DOI:
10.1039/C5CC08182C
(Communication)
Chem. Commun., 2016,
52, 2023-2026
Tetra-cationic imidazoliumyl-substituted phosphorus–sulfur heterocycles from a cationic organophosphorus sulfide†
Received
30th September 2015
, Accepted 17th November 2015
First published on 1st December 2015
Abstract
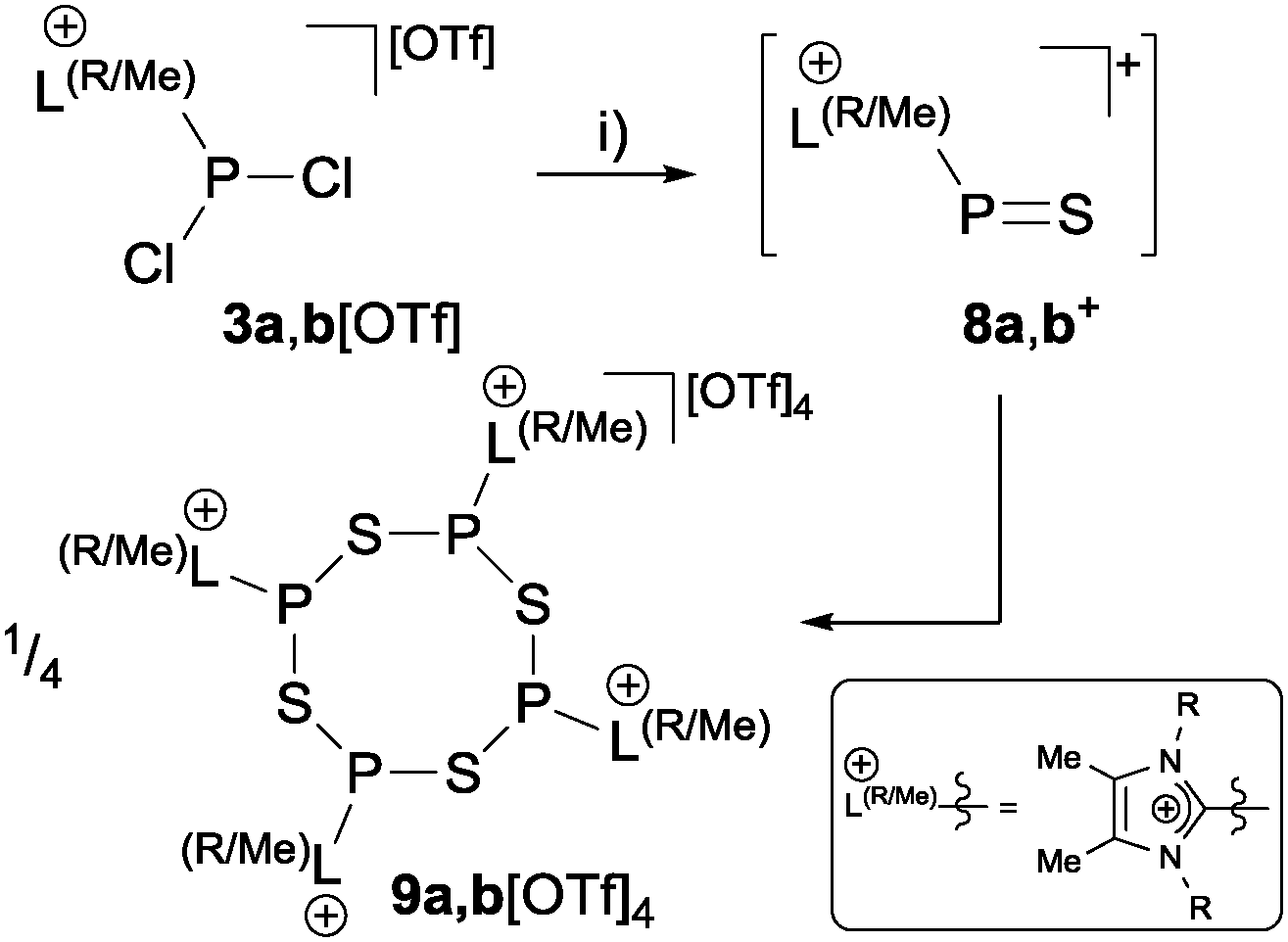
The reaction of imidazoliumyl-substituted P(III) cations of type [L(R,Me)PCl2]+ (3a,b+; LR,Me = imidazolium-2-yl a: R = Me; b: R = iPr) with (Me3Si)2S leads to the formation of tetra-cationic, eight-membered phosphorus sulfur heterocycles [L(R,Me)PS]44+ (9a,b4+), which can be explained by the tetramerization of the intermediately formed cationic phosphorus monosulfide [L(R,Me)PS]+ (8a,b+). The P4S4 ring adopts a crown conformation as observed for cyclo-S8. The Lewis base DMAP (4-dimethylaminopyridine) initiates a deoligomerization- and dismutation reaction of 9a,b4+ to give P(I) centered cation [L(R,Me)2P]+ (12a,b+) and phosphorus disulfide [(DMAP)2PS2]+ (14+).
