronic acid counterparts that engage in the Nef reaction.12 Despite the intermediacy of a nitrolic acid (existing at aldehyde or carboxylic acid oxidation states, depending on tautomer) and the two carbon–nitrogen bonds in this intermediate, carboxylic acids and esters – not amides – are produced in subsequent conversions. Our own analysis (Scheme 1, eqn (3)–(5)) of terminal nitroalkane (1) and nitrolic acid (4) behavior confirmed that the carboxylic acid (2) is the predominant, or only product, even when a nucleophilic amine is present.
Oxidative methods to convert secondary nitroalkanes to ketones, and primary nitroalkanes to aldehydes and carboxylic acids using permanganate13 or oxygen14 (and base) provide additional chemoselective alternatives, highlighted by applications in complex target synthesis.15 The conversions of α-substituted nitroalkanes to carboxylic acid derivatives were established by Barrett using α-oxy and α-thionitroalkanes, leading to esters and amides by ozonolysis of their conjugate base.
Based on the knowledge that α-bromo nitroalkanes engage amines in umpolung amide synthesis (UmAS),4 we sought a protocol wherein a terminal nitroalkane can be sequentially halogenated and transformed to amide in one pot, which would require the identification of a base and halonium reagent combination compatible with one another and effective in their respective roles for each step. Hayashi's realization of a similar goal using a different, but functionally equivalent protocol has stimulated us to report our success at this time.5 Together, these findings extend the utility of UmAS by virtue of the substantially larger number of enantioselective transformations of nitromethane,16–18 relative to bromonitromethane.19
Our attempts began with the most straightforward embodiment (Scheme 2), wherein two representative primary nitroalkanes (1, 5) were treated using the standard UmAS conditions; one equivalent of halogen would be required for α-halogenation, and as little as 5 mol% is required for efficient amidation (based on α-bromonitroalkanes).20 Both cases led to low yields of the desired amide product, but established the viability of a single-pot amidation of primary nitroalkanes. In addition to unreacted nitroalkane in these attempts, varying amounts of carboxylic acid were noted, consistent with the findings of Kornblum. It was unclear from these results whether the conditions were suboptimal for the nitronate halogenation step, or subsequent UmAS, leading us to scrutinize further the nitronate formation and halogenation.
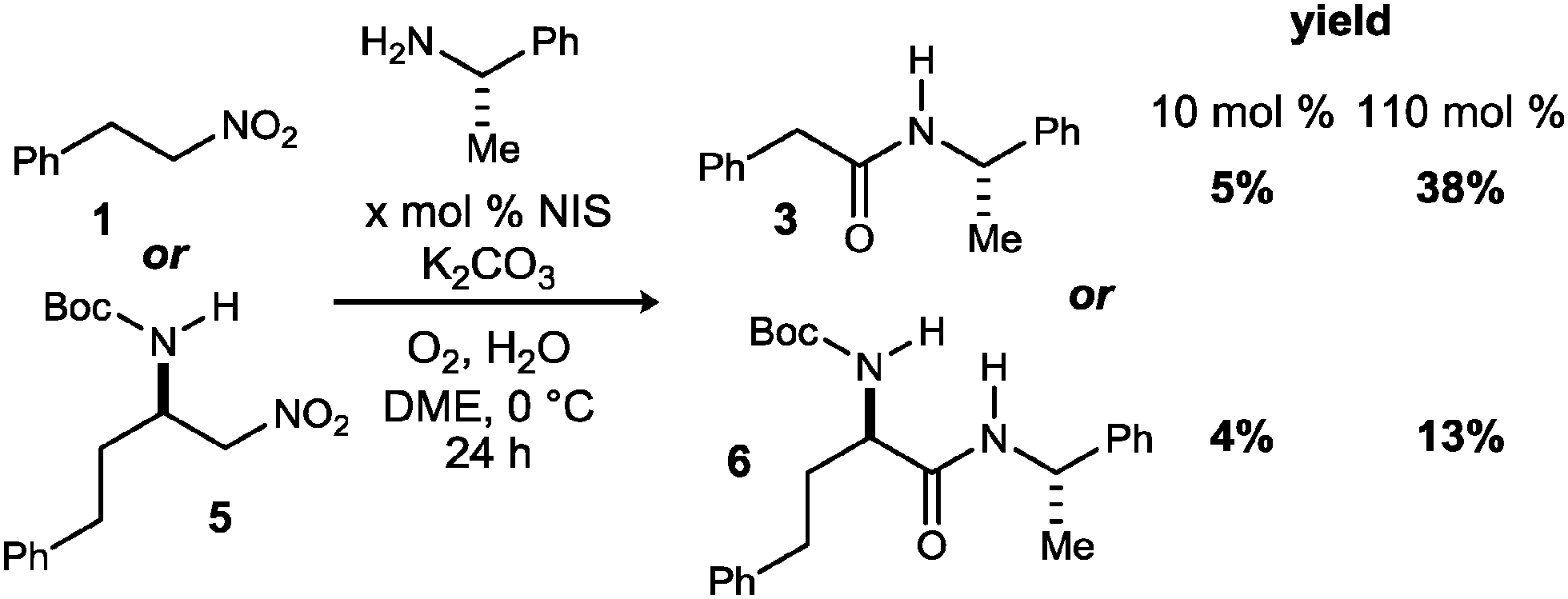 | ||
| Scheme 2 Initial results to convert primary nitroalkanes to amides using UmAS. | ||
