
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific conjugation of drug-like fragments to an antimiR scaffold as a strategy to target miRNAs inside RISC†
A.
Brunschweiger‡§
a,
L. F. R.
Gebert‡¶
a,
M.
Lucic
a,
U.
Pradère
a,
H.
Jahns
a,
C.
Berk
a,
J.
Hunziker
b and
J.
Hall
*a
aInstitute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland. E-mail: jonathan.hall@pharma.ethz.ch
bNovartis Institutes for BioMedical Research, Global Discovery Chemistry, Novartis Campus, 4056 Basel, Switzerland
First published on 27th October 2015
Abstract
We synthesized a miR-122 antimiR library in which drug-like fragments were site-specifically introduced to short 2′-O-methyl-RNAs. At some sites selected fragments elevated cellular antimiR activity to that of an unmodified 23mer antimiR, whereas at others the same fragments abolished activity. The potency of the antimiRs correlated with uptake into miRISC.
MicroRNAs (miRNAs) are a family of 20–23 nucleotide (nt) non-coding RNAs that regulate gene expression post-transcriptionally.1 They bind their mRNA targets via a stretch of approximately 7 nt at their 5′ end (the “seed”) in an Argonaute (AGO)-containing ribo-nucleoprotein complex termed miRISC (microRNA induced silencing complex).2 Some miRNAs are linked to disease and are drug targets,3e.g. the liver-specific miR-122 which is a host-factor for hepatitis C (HCV) viral life-cycle.4 The targeting of miRNAs with complementary oligonucleotides (antimiRs) has advanced to clinical trials.5 AntimiRs have exceptional affinity and selectivity for their miRNA targets, and their chemical modification provides essential improvements to their pharmacokinetic (PK) properties.3,6 State of the art in antimiR medicinal chemistry is the locked nucleic acids (LNA)7 and constrained ethyl (cEt) derivatives.8 Recently, we compared the properties of two miR-122 antimiRs:9 a previously described 23 nt 2′-O-methyl (2′-O-Me) sequence (AMO122) and the 15 nt mixed DNA/locked nucleic acid (LNA) drug miravirsen (SPC3649). 2′-O-Me RNAs are a readily-accessible class of antimiRs, but they suffer from a relatively weak affinity for miRNAs and therefore are effective only when they are relatively long,10,11 which because of their size negatively impacts their PK properties.
As part of a program to develop new superior classes of antimiRs, we investigated whether intermittent modification of 2′-O-Me antimiRs would enable them to be shortened without loss of inhibitory activity.12,13 Recently, a fragment-based strategy to inhibit miRNA-specific AGO2 function in vitro was described in which a tetranucleotide complementary to the seed region of miR-122 served to project 5′-end-conjugated fragments into the active site of AGO2.14 In another example, the conjugation of an aromatic fragment close to the termini of 2′-O-Me antimiRs enhanced their cellular activity.15,16 All of these works pre-date a recent report that arguably transformed antimiR medicinal chemistry: it showed that affinity-enhancing modifications require careful positioning on the antimiR so as to interact with both miRNA and the proteins of the miRISC.17
As a “lead” structure we selected a relatively short 16 nt antimiR (1, (Table 1)) complementary to 1–16 nts of miR-122. Our objective was to use fragment-based modification of the 16 nt antimiR in order to raise its inhibitory activity to that of the longer AMO122. The 2′-O-pent-4-ynyladenosine phosphoramidite building block 2 (Scheme S2, ESI†) was used to site-specifically introduce alkyne moieties into 1. We appended small molecule fragments at various positions of 1 by copper-catalyzed alkyne–azide cycloaddition to generate a library of antimiR-fragment conjugates for structure–activity relationship studies. Building block 2 was introduced into three internal positions (P3, P9, P11) and at the 3′-terminal position of 1 (P16) using a standard oligonucleotide synthesis protocol (50 nmol scale), giving rise to sequences 3–6a (Table 1).