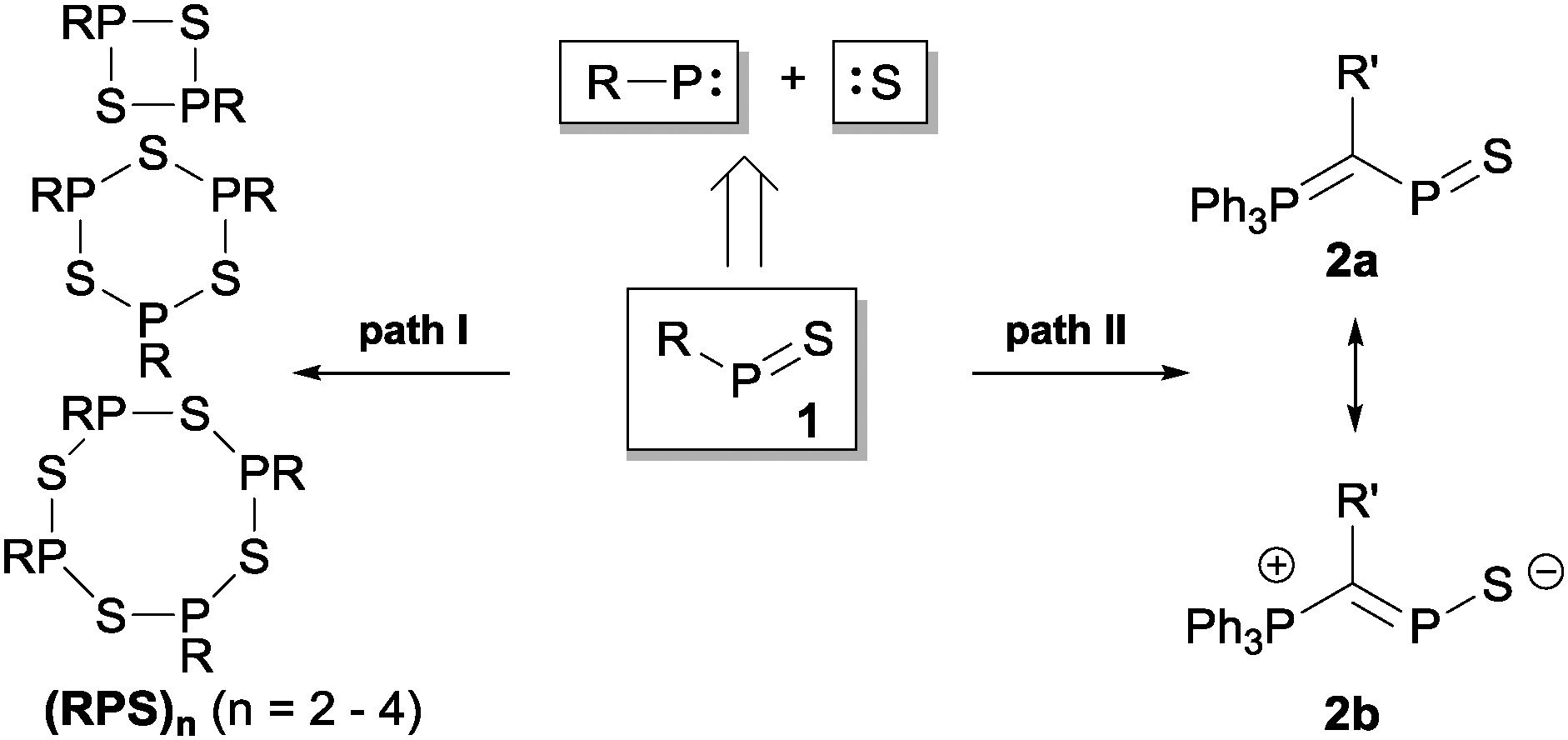
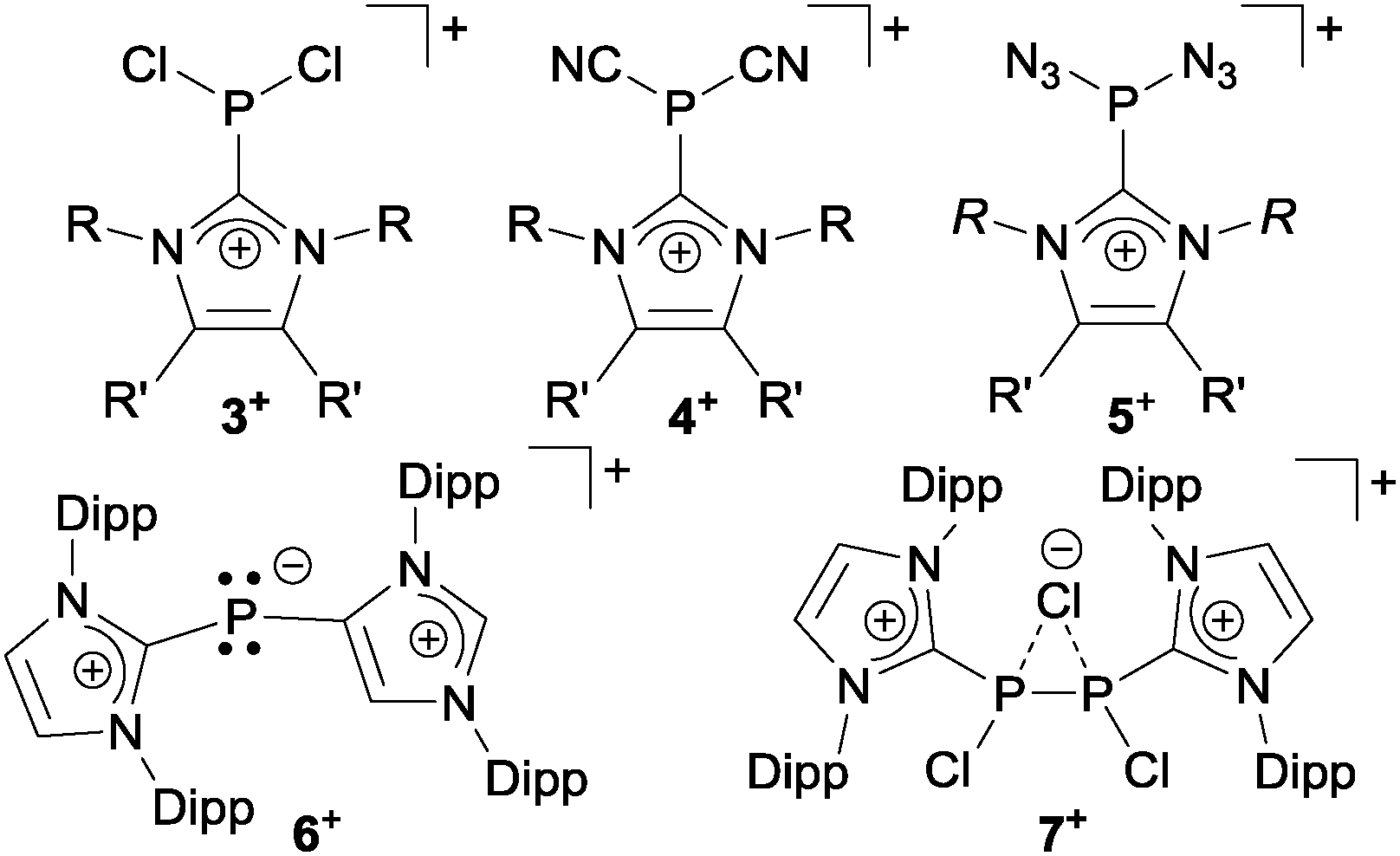
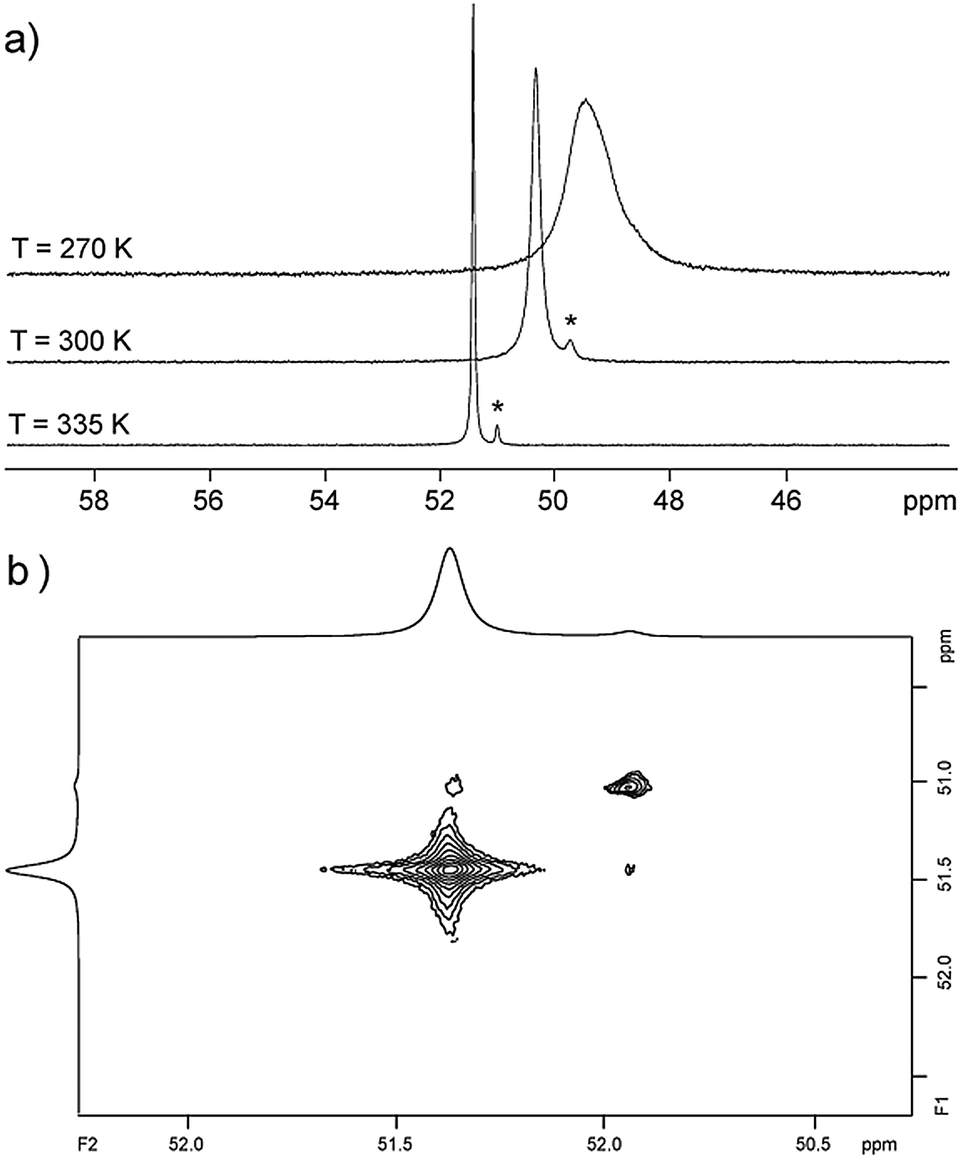
Organophosphorus–sulfur heterocycles with the general constitution (RPS)n (n = 2–4) and phosphorus in the oxidation state +III remain scarce, since their main access is from the reaction of a dichlorophosphane RPCl2 (R = Aryl) and a source of sulfide (S2−; e.g. M2S (M = Li, Na) or (Me3Si)2S).1 The formation of these compounds can formally be viewed as a combination of divalent RP: and S: units to give monomeric phosphorus mono-sulfides such as 1, which then can either yield oligomerization products (Fig. 1; path I) or ylidylphosphorus sulfide 2a (path II).12a represents a rare example of a stable and structurally confirmed monomeric ylidylphosphorus monosulfide. Its stability is rationalized by a high contribution of the zwitterionic resonance formula 2b (path II).2 Aiming at the synthesis of new cationic phosphorus species, we are investigating reactions of imidazoliumyl-substituted P-centered cations such as [L(R,R′)PCl2]+3+ (LR,Me = imidazolium-2-yl, R = Aryl, Alkyl; R′ = H, Me, Cl)3 towards substitution3,4 (e.g.4+,5+), coordination,5 oxidation6 or reduction7 (e.g.6+,7+) and