Bromonitromethane derivatives are typically prepared by bromination of the nitronate salt with elemental bromine,21 or increasingly through catalyzed additions of bromonitromethane to carbon–carbon and carbon–nitrogen π-bonds.19,22 The nitronate nucleophile for bromination is typically prepared by deprotonation using aqueous potassium hydroxide, while the UmAS step employs a combination of amine and aqueous potassium carbonate. Therefore, the nitronate bromination step was examined using aqueous base (Method A) and amine base (Method B).
We hypothesized that although iodonium (from NIS) might be favored for N-halamine formation in UmAS, the nitronate of 1 or 5 may exhibit very different reactivity toward bromonium. This was confirmed by a comparison of common electrophilic bromine sources (Table 1). The traditional bromination procedure (Method A) was compared to a ‘mix-and-stir’ procedure (Method B). Preformation of the potassium nitronate allowed the α-bromonitroalkane to be prepared in each case, however, NBS and elemental bromine were superior (Table 1, entries 1 and 3). In stark contrast, when triethyl amine was used as the base – a choice made to simulate the basicity of the amine in UmAS without amide formation – in DME (no nitronate preformation), only dibromo tetrachloroethane was effective in α-bromination (Table 1, entry 2). This behavior may reflect both compatibility of DBTCE with the basic reagent, as well as an alignment of reactivity between the nitronate and brominating agent.
|
|
|||
|---|---|---|---|
| Entrya | Bromonium source | Method Aa yieldc (%) | Method Bb yieldc (%) |
a Reactions are 0.4 M (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/MeOH) in nitroalkane and employ 1 equiv. of Br+ source (see ESI for reaction details).
b Reactions are 0.2 M in nitroalkane and employ 3 equiv. Et3N and 1 equiv. Br+ source and were run for 24 h.
c Isolated yield. :1 H2O/MeOH) in nitroalkane and employ 1 equiv. of Br+ source (see ESI for reaction details).
b Reactions are 0.2 M in nitroalkane and employ 3 equiv. Et3N and 1 equiv. Br+ source and were run for 24 h.
c Isolated yield.
|
|||
| 1 | NBS | 78 | <5 |
| 2 | DBTCE | 27 | 57 |
| 3 | Br2 | 74 | <5 |
| 4 | DBDMH | 57 | <5 |
| 5 | TBCO | 14 | <5 |

The development of a mix-and-stir protocol for the one-pot amidation of primary nitroalkanes was attempted next (Table 2). The UmAS step requires an electrophilic halogen, and this protocol employs 10 mol% NIS, but we have shown that a bromonium source (including the α-bromonitroalkane itself) is sufficient to produce some amide product.20 In the event, significant formation of amide product was observed in each case. While the conditions of Method B (Table 1) were designed to probe the efficiency of bromination using homogeneous conditions, the semiaqueous, heterogeneous conditions of a standard UmAS reaction were applied in Table 2. The superiority of DBTCE appeared to carry through in this protocol, providing the desired amide in 71% yield (Table 2, entry 2). An important aspect of these results (Scheme 2, Tables 1 and 2) is the dual role that halogens play, and it is the parallel optimization of these roles that leads to the highest yield of amide.