| No. | Sequence | Mass (obs) | Mass (calcd) |
|---|---|---|---|
| a Phosphorothioate in lower case, 2′-O-Me. b LNA in upper case, DNA in lower case, fully phosphorothioated backbone. c Phosphorothioate in lower case, 2′-O-Me, 2′-O-pent-4-yn-1-yladenosine in bold. d Phosphorothioate in lower case, 2′-O-Me, 2′-O-propyltriazolyl-linked fragment (Fig. 1) in bold, fragment in bold and superscript. | |||
| miR122 | GUUUGUGGUAACAGUGUGAGGU-5′ | — | — |
| AMO122 | acAAACACCAUUGUCACACucca-3′a | — | — |
| SPC3649 | CcAttGTcaCaCtCC-3′b | — | — |
| 1 | ccAUUGUCACACUCca-3′a | — | — |
| 3a | ccAUUGUCACACUCca-3′c | 5301.0 | 5301.8 |
| 4a | ccAUUGUCACACUCca-3′c | 5301.0 | 5301.8 |
| 5a | ccAUUGUCACACUCca-3′c | 5301.0 | 5301.8 |
| 6a | ccAUUGUCACACUCca-3′c | 5301.1 | 5301.8 |
| 7 | ccAhUUGUCAiCACUCcai-3′d | 6060.4 | 6060.6 |
| 8 | ccAiUUGUCAiCAhCUCca-3′d | 6060.4 | 6060.6 |
The library was furnished with a set of fragments b-j (Fig. 1), structurally similar to members of an RNA-directed fragment library.18 We performed the CuAAC on the solid support-bound oligonucleotides to facilitate product purification by HPLC. As only a small number of azides are commercially available, we developed a robust and convenient one-pot protocol which yielded azides from readily available aliphatic and benzylic halides in situ (Fig. 1). Four batches of CPG solid support, each one containing an alkynyl-modified DMT-protected antimiR 3a–6a were distributed each into 9 wells of a 96-well plate for conjugation reactions. To each well was added an in situ-generated azide b-j, yielding a 40-membered library with 10 fragments attached to four positions of 1. HPLC-purification of the oligonucleotide-conjugates indicated in all cases full conversion to the desired triazole-linked libraries 3b-j–6b-j. This new synthesis strategy represents a straightforward access to a diverse library of systematically modified antimiRs which facilitates SAR studies. Given the findings of this work, it should provide further insights into the requirements for targeting miRNAs in miRISC.17
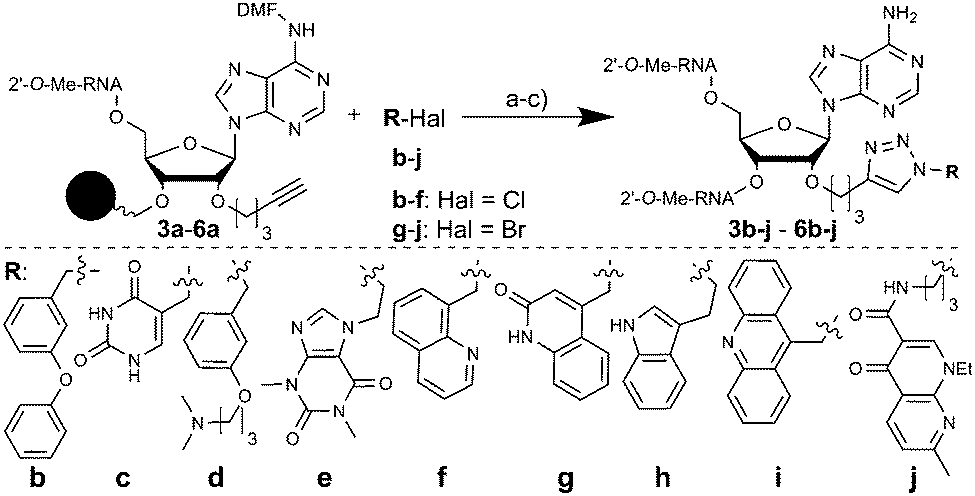 | ||
| Fig. 1 Conjugation of fragments b-j to 3–6. Conditions: (a) NaN3, TBAI, DMF, H2O, 1 h, RT (b-d, f-i) or 70 °C (e, j); (b) CuSO4, Na-ascorbate, TBTA, H2O, MeOH, DMF, 16 h, 45 °C; (c) gas. MeNH2, 1.5 h, 70 °C; DMF: dimethylformimidamide. | ||
The library was screened at four concentrations for inhibition of endogenously expressed miR-122 acting on a sensitive luciferase reporter.9SPC3649 served as a control, and as expected was the most active of all molecules tested (Fig. 2). An unrelated antimir sequence (CON) served as a control to help identify artifacts associated with toxicity of the cell transfections. The 2′-O-Me-per-modified 16 nt 1 and the 23 nt long AMO122 served as the reference compounds to compare the activities of library members. The reference compounds were added to each screening plate to account for plate-to-plate variations in efficiency of the antimir transfection. The linker-bearing sequences (3a–6a) already showed a remarkable position-dependent pattern of activity (Fig. 2).