successfully isolated a series of novel cationic derivatives with intriguing bonding motives (Fig. 2). In this contribution we present the results of the attempted preparation of the imidazoliumyl-substituted [L(R,Me)PS]+ cations 8a,b+ (a: R = Me; b: R = iPr) from the reaction of 3a,b+ with (Me3Si)2S and the respective oligomerization to tetra-cations 9a,b4+ which can be isolated as triflate salts (Scheme 1). Compounds 3a,b[OTf] (ref. 3) were reacted with 1 eq. (Me3Si)2S in fluorobenzene for 5 h at ambient temperature, accompanied by the formation of colorless precipitates. After workup, compounds 9a,b[OTf]4 were isolated in excellent yields (>90%; Scheme 1). The 31P{1H} NMR spectra of the dissolved compounds in d3-MeCN display one major resonance next to a minor singlet in the typical region of tri-coordinate phosphorus derivatives (9a4+: δ(P)major/minor = 48.2 ppm/48.0 ppm; 9b4+: δ(P)major/minor = 50.5 ppm/50.3) indicating the oligomerization of the intermediately formed cationic phosphorus monosulfide 8a,b+. However, no evidence for the formation of other ring sizes was found, showing a high selectivity of the oligomerization process. It can be assumed that, under these conditions, the formation of the P4S4 ring is thermodynamically (considering ring strain and steric effects) favored. The variable-temperature (VT) 31P NMR spectra for 9b[OTf]4 are depicted in Fig. 3a, evidencing a dynamic behavior and the presence of two highly symmetric conformational isomers (crown (C4v) vs. boat-chair (Cs) conformer) of the P4S4 ring in solution (Scheme 2). A significant broadening of the two resonances upon cooling is observed. The VT 1H NMR spectra also show additional dynamic behavior of the iPr-groups. We thus confirmed the existence of two conformational isomers independently by 31P EXSY NMR experiments (Fig. 3b), although quantitative accuracy (to determine kinetic processes or exchange rates) was limited by the narrow temperature range in which useful spectra could be measured. The 31P EXSY NMR experiments suggest exchange of two conformers in which one of the sulfur-bridges reversibly changes its relative position, giving either the approximate C4v or Cs symmetric cations 9a,b4+ (Scheme 2). The cross peaks in the 2D spectrum demonstrate the exchange between the two symmetric conformers in solution, consistent with the observation of singlet resonances as expected for an A4 spin-system for both isomers.