Whether the amide is produced through an iodonium-only, bromonium-only or mixed-halonium pathway is less important, and ultimately determined by equilibria at play under specific conditions. It is of interest, however, to know the extent to which a specific halogen combination and order might affect the yield of amide. Furthermore, the experiments in Table 1 highlighted the need to avoid ineffective combinations of base and halogen source. This was probed further using the sequence outlined in Table 3. This protocol employed the amine to be used in the subsequent amide synthesis step as a base for the first step, as a variation on Method B (Table 1). Hence, primary nitroalkane and amine were stirred for 5 hours, then treated with a stoichiometric amount of halogenating agent. This mixture was then formulated using the standard heterogeneous UmAS conditions, specifically using conditions for halonium turnover.20 Use of NIS for each step resulted in a 29% yield of amide 3 (Table 3, entry 1), in close parallel to the analogous, non-stepwise addition protocol utilized earlier (Scheme 2, 38% yield). With this protocol, NBS was similarly effective when used for both steps (Table 3, entry 2). When an NBS/NIS sequence was employed, a significant, but still low yield of the amide was produced (33% yield, Table 3, entry 3). Collectively, results to this point seemed to emphasize two features: (1) the yield of amide is directly related to the efficiency of nitronate bromination, and the specific combination of α-halo nitroalkane and halamine created for amidation, and (2) the various halogenated species may be in equilibrium despite the physical sequencing characteristic of the protocol in Table 3. This reasoning led to further examination of DBTCE, since it proved more effective in the nitronate bromination using an amine base (Method B), and was somewhat tolerant of aqueous base (Method A) (Table 1, entry 2). In the event, the yield of amide increased dramatically (70%, Table 3, entry 4), in line with the yield of amide beginning from α-bromo nitroalkane 7. When NIS and DBTCE roles were reversed, the yield returned to 29% (Table 3, entry 5). The benefit of iodonium in the UmAS step was reaffirmed when it was not added, an experiment that provided a 37% yield of amide (Table 3, entry 6).

Based on these studies, a one-pot mix-and-stir protocol was developed for the amidation of primary nitroalkanes. An aqueous DME solution of nitroalkane and amine is treated with potassium carbonate, DBTCE (100 mol%) and NIS (10 mol%) under an oxygen atmosphere (balloon). This procedure resulted in significant improvement in yield for amides 3 and 6 (Scheme 2vs.Table 4) and was applied to a range of primary nitroalkanes resulting in moderate to good yields of the derived amides (Table 4). Notable examples include the use of a γ-nitro ester to prepare succinate 9b and amidation of a hindered nitroalkane to give β-cyano amide 9d. To illustrate the value and complementarity of the approach, several terminal nitroalkanes were prepared using enantioselective catalysis, thereby providing a more direct entry to enantioenriched amides. α-Amino amides 6, 9c, 9e, and 9f were derived from the respective nitromethane aza-Henry adducts. The use of an enantioselective conjugate reduction provided the nitroalkane precursor to 9g. Unprotected valine (9j) and phenylalanine (9l) were successfully coupled demonstrating the orthogonality of the current method with condensative peptide couplings. Furthermore, γ-hydroxy amide 9k was prepared without lactone formation that would otherwise compete with active ester-based amide synthesis.