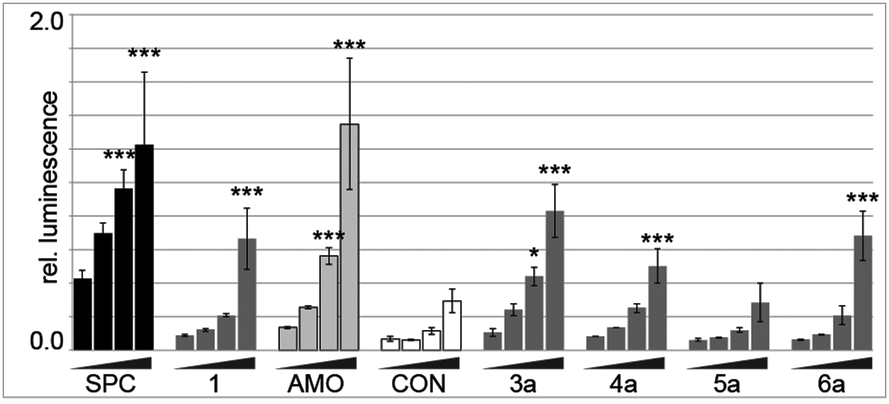 | ||
| Fig. 2 AntimiR activity in a luciferase reporter assay performed in Huh-7 cells after treatments at 0.6, 2.5, 10 and 40 nM. Bars indicate relative Renilla luciferase activity (compared to firefly control) from inhibition of endogenous miR-122. Error bars are SD (n = 3). Statistical analysis against the lowest dose: *P < 0.05; **P < 0.01; ***P < 0.001. (SPC: SPC3649; AMO: AMO122). | ||
Sequences 4a and 6a (linker at P9 and P16, respectively, base-pairing with positions 8 and 1 of miR-122, respectively) showed similar activity to the parent 1. In contrast, 3a (linker at P3, opposite position 14 of miR-122) had substantially higher potency than 1, approaching that of AMO122 at two treatment concentrations. Surprisingly, a pent-4-ynyl fragment abolished antimiR activity when placed opposite to position 6 (P11 of the antimiR) in the seed of miR-122 (5a). While hints for positional effects of modifications are present in literature,17,19 we demonstrated for the first time how a single modification could increase or decrease antimiR activity by changing its position. Extending the linker with other fragments confirmed this trend (Fig. 3). Indeed, we found that most modifications placed at P3 increased antimiR activity as compared to 1, with the nalidixic acid fragment j yielding probably the greatest increase in activity, similar to, or only slightly less, at all concentrations than the 23 nt AMO122. Compared to 1, it showed a three to four-fold increase in potency. Previously, nalidixic acid was shown to bind to DNA and RNA.13,20 The flat, lipophilic, aromatic acridine and indole fragments (h, i) also performed well in the assay. Less pronounced effects were observed with all other fragments. Taken together, the data showed that a loss of inhibitory activity which results from a shortening of the antimiR can be compensated by fragment-based modification, in line with the initial hypothesis.13,15,16 Importantly, the data showed position-dependent effects of the fragments, exemplified most strongly by all modifications at P11 of the antimiR, which abolished the inhibitory activity.