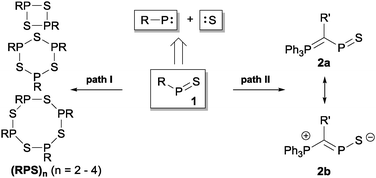 |
| | Fig. 1 Formation of oligomeric phosphorus sulfides (RPS)n (n = 2–4; R = Aryl, R′ = Alkyl; path I) and phosphorus monosulfide 2a,b (path II). | |
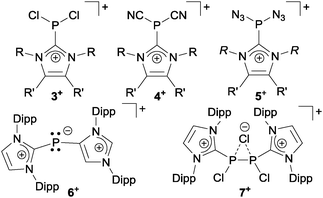 |
| | Fig. 2 Selected P-centered cations 3–7+ featuring imidazoliumyl-substituents (Dipp = 2,6-diisopropylphenyl). | |
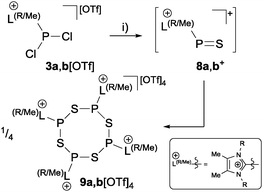 |
| | Scheme 1 Preparation of tetrameric imidazoliumyl-substituted phosphorus–sulfur heterocycles 9a,b[OTf]4 from the intermediately formed cation 8a,b+ (a: R = Me; b: R = iPr); (i) +(Me3Si)2S, C6H5F, rt, −2Me3SiCl. | |
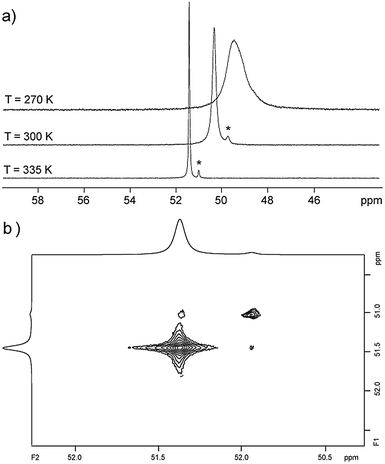 |
| | Fig. 3 (a) VT 31P NMR spectra of 9b[OTf]4 recorded in CD3CN (see ESI†) * indicates minor amounts of the conformational isomer of 9b+; (b) 31P EXSY NMR spectrum of 9b[OTf]4 recorded at 335 K with a mixing time of tm = 0.55 s. | |
 |
| | Scheme 2 Interconversion process of cations 9a,b4+ from the conformational isomer with crown shape (C4v) to the boat-chair (Cs) conformer without considering the imidazoliumyl substituents. | |
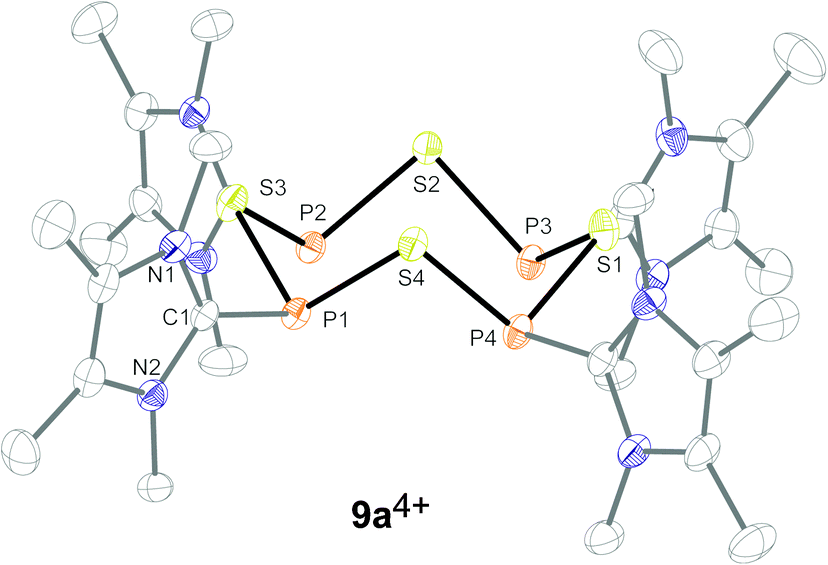
Satisfactory crystal structure analyses could be performed confirming the formation of the suggested eight-membered organophosphorus–sulfur heterocycles (Fig. 4). In the case of compound 9b[OTf]4 the refinement of the X-ray data confirmed the crown-shaped P4S4 moiety, similarly to cation 9a4+, however, due to severe disorder caused by alternating ring orientation full refinement was only possible after treatment by an appropriate disordered model.8 The PIII–S bond lengths (av. 2.129 Å) in 9a4+ are in the typical range for PIII–S single bonds and compare well with those reported by Sheldrick et al. for the neutral derivative (MesPS)4 (av. 2.117 Å; Mes = 2,4,6-trimethylphenyl).1b The pyramidalization of the P atoms, as well as the lengths of the C–P bonds (av. 1.817 Å), together with the internal angle N1–C1–N2 of the imidazoliumyl moieties (9a4+: av. 106.9° vs. ∼101° in NHCs)9 are in agreement with the ability of the imidazoliumyl fragment to (a) delocalize the positive charge, (b) reduce the nucleophilicity of a directly bonded P atom and thus may account for the stability of these cations.5,10,11
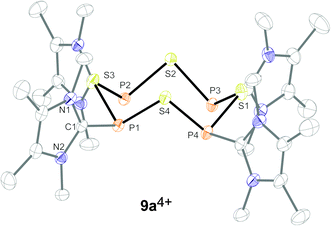 |
| | Fig. 4 Molecular structure of tetracation 9a4+ in 9a[OTf]4. All hydrogen atoms and triflate anions are omitted for clarity. Selected bond lengths in Å and angles in °: P1–S1 2.1306(9), P1–S4 2.1364(9), P2–S1 2.1234(9), P1–C1 1.820(3), S1–P1–S4 101.99(4), S1–P1–C1 99.31(8), S4–P1–C1 101.26(9), N1–C1–N2 101.26(9). | |
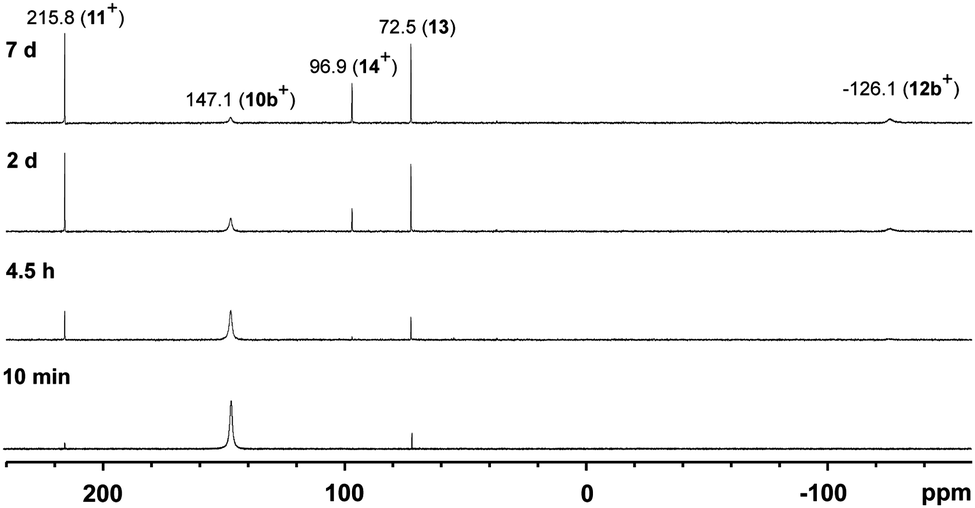
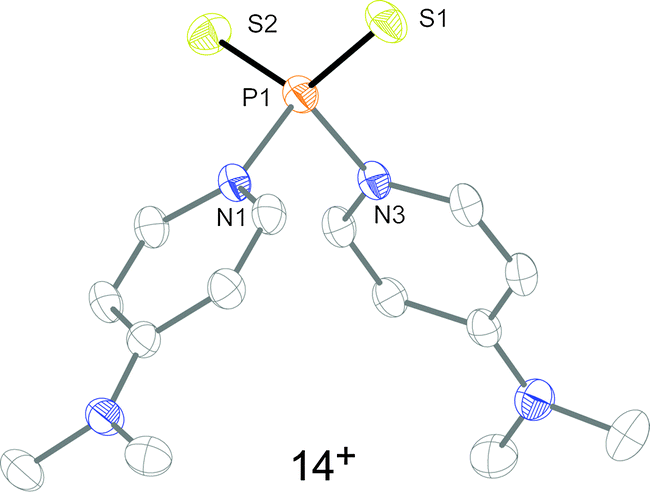