|
|
|---|
| a Reactions are 0.2 M in nitroalkane and employ 2 equiv. amine, 2 equiv. K2CO3, and 5 equiv. H2O. Isolated yields reported. |
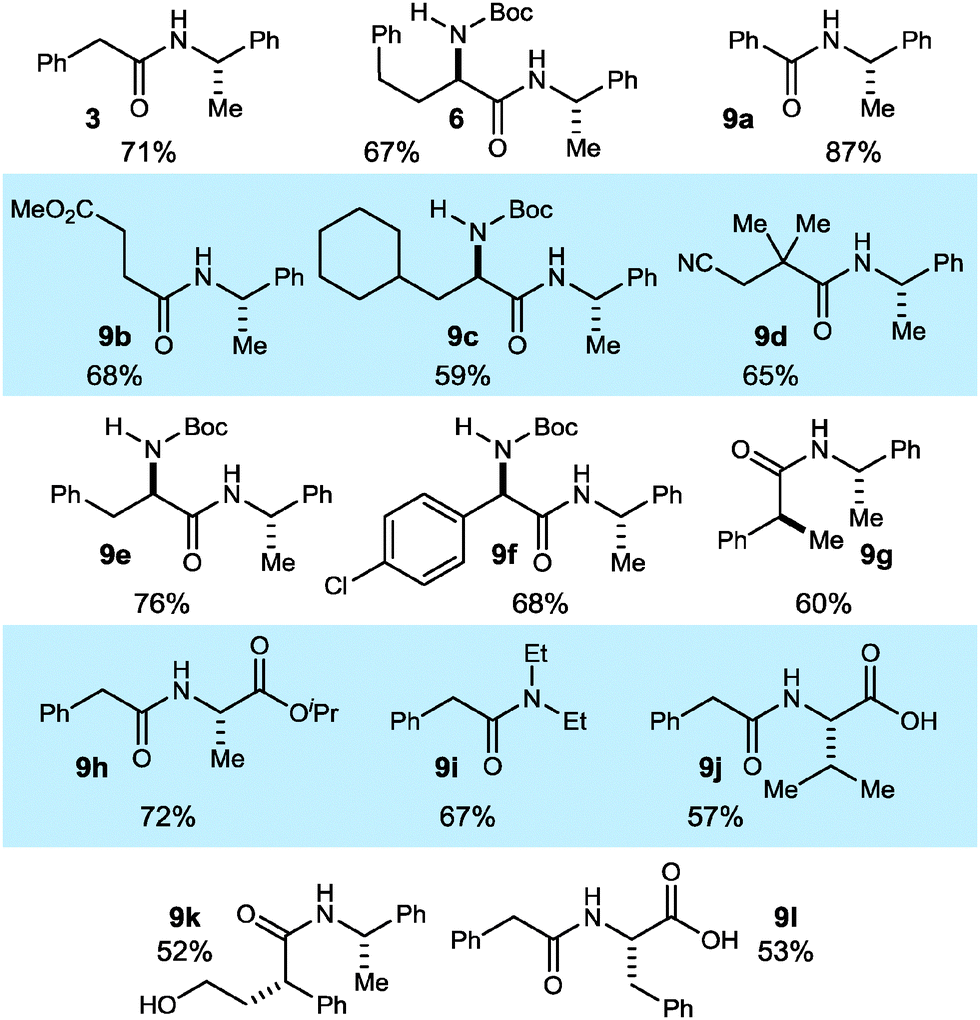
|
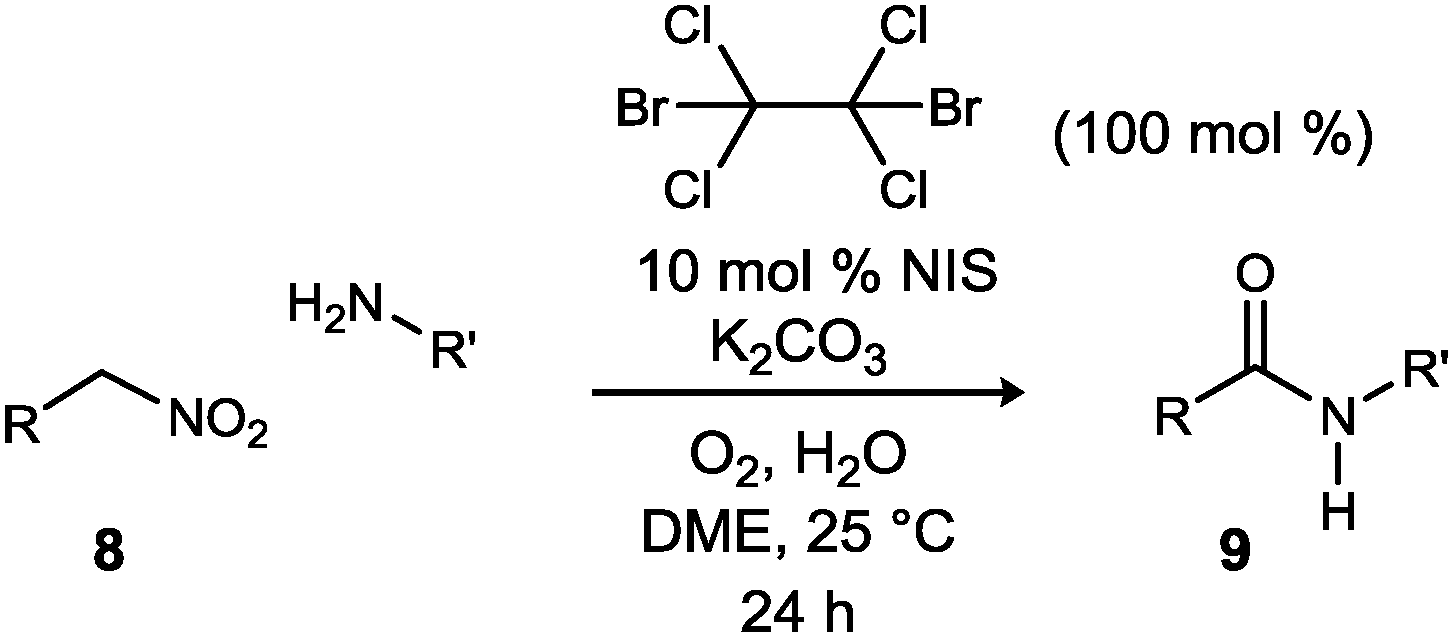
In closing, a detailed evaluation of nitronate bromination and iodination, and comparative analysis to the UmAS step leading to amide, has resulted in a convenient one-pot, mix-and-stir protocol to convert primary nitroalkanes to amides. Select examples of enantioenriched nitroalkanes were converted to their amide products in moderate to good yield. In principle, the advances in nitromethane-based enantioselective synthesis can now be generally leveraged to prepare a broad selection of amides, for which nitroalkanes serve as an alternative to carboxylic acids and their precursors, particularly terminal alcohols.
Notes and references
- Reviews: V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- Reviews: C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; K. Ekoue-Kovi and C. Wolf, Chem. – Eur. J., 2008, 14, 6302–6315 CrossRef CAS PubMed.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed; J. P. Shackleford, B. Shen and J. N. Johnston, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 44–46 CrossRef PubMed.