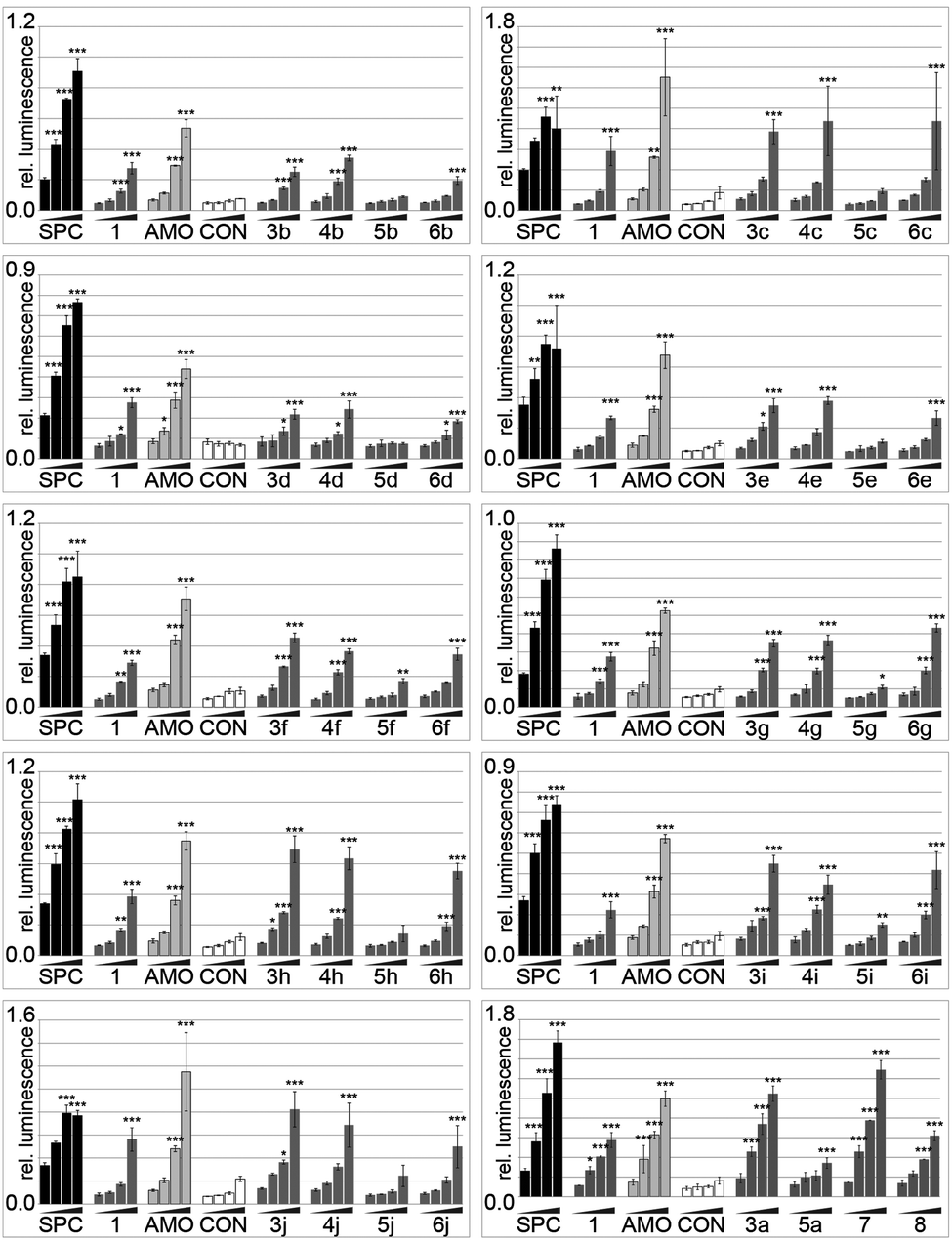 | ||
| Fig. 3 Inhibitory activity of 40 antimiR conjugates in a miR-122 luciferase reporter. Assay conditions and statistics as described in Fig. 2. | ||
In order to determine whether the aforementioned positional effects of fragment substitutions were additive we prepared two antimiRs modified at multiple positions. New chemistry which we specifically developed for modification of pre-miRNAs,21 allowed us to synthesize the first examples of antimiRs that display a combination of different fragments at selected positions. This enabled us to probe the effect of simultaneous modification of internal positions P3, P9, and P16 (7, Table 1) with fragments that had shown a beneficial effect at the respective positions (Fig. 3). Thus, we prepared a tri-substituted antimiR with groups appended to P3, P9, and P11 (8, Table 1) to determine whether the negative effects of the substitution at P11 would apply to more highly substituted antimiRs.
Indeed, 7 was more potent than 1 and AMO122 (at 40 and 10 nM concentrations). Disappointingly however, it was only slightly more active than the singly-modified 3a. Shifting the conjugated fragment from P16 to P11 (antimiR 8) reduced activity by approximately two-fold, demonstrating again the negative effects on activity for a group larger than methoxy at this site.