To confirm that the formation of tetracations 9a,b4+ proceeds via monomeric phosphorus mono-sulfides 8a,b+, we reacted 9a,b[OTf]4 with DMAP in MeCN, since this Lewis base has been widely used to stabilize low-coordinate phosphorus species.12 The σ-donor strength of DMAP should be high enough to deoligomerize tetracations 9a,b4+ to cations 10a,b+ which represent DMAP adducts of the elusive cations 8a,b+ (Scheme 3). The reaction of 4 equivalents DMAP in MeCN proceeds comparably clean with 9b[OTf]4 at ambient temperature giving pale-yellow solutions. The 31P{1H} NMR spectra of the reaction mixture of 9b[OTf]4 and DMAP after reaction times of 10 min to 7d are depicted in Fig. 5. After 10 min a broad resonance at δ(P) = 147.1 ppm is observed which splits into 2 singlets at low temperature (δ(P) = 140.3 ppm and δ(P) = 144.4 ppm; 253 K). We believe that the splitting of the resonances results from two rotational isomers caused by a restricted rotation of the iPr groups at low temperature.8,13 From the distinctive chemical shift a dimeric derivative with tetra-coordinate phosphorus atoms (cf. [(Et2N)2PS2][AlCl4]2: δ(P) = 21.0 ppm)14 can be excluded and we thus propose the formation of DMAP adduct 10b+ which is also supported by our NMR investigation.8 We were not able to isolate 10b[OTf], however, we assume that cation 10b+ readily dismutates to cations 11+ (δ(P) = 215.8 ppm) and 12b+ (δ(P) = −126.1 ppm) via an intermolecular scrambling reaction, i.e. an intermolecular exchange of imidazolium-2-yl and sulfur substituents. Related exchange reactions were discussed for the DMAP induced disproportionation of POCl315 and scrambling reactions of imidazoliumyl-substituted [L(Me,Me)PCl2]+ cations.4 PI centered cation 13b+ (δ(P) = −126.1 ppm) was recently reported by Macdonald et al. and unambiguously confirmed by its characteristic chemical shift (cf. lit: δ(P) = −124.2 ppm, CD2Cl2).16 Cations of type 12a+ are also known and reported by Schmidpeter et al. as ylidylphosphorusdisulfide containing a tri-coordinate R–PS2 moiety. For these types of compounds the chemical shift strongly depends on the nature of the supporting substituent R and is typically observed in the range of δ(P) = 170–240 ppm (cf. Ph3PCMe–PS2: δ(P)PS2 = 243.4 ppm, d8-THF).2 In an equilibrium reaction of cation 11+ with the triflate anion the formation of 13 is explained and supported by the pronounced upfield shifted triplet resonance due to the coupling to the ortho-protons of the DMAP ligand (eqn (1); δ(P) = 72.5 ppm, triplet, 3JPH = 9.0 Hz; cf. PyPS2Br: δ(P) = 65.5 ppm, Py = pyridine; d3-MeCN).17 The formation of cation 14+ results either from the equilibrium reaction of 11+ (eqn (2)) or 13 (eqn (3)) with DMAP which is liberated during the dismutation of cation 10b+ (Scheme 3). The 31P NMR spectrum displays a quintet resonance which is indicative for the presence of two DMAP substituents consistent with the C2v symmetry of cation 14+ (δ(P) = 96.9 ppm; cf. [Py2PS2]+: δ(P) = 104.7 ppm, quintet, 3JPH = 9.6 Hz; Py = pyridine; d3-MeCN). Similar equilibria and cations have been observed by Meisel et al. who reported on the pyridine (Py) stabilized phosphorus disulfide [Py2PS2]+ cation.18 Two crystalline polymorphs of 14[OTf] were obtained after layering the reaction mixtures with Et2O (Fig. 6).19 The obtained structural parameters compare well with those reported for the related pyridine cation [Py2PS2]+ by Meisel et al.18