- During the completion of our work, the first example of this conversion appeared. We are grateful to Y. Hayashi for sharing a preprint of his work in this area: J. Li, M. J. Lear, Y. Kawamoto, S. Umemiya, A. R. Wong, E. Kwon, I. Sato and Y. Hayashi, Angew. Chem., Int. Ed., 2015, 54, 12986–12990 CrossRef CAS PubMed.
- N. Kornblum and P. A. Wade, J. Org. Chem., 1973, 38, 1418–1420 CrossRef CAS.
- S. Umemiya, K. Nishino, I. Sato and Y. Hayashi, Chem. – Eur. J., 2014, 20, 15753–15759 CrossRef CAS PubMed.
- See the ESI† for the mechanism proposed. N. Kornblum, R. K. Blackwood and D. D. Mooberry, J. Am. Chem. Soc., 1956, 78, 1501–1504 CrossRef CAS; N. Kornblum, H. O. Larson, R. K. Blackwood, D. D. Mooberry, E. P. Oliveto and G. E. Graham, J. Am. Chem. Soc., 1956, 78, 1497–1501 CrossRef.
- C. Matt, A. Wagner and C. Mioskowski, J. Org. Chem., 1997, 62, 234–235 CrossRef CAS PubMed.
- V. Meyer, Justus Liebigs Ann. Chem., 1875, 175, 88–140 CrossRef.
- The putative nitrolic acid in the Kornblum-Mioskowski procedure was later isolated, characterized, and shown to convert to nitrile oxide and carboxylic acid under the conditions of the conversion: C. Matt, A. Gissot, A. Wagner and C. Mioskowski, Tetrahedron Lett., 2000, 41, 1191–1194 CrossRef CAS; A. Gissot, S. N'Gouela, C. Matt, A. Wagner and C. Mioskowski, J. Org. Chem., 2004, 69, 8997–9001 CrossRef PubMed.
- R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS.
- Leading reference: N. Kornblum, A. S. Erickson, W. J. Kelly and B. Henggeler, J. Org. Chem., 1982, 47, 4534–4538 CrossRef CAS.
- S. Umemiya, K. Nishino, I. Sato and Y. Hayashi, Chem. – Eur. J., 2014, 20, 15753–15759 CrossRef CAS PubMed.
- Y. Hayashi and S. Umemiya, Angew. Chem., Int. Ed., 2013, 52, 3450–3452 CrossRef CAS PubMed; N. Ono and A. Kaji, J. Synth. Org. Chem., Jpn., 1980, 38, 115–127 CrossRef; D. Seebach, E. W. Colvin, F. Lehr and T. Weller, Chimia, 1979, 33, 1–18 Search PubMed.
- To illustrate, a search of the literature for the use of these two reagents in enantioselective synthesis revealed 60 citations for nitromethane relative to 3 citations for bromonitromethane in 2014. See the ESI† for a graph of the annual trend.
- Reviews: A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed; B. Westermann, Angew. Chem., Int. Ed., 2003, 42, 151–153 CrossRef PubMed; E. Marques-Lopez, P. Merino, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2009, 2401–2420 CrossRef.
- Selected examples: T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625–627 CrossRef CAS PubMed; B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418–3419 CrossRef PubMed; K. Yamada, S. J. Harwood, H. Groger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 3504–3506 CrossRef; K. R. Knudsen, T. Risgaard, N. Nishiwaki, K. V. Gothelf and K. A. Jorgensen, J. Am. Chem. Soc., 2001, 123, 5843–5844 CrossRef; T. P. Yoon and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 466–468 CrossRef PubMed.
- M. C. Dobish, F. Villalta, M. R. Waterman, G. I. Lepesheva and J. N. Johnston, Org. Lett., 2012, 14, 6322–6325 CrossRef CAS PubMed; M. W. Leighty, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 15233–15236 CrossRef PubMed; D. M. Makley and J. N. Johnston, Org. Lett., 2014, 16, 3146–3149 CrossRef PubMed; K. E. Schwieter and J. N. Johnston, Chem. Sci., 2015, 6, 2590–2595 RSC.
- K. E. Schwieter, B. Shen, J. P. Shackleford, M. W. Leighty and J. N. Johnston, Org. Lett., 2014, 16, 4714–4717 CrossRef CAS PubMed.
- A. S. Erickson and N. Kornblum, J. Org. Chem., 1977, 42, 3764–3765 CrossRef CAS; S. Trippett and D. M. Walker, J. Chem. Soc., 1960, 2976–2978 RSC.
- L.-T. Dong, R.-J. Lu, Q.-S. Du, J.-M. Zhang, S.-P. Liu, Y.-N. Xuan and M. Yan, Tetrahedron, 2009, 65, 4124–4129 CrossRef CAS; J. Izquierdo, C. Ayats, A. H. Henseler and M. A. Pericas, Org. Biomol. Chem., 2015, 13, 4204–4209 Search PubMed; R. Manzano, J. M. Andrés, R. Álvarez, M. D. Muruzábal, Á. R. de Lera and R. Pedrosa, Chem. – Eur. J., 2011, 17, 5931–5938 CrossRef PubMed; S. Piovesana, D. M. S. Schietroma, L. G. Tulli, M. R. Monaco and M. Bella, Chem. Commun., 2010, 46, 5160–5162 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all new compounds. See DOI: 10.1039/c5cc08415f |
| This journal is © The Royal Society of Chemistry 2016 |