Variations in activity among the modified antimiRs might conceivably have been due to changes in various parameters. However, when for a given fragment the difference between biological activity and inactivity of an antimiR was the site of fragment conjugation, then variances in binding affinity, in stability to exonucleases, or in cellular uptake seemed unlikely to explain the phenomenon. In order to provide further insight on these factors we performed additional experiments. Hence, we measured the melting temperatures (Tm′s) of compounds 3b-i and the nalidixic acid conjugates (3–6j) to miR-122, comparing them with that of parent compound 1. Across the 3-series of compounds, the Tm′s varied widely (Fig. S1, ESI†). In some cases a high Tm correlated with high cellular activity (e.g.3h, 3i), but not always (e.g.3g) (Fig. 3). As expected, conjugation of a single nalidixic acid fragment at four different sites yielded a much smaller range of Tm′s (1.0–2.3 °C) (Fig. S1, ESI†). Together, this provided strong evidence that differences in hybridization affinity were not responsible for the opposing activities of the 3- and 5-series of compounds.
We then turned to possible differences in the transport of antimiRs into cells and into the miRISC. As modified oligonucleotides are generally not assayable by conventional quantitative real-time PCR (qPCR), we developed a new variant of chemical-ligation PCR (CL-qPCR) to quantify antimiR delivery into cells and the miRISC.22 In the CL-qPCR method the antimiR templates chemical ligation of two DNAs, the product of which can then be quantified by qPCR (Scheme S1, ESI†). In the absence of template no ligation/PCR occurs. First, we generated standard concentration curves for 1, 3a and 5a (Fig. S2–S4, ESI†) and then transfected them into cells and isolated lysates. In two experiments the transfection efficiency of the antimiRs was not reproducibly affected by the pent-4-ynyl group (Fig. 4a) and thus did not explain differences in activity.
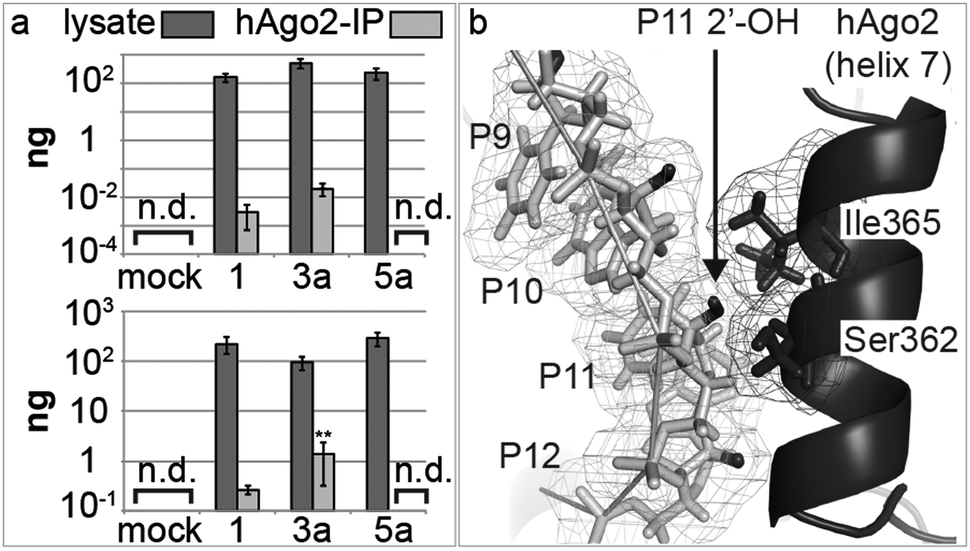 | ||
| Fig. 4 (a) Detection of antimiRs in Huh-7 cell lysates 25 h post-transfection, and in miRISC, immunoprecipitated using an anti-AGO2 antibody. Error bars are SEM; (n = 3); [n.d.: not detected]. Statistical analysis against 1: **P < 0.01; (b) structure of AGO2 with a guide miRNA and a complementary RNA (miRNA is removed for clarity). RNA and protein are light and dark grey, respectively; antimiR 2′-OH groups are black. The 2′-OH at P11 is tightly bound between the P10 ribose and Ser362/Ile365 of helix 7. PDB ID: 4W5O.23 Image generated with PyMOL (PyMOL Molecular Graphics System, Version 1.6 Schrödinger, LLC). | ||
Next, we immunoprecipitated AGO2 protein from transfected cells, and quantified the antimiRs in the immunoprecipitates: 3a showed the highest association with AGO2, whereas no signal was detected for 5a (Fig. 4a). This trend mirrors their inhibitory activity in cells, i.e. that 3a was the most active, and that 5a was inactive. Together, the data suggested that the differences in activity of 1, 3a and 5a and possibly those of other library members was at least partly due to differences in association of the compounds with the miRNA–AGO2 complex. We hypothesized that the loss of activity seen in passing from a 2′-O-Me substituent (1) at P11 of the antimiR to a 2′-O-pent-4-ynyl group (5a), or even larger substituents (5b-j; Fig. 3) was possibly due to clashes with amino-acid residues of AGO2 in miRISC. Therefore, we studied the recently-published structure of AGO2 bound to a model miRNA/RNA-target duplex.23 The structure shows that the protein makes extensive contacts with the miRNA/mRNA-target in the miRNA seed region, through α-helix 7 (Fig. 4b). We considered the RNA target as a surrogate for an antimiR. According to the structure, the 2′-position of the nucleotide P11 in the antimiR that pairs with nucleotide 6 in the miRNA seed region is closely associated with Ile365 and Ser362 of AGO2. This suggests that introduction of large substituents to antimiRs at that position may prevent their interaction with miR-122 when complexed to AGO2, which is in agreement with the observed loss of activity of the 5-series of antimiRs.
The 4-series of antimiRs carry fragments on the P9 nucleotide, in an area devoid of minor groove interactions with AGO2. This may explain the difference in cellular activity of the 4- and the 5-series of antimiRs. As the crystallographic structure was determined using an RNA target of 11 nt, it was unable to help explain the improved activities of some of the 3-series antimiRs. However, we cannot rule out that the enhanced activity of these antimiRs might be partly due to increases in hybridization affinity, as indicated.
Recent evidence17 confirmed early indications11 that antimiR drugs sequester the miRNA in the AGO/miRNA ternary complex. The findings imply that affinity-enhancing chemical modifications for antimiRs require careful positioning to ensure favourable interactions with both the miRNA and the associated protein(s). Beginning with a 16 nt 2′-O-Me antimiR targeting miR-122, we developed a pragmatic chemistry-based approach to incorporate multiple drug-like fragments (e.g.h, i, j) into the antimiR so as to elevate its potency almost to that of a 23 nt homologue. We showed that careful positioning of flat aromatic hydrophobic fragments were the most effective. Surprisingly, more important than the fragments' structure was its position: activity was generally higher for antimiRs modified at P3, whereas inclusion at P11 abolished activity. Using a novel variant of a CL-qPCR assay, we quantified the antimiR in miRISC and showed that these effects were likely due to steric clashes between the added fragments and the AGO2 protein. These findings are of high importance for the future design of antimiR drugs because they demonstrate the potentially radical effects of regioselective incorporation of modified nucleotides upon antimiR activity.
This work was supported by grants from the Swiss National Science Foundation (CRSII3_127454; 205321_124720) to JH. We thank S. Forli, S. Crivelli and I. Beuwink for help with PyMOL, luciferase assays and the CL-qPCR protocol, resp.
Notes and references
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- M. R. Fabian and N. Sonenberg, Nat. Struct. Mol. Biol., 2012, 19, 586–593 CAS.
- Z. Li and T. M. Rana, Nat. Rev. Drug Discovery, 2014, 13, 622–638 CrossRef CAS PubMed.
- C. L. Jopling, M. Yi, A. M. Lancaster, S. M. Lemon and P. Sarnow, Science, 2005, 309, 1577–1581 CrossRef CAS PubMed.
- H. L. Janssen, H. W. Reesink, E. J. Lawitz, S. Zeuzem, M. Rodriguez-Torres, K. Patel, A. J. van der Meer, A. K. Patick, A. Chen, Y. Zhou, R. Persson, B. D. King, S. Kauppinen, A. A. Levin and M. R. Hodges, N. Engl. J. Med., 2013, 368, 1685–1694 CrossRef CAS PubMed.
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef CAS PubMed.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- P. P. Seth, A. Siwkowski, C. R. Allerson, G. Vasquez, S. Lee, T. P. Prakash, G. Kinberger, M. T. Migawa, H. Gaus, B. Bhat and E. E. Swayze, Nucleic Acids Symp. Ser., 2008, 2004, 553–554 CrossRef PubMed.
- L. F. Gebert, M. A. Rebhan, S. E. Crivelli, R. Denzler, M. Stoffel and J. Hall, Nucleic Acids Res., 2014, 42, 609–621 CrossRef CAS PubMed.
- J. Krutzfeldt, S. Kuwajima, R. Braich, K. G. Rajeev, J. Pena, T. Tuschl, M. Manoharan and M. Stoffel, Nucleic Acids Res., 2007, 35, 2885–2892 CrossRef CAS PubMed.
- S. Davis, B. Lollo, S. Freier and C. Esau, Nucleic Acids Res., 2006, 34, 2294–2304 CrossRef CAS PubMed.
- N. Kalra, M. C. Parlato, V. S. Parmar and J. Wengel, Bioorg. Med. Chem. Lett., 2006, 16, 3166–3169 CrossRef CAS PubMed.
- A. Bugaut, K. Bathany, J.-M. Schmitter and B. Rayner, Tetrahedron Lett., 2005, 46, 687–690 CrossRef CAS.
- M. F. Schmidt, O. Korb and C. Abell, ACS Chem. Biol., 2013, 8, 2122–2126 CrossRef CAS PubMed.
- K. A. Lennox, R. Owczarzy, D. M. Thomas, J. A. Walder and M. A. Behlke, Mol. Ther. – Nucleic Acids, 2013, 2, e117 CrossRef PubMed.
- J. Blechinger, H. Pieper, P. Marzenell, L. Kovbasyuk, A. Serva, V. Starkuviene, H. Erfle and A. Mokhir, Chem. Commun., 2013, 49, 7397–7399 RSC.
- D. J. Hogan, T. M. Vincent, S. Fish, E. G. Marcusson, B. Bhat, B. N. Chau and D. G. Zisoulis, PLoS One, 2014, 9, e100951 Search PubMed.
- K. Bodoor, V. Boyapati, V. Gopu, M. Boisdore, K. Allam, J. Miller, W. D. Treleaven, T. Weldeghiorghis and F. Aboul-ela, J. Med. Chem., 2009, 52, 3753–3761 CrossRef CAS PubMed.
- J. Elmen, M. Lindow, S. Schutz, M. Lawrence, A. Petri, S. Obad, M. Lindholm, M. Hedtjarn, H. F. Hansen, U. Berger, S. Gullans, P. Kearney, P. Sarnow, E. M. Straarup and S. Kauppinen, Nature, 2008, 452, 896–899 CrossRef CAS PubMed.
- L. L. Shen, L. A. Mitscher, P. N. Sharma, T. J. O'Donnell, D. W. Chu, C. S. Cooper, T. Rosen and A. G. Pernet, Biochemistry, 1989, 28, 3886–3894 CrossRef CAS PubMed.
- U. Pradere, A. Brunschweiger, L. F. Gebert, M. Lucic, M. Roos and J. Hall, Angew. Chem., Int. Ed. Engl., 2013, 52, 12028–12032 CrossRef CAS PubMed.
- J. A. Boos, D. W. Kirk, M. L. Piccolotto, W. Zuercher, S. Gfeller, P. Neuner, A. Dattler, W. L. Wishart, F. Von Arx, M. Beverly, J. Christensen, K. Litherland, E. van de Kerkhof, P. J. Swart, T. Faller, A. Beyerbach, D. Morrissey, J. Hunziker and I. Beuvink, Nucleic Acids Res., 2013, 41, e145 CrossRef CAS PubMed.
- N. T. Schirle, J. Sheu-Gruttadauria and I. J. MacRae, Science, 2014, 346, 608–613 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds; luciferase assays; RNP chemical-ligation PCR. See DOI: 10.1039/c5cc07478a |
| ‡ Equal contribution. |
| § Current address: Faculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn-Str.6, 44227 Dortmund, Germany. |
| ¶ Current address: Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA 92037, USA. |
| This journal is © The Royal Society of Chemistry 2016 |