| | | 11[OTf] + DMAP ⇌ 14[OTf] | (2) |
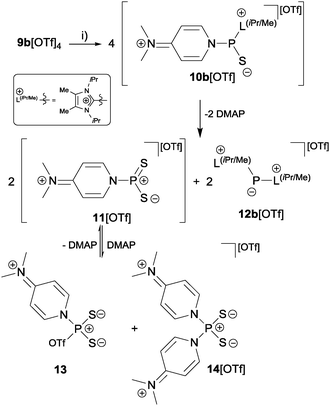 |
| | Scheme 3 Deoligomerization of 9b[OTf]4 with DMAP and proposed intermediates; (i) +4 DMAP, MeCN, rt. | |
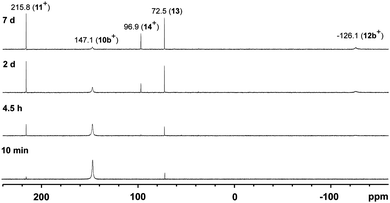 |
| | Fig. 5
31P{1H} NMR spectra of the 1:4 reaction of 9b[OTf]4 and DMAP in CD3CN showing the scrambling reaction to cations 11+, 12b+, 14+ and adduct 13. | |
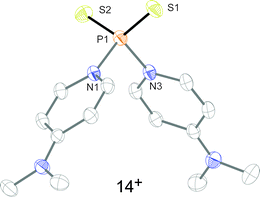 |
| | Fig. 6 Molecular structure of 14+ in 14[OTf]. All hydrogen atoms and the triflate anion are omitted for clarity. Selected bond lengths in Å and angles in °: P1–S1 1.9309(8), P1–S2 1.9321(8), P1–N1 1.782(2), P1–N2 1.798(2), S1–P1–S2 124.09(4), N1–P1–N2 96.77(8), S–P1–N av. 108.14. | |
In order to confirm our findings and support the suggested dismutation, quantum chemical calculations have been performed from the reaction of 9a4+ with 4 eq. DMAP to derive geometry and Gibbs free energy of the involved species as well as the suggested reaction intermediate 10a+.20 The density functional theory (DFT) hybrid model B3LYP21 was used in combination with Grimme’s atom-pairwise dispersion correction (D3).22Fig. 7 illustrates the Gibbs free energy ΔG of educts (9a4+, DMAP), products (12b+, 14+) and the proposed intermediate (10a+) calculated at the B3LYP-D3/def-SVP level of theory. The whole reaction pathway is thermodynamically favored (ΔG = −284.4 kcal mol−1). The proposed reaction intermediate 10a+ is energetically less favorable by 22 kcal mol−1 compared to the final products, but 262 kcal mol−1 lower in energy than the educt molecules (ΔG = −262.4 kcal mol−1). This observation is perfectly in line with the assumption of 10a+ being the main reaction intermediate.
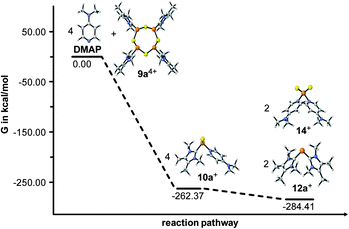 |
| | Fig. 7 Gibbs free energy in kcal mol−1 of the observed and proposed phosphorus species. | |
In summary, we reported on the oligomerization reaction of cationic phosphorus monosulfides 8a,b+ which were formed in situ from the reaction of [L(R,Me)PCl2]+ cations 3a,b+ (LR,Me = imidazolium-2-yl a: R = Me; b: R = iPr) and (Me3Si)2S. The obtained tetra-cationic, eight-membered phosphorus-sulfur heterocycles [L(R,Me)PS]44+9a,b4+ primarily exist as crown conformers similar to cyclo-S8, however, the boat-chair (Cs) conformers of the P4S4 rings also exist in solution according to 2D-EXSY 31P NMR experiments. Cations 9a,b4+ can be deoligomerized by DMAP to yield adducts 10a,b+ of the elusive cations 8a,b+. Subsequent dismutation of 10a,b+ gives cations [L(R,Me)2P]+ (12a,b+) and [(DMAP)2PS2]+ (14+) as final products. The suggested reaction pathway was supported by DFT calculations.
This work was supported by the Fonds der Chemischen Industrie (FCI, Kekulé scholarship for F. H.), the German Science Foundation (DFG, WE 4621/2-1) and the ERC (SynPhos 307616). Financial support of M. K. by a POSTUP2 grant (European social fund in the Czech Republic, European Union, Czech ministry of education and sports, operational program for the advancement of the competitive capacity) is gratefully acknowledged (CZ.1.07/2.3.00/30.0041). We thank the Center for Information Services and High Performance Computing (ZIH) for the generous allocation of computation time.
References
-
(a)
T. Chivers and I. Manners, Inorganic Rings and Polymers of the p-Block Elements, RSC Publishing, 2009 Search PubMed;
(b) C. Lensch and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1984, 2855–2857 RSC;
(c) B. Cetinkaya, P. B. Hitchcock, M. F. Lappert, A. J. Thorne and H. Goldwhite, J. Chem. Soc., Chem. Commun., 1982, 691–693 RSC;
(d) M. Baudler, D. Koch, T. Vakratsas, E. Tolls and K. Kipker, Z. Anorg. Allg. Chem., 1975, 413, 239–251 CrossRef CAS.
- G. Jochem, K. Karaghiosoff, S. Plank, S. Dick and A. Schmidpeter, Chem. Ber., 1995, 128, 1207–1219 CrossRef CAS.
- J. J. Weigand, K.-O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321–16323 CrossRef CAS PubMed.
- F. D. Henne, A. T. Dickschat, F. Hennersdorf, K. O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849–6861 CrossRef CAS PubMed.
- K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 14204–14208 CrossRef CAS PubMed.
- K. Schwedtmann, S. Schulz, F. Hennersdorf, T. Strassner, E. Dmitrieva and J. J. Weigand, Angew. Chem., Int. Ed., 2015, 54, 11054–11058 CrossRef CAS PubMed.
- F. D. Henne, E.-M. Schnöckelborg, K.-O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674–6680 CrossRef CAS.
- See ESI†.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078–11082 CrossRef CAS PubMed.
- D. J. D. Wilson, S. A. Couchman and J. L. Dutton, Inorg. Chem., 2012, 51, 7657–7668 CrossRef CAS PubMed.
-
(a) R. J. Davidson, J. J. Weigand, N. Burford, T. S. Cameron, A. Decken and U. Werner-Zwanziger, Chem. Commun., 2007, 4671–4673 RSC;
(b) J. J. Weigand, N. Burford, A. Decken and A. Schulz, Eur. J. Inorg. Chem., 2007, 4868–4872 CrossRef CAS;
(c) M. Blättner, M. Nieger, A. Ruban, W. W. Schoeller and E. Niecke, Angew. Chem., Int. Ed., 2000, 39, 2768–2771 CrossRef;
(d) E. Rivard, K. Huynh, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2004, 126, 2286–2287 CrossRef CAS PubMed;
(e) N. Burford, H. A. Spinney, M. J. Ferguson and R. McDonald, Chem. Commun., 2004, 2696–2697 RSC;
(f) J. J. Weigand, N. Burford, D. Mahnke and A. Decken, Inorg. Chem., 2007, 46, 7689–7691 CrossRef CAS PubMed.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1970, 9, 219–235 CrossRef CAS.
- N. Burford, R. E. v. H. Spence and R. D. Rogers, J. Chem. Soc., Dalton Trans., 1990, 3611–3619 RSC.
- P. Rovnaník, L. Kapička, J. Taraba and M. Černík, Inorg. Chem., 2004, 43, 2435–2442 CrossRef PubMed.
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, Chem. Commun., 2005, 1965–1967 RSC.
- The low symmetry of 13 and the presence of the triflate anion encourage an effective quadrupole relaxation and prevents the observation of the 1J(31P, 14N) coupling. The same accounts for the more symmetric cation 14+.
- M. Meisel, P. Lönnecke, A.-R. Grimmer and D. Wulff-Molder, Angew. Chem., Int. Ed., 1997, 36, 1869–1870 CrossRef CAS.
- Similarly to the highly reactive intermediate 10b+, the tetramethyl substituted derivative 10a+ was also observed in the reaction of 9a[OTf]4 and 4 eq. DMAP at δ(P) = 144.7 ppm. From the reaction spectra, chemical shifts as δ(P) = 216.6 ppm, 97.3 ppm, 70.9 ppm and −113.2 ppm can be assigned to 11+, 14+, 13 and 12a+, respectively. The reaction NMR is depicted in the supporting information; Details of the two polymorphs are given in the ESI†.
- Cation 9a4+ was choosen as model system.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS;
(c) P. J. Stephens, E. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS;
(d) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
-
(a) T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 4398–4401 RSC;
(b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For detailed experimental procedures and characterization details of new compounds, NMR spectra, crystallographic details and computational data. CCDC 1424341–1424343 and 1426199. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc08182c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence