
Rhenium(V)–oxo corrolazines: isolating redox-active ligand reactivity†
Jan Paulo T.
Zaragoza
,
Maxime A.
Siegler
and
David P.
Goldberg
*
Department of Chemistry, The Johns Hopkins University, 3400 N. Charles Street, Baltimore, MD 21218, USA. E-mail: dpg@jhu.edu
First published on 28th October 2015
Abstract
The synthesis of the first example of a third-row metallocorrolazine characterized by single crystal X-ray diffraction is reported. This ReV(O) porphyrinoid complex shows an exclusively ligand-based reactivity with strong acids and oxidizing agents. The one-electron oxidized π-radical-cation complex is capable of H-atom abstraction.
The synthesis of rhenium complexes is of interest due to their potential use as catalysts for oxygen-transfer,1 X–H (X = Si, B, P and H) bond activations,2 and in CO2 photoreduction reactions.3 In addition, certain isotopes of rhenium are used in medical imaging, diagnostics, and therapeutics.4 Although not found in natural heme systems, a few examples of rhenium porphyrins have been synthesized, including Buchler's first report of high-valent ReV(O)(X) porphyrins.5 A decade later, the ring-contracted high-valent metal–oxo ReV(O)(TCF3C) (TCF3C = 5,10,15-tris(trifluoromethyl)corrole) was serendipitously prepared in an attempted porphyrin metallation reaction, providing the only example of a rhenium corrole.6
High-valent metal–oxo porphyrinoid complexes are of significant interest because of their role in synthetic and biological oxidation catalysis.7 Our group described the characterization of high-valent MnV(O) and CrV(O) corrolazine (Cz) complexes by single crystal X-ray diffraction (XRD), and showed that these complexes exhibited dramatically different abilities to abstract hydrogen atoms from X–H (X = O, C) bonds.8 In earlier efforts, it was also shown that the MnV(O)(Cz) complex could be chemically oxidized to give MnV(O)(Cz˙+), the first example of an MnV(O) π-radical cation complex. The latter complex showed greatly enhanced O-atom transfer (OAT) reactivity compared to its neutral precursor.9 Lewis/Brønsted acids (LA) were also shown to have a profound influence on MnV(O)(Cz), stabilizing the valence tautomer MnIV(O-LA)(Cz˙+) and providing a rare example of a chemically driven, reversible valence tautomerization. The reactivity of MnIV(O-LA)(Cz˙+) was significantly different from MnV(O)(Cz), with the metal–oxo unit and redox-active Cz ligand functioning together to carry out H-atom transfer (HAT) and OAT reactions.10 Recently, much attention has been given to redox-active ligands and how they operate in conjunction with a metal ion to mediate various chemical transformations.11
Herein we report the synthesis and characterization by XRD of an ReV(O)(Cz) complex, the first metallocorrolazine containing a third-row metal ion, and a rare example of a structurally characterized rhenium porphyrinoid complex. This complex was prepared as an isoelectronic analog of MnV(O)(Cz). Addition of strong Brønsted acids does not lead to stabilization of a valence tautomer in this case, but rather the reversible protonation of a remote site on the ligand. It is shown that the redox-active Cz ring can participate in electron-transfer and H-atom transfer reactions without the involvement of the metal–oxo unit. The H-atom transfer reactivity for the one-electron-oxidized ReV(O)(Cz˙+) is also shown to be strongly dependent on the nature of the external one-electron oxidant through a surprising observation of “zero-order” kinetics.
The synthesis of the ReV(O) corrolazine complex was accomplished by metallation of octakis(p-tert-butylphenyl)corrolazine (TBP8CzH3) (1) with excess ReCl5 in refluxing decalin (Scheme 1). The product was purified by chromatography and gave ReV(O)(TBP8Cz) (2) as a dark green solid (99% yield). The UV-vis spectrum of 2 exhibits a Soret band at 460 nm, which is the most red-shifted for any metallocorrolazine, and a Q-band at 670 nm. The 1H NMR spectrum of this complex is diamagnetic, consistent with a low-spin ReV ion. Eight doublets appear between 8.44–7.26 ppm, which can be assigned to the peripheral para-substituted phenyl substituents (Fig. S1, ESI†). An LDI-TOF mass spectrum gives M+ = 1557.38 m/z, in good agreement with the Re(O)(TBP8Cz) formulation.
 | ||
| Scheme 1 Synthesis of ReV(O)(TBP8Cz) (2). | ||
Confirmation of the structure of 2 comes from X-ray crystallography. Single crystals of 2 were obtained by vapor diffusion of acetonitrile into toluene. The molecular structure is shown in Fig. 1, revealing a 5-coordinate Re center, with a terminal oxo ligand at a Re–O distance of 1.682(5) Å. This distance is consistent with an Re–O triple bond.1e,12 The Re ion is displaced by ca. 0.74 Å from the plane defined by the four Npyrrole atoms. For comparison, ReV(O)(TCF3C) exhibits an Re–O bond length of 1.662(2) Å and an out-of-plane distance of 0.701 Å.6 The Re–O distance in 2 is significantly elongated compared to MnV(O) (1.5455(18) Å) and CrV(O) (1.553(2) Å) corrolazines,8a as expected for a third-row metal ion. The Re ion is also much further out of plane, by at least 0.2 Å, compared to either MnV or CrV complexes.
 | ||
| Fig. 1 Displacement ellipsoid plot (30% probability level) of ReV(O)(TBP8Cz) (2). Selected bond distances (Å): Re–O, 1.682(5), Re–Nave, 1.976, Re–Nplane, 0.739. (a) Top view; (b) side view (peripheral aryl groups were omitted). In both cases, the disorder and H-atoms were removed for clarity. | ||
We previously found that MnV(O)(TBP8Cz) reacts with Lewis and Brønsted acids at 23 °C to give MnIV(O-LA)(TBP8Cz˙+) (LA = H+, ZnII, B(C6F5)3), in which the paramagnetic MnIV valence tautomer is stabilized over the diamagnetic, low-spin MnV species through a proposed weakening of the Mn–O π-bonding from coordination of LA to the terminal oxo group.10 In contrast, the addition of the Brønsted acid [H(OEt2)2][B(C6F5)4] (HBArF) to MnV(O)(TBP8Cz) at low temperature (−60 °C) led to protonation of a remote meso-nitrogen atom on the Cz ring and retention of the low-spin MnV configuration. Protonation of the meso-N atoms in MnIII corrolazines was also definitively confirmed by XRD.13 To determine the reactivity of the isoelectronic ReV(O) analog toward Brønsted acids, the reaction of 2 with HBArF was examined. Addition of one equiv. of HBArF to 2 in CH2Cl2 causes clear shifts in the UV-vis spectrum (Fig. 2a), although these changes are not consistent with formation of a Cz π-radical-cation. The final spectrum is more typical of protonation at the remote site on the Cz ligand.13 Spectral titration with HBArF supports a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry (inset, Fig. 2a) to form [ReV(O)(TBP8Cz)(H)]+ (3).
:1 binding stoichiometry (inset, Fig. 2a) to form [ReV(O)(TBP8Cz)(H)]+ (3).
 | ||
| Fig. 2 (a) UV-vis spectral changes upon addition of HBArF (0–2 equiv.) to 2 in CH2Cl2. Inset: Spectral titration at 723 nm showing maximal formation of product at 1 equiv. HBArF. (b) Comparison of 1H NMR spectra (400 MHz) near the aryl region of 2 (black) and 3 (red) in CD2Cl2 at 23 °C. | ||
Characterization of the monoprotonated complex 3 was performed by 1D (Fig. 2b) and 2D (Fig. S3 and S4, ESI†) NMR spectroscopy. The 1H NMR spectrum of 3 is diamagnetic, confirming that the paramagnetic valence tautomer ReIV(O)(Cz˙+) is not observed. Spectra for 2 and 3 are shown in Fig. 2b (see Fig. S2, ESI†). A new peak is observed at 13.45 ppm for the protonated complex, and integrates to 1H. This peak can be assigned to protonation of a meso-N of the corrolazine ring, shifted downfield by the ring current effect. For comparison, the meso C–H proton of Zn octaethylporphyrin appears at ca. 10 ppm.14 The aromatic pattern is complex, consistent with addition of H+ to one of the meso-N atoms that does not lie on the mirror plane bisecting the pyrrole–pyrrole linkage. The NMR spectrum for 2 at 23 °C is similar to what was observed for protonation of MnV(O)(TBP8Cz) at −60 °C.13
The IR spectrum of 2 shows an intense peak at 997 cm−1 (Fig. 3a), in the region expected for the stretching frequency of an ReV–O triple bond. However, an intense peak at ∼997 cm−1 has also been observed in several other metallocorrolazines, and may arise from Cz vibrational modes.15 To conclusively identify the Re–O stretch, we synthesized the isotopically labeled ReV(18O)(TBP8Cz) (2–18O) by addition of excess H218O (80 equiv.) to the Re metallation reaction involving TBP8CzH3 (1) and ReCl5 in decalin. This method yielded the ReV(O) complex with >99% 18O isotopic incorporation as seen by LDI-MS (Fig. S5, ESI†), and indicates a mechanism of oxygen incorporation that involves hydrolysis of an ReV(Cl)2(TBP8Cz) precursor.5 The appearance of a new band upon 18O substitution at 945 cm−1 is accompanied by a slight decrease in intensity of the band at 997 cm−1 (Fig. 3). These spectral changes are consistent with the v(Re16O) mode overlapping with the intense peak at 997 cm−1. The predicted isotopic shift for 18O substitution in an isolated Re–O diatomic oscillator is 52 cm−1, in excellent agreement with the predicted value of v(Re16O) = 997 cm−1. The Re–O stretch is close to that observed for ReV(O) corrole (994 cm−1),6 but higher than most ReV(O) porphyrins, phthalocyanines5,12b or N-confused porphyrin.12a
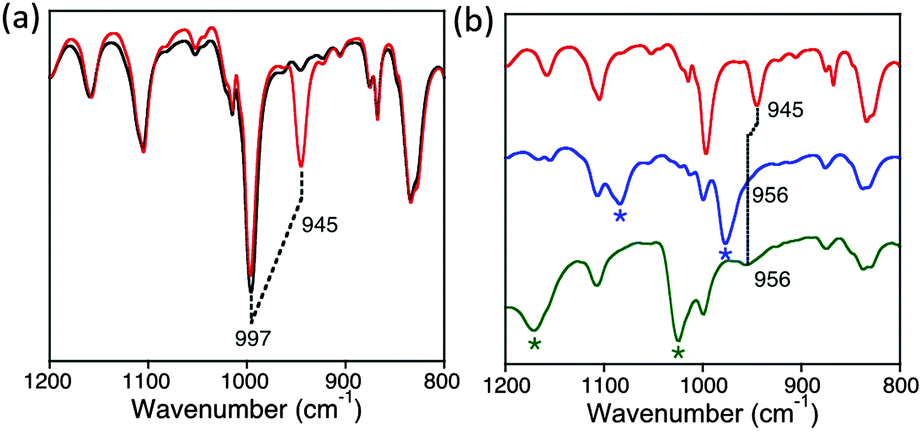 | ||
| Fig. 3 ATR-IR spectra (800–1200 cm−1) of (a) 2–16O (black) and 2–18O (red), (b) 2–18O (red), (3–18O)(BArF−) (blue), and (3–18O)(OTf−) (green). Asterisk (*) = peaks associated with OTf− or BArF− counterions. | ||
The effect of protonation on the Re–O bond can be probed by IR spectroscopy (Fig. 3b). Upon protonation of 2 with HBArF, a sharp peak at 3280 cm−1 appears, consistent with the N–H stretch of a meso-NH group (Fig. S6, ESI†). At the same time, the v(Re18O) peak at 945 cm−1 disappears, and only a broad band associated with BArF− is present at 975 cm−1 without any clear evidence for a new Re–18O stretch. However, replacing HBArF with HOTf causes the broad peak at 975 cm−1 to disappear, revealing a new peak for v(Re18O) = 956 cm−1 (Fig. 3b). These data indicate that an 11 cm−1 blue shift occurs for the metal–oxo stretch, implicating a strengthening of the metal–oxo bond upon protonation of a remote site on the ligand.
Density functional theory (DFT) calculations were performed on complexes 2 and 3 to support structural and spectroscopic assignments. Geometry optimizations were performed at the PBE0/LANL2TZ/6-31G** level of theory, beginning with the crystal structure coordinates for 2. The peripheral aryl substituents were replaced by H atoms to facilitate the calculations. The optimized geometry for 2 matched well with the experimentally determined crystal structure. Geometry optimizations for protonated 2 were performed with the H+ attached at either the meso-N or the terminal oxo ligand. These calculations showed that the O–H tautomer is +29 kcal mol−1 higher in energy than the N–H tautomer 3 (Fig. S7 and Table S2, ESI†), and indicate that the meso-N is the preferred site of protonation. Frequency calculations give v(Re–O) = 1055 cm−1 for 2 and 1064 cm−1 for 3, which are both higher than the corresponding experimental values. DFT is known to overestimate vibrational frequencies due to systematic errors.16 However, the difference between the calculated v(Re–O) values for 2 and 3 (Δv(ReO) = 9 cm−1) is in excellent agreement with experiment (11 cm−1).
The MnV(O)(TBP8Cz) complex reacts rapidly with phosphine derivatives through an O-atom transfer mechanism.10c Attempts to react 2 with the phosphine derivatives PPh3, PMe3, or PEt3 in CH2Cl2 at 23 °C showed no reaction even over prolonged reaction times (5 d). Similarly, H-atom donors such as TEMPOH or 2,4-di-tert-butylphenol were unreactive toward 2, although the isoelectronic MnV(O) complex readily abstracts H-atoms from both of these substrates.8b Reduction of 2 with strong one-electron donors such as cobaltocene (Ered = −1.33 V vs. SCE)17 was also unsuccessful. We speculated that 3, with an additional full unit of positive charge, might show enhanced oxidative reactivity compared to 2, but reactions with PR3 or ArOH substrates led only to deprotonation and recovery of 2. Both the neutral and monoprotonated ReV(O) complexes appear inert to either H-atom or O-atom transfer reactions, suggesting a greatly enhanced stability for ReV(O) compared to MnV(O) in the corrolazine environment.
To gain further insights into the reactivity of 2 and 3, cyclic voltammetric measurements were performed (Fig. 4). Complex 2 exhibits a single reversible wave at 1.04 V vs. SCE, which is close to an assigned Cz ring oxidation for MnV(O)(TBP8Cz),9 as well as other metallocorrolazines.15a However, there are no other redox events observed for 2, in contrast to the MnV(O) complex, which exhibits an MnV/MnIV couple near −0.05 V.15 Upon protonation of 2, the ring oxidation wave disappears, leaving only an irreversible reduction at −0.17 V as seen in the CV for 3. Thus for 2 and 3 there are no clearly accessible metal-based redox couples, consistent with a lack of HAT and OAT reactivity. However, the reversible oxidation seen for 2 at 1.04 V suggested that a one-electron oxidized ReV(O)(Cz˙+) π-radical-cation complex might be accessible by chemical oxidation.9,18
 | ||
| Fig. 4 (a) Cyclic voltammograms of 2 (black line) and 3 (red line) in CH2Cl2, with 0.1 M TBAPF6 supporting electrolyte, 150 mV s−1. (b) UV-vis spectral changes of 2 observed upon addition of [Ar′3N˙+][SbCl6−] (0–1 equiv.) in CH2Cl2. Inset: X-band EPR of ReV(O)(TBP8Cz˙+) (4) (3 mM) at 298 K in CH2Cl2. | ||
Reaction of ReV(O)(TBP8Cz) with [Ar′3N˙+][SbCl6−] (Ar′ = 4-BrC6H4, Ered = 1.16 V vs. SCE)17 resulted in isosbestic conversion to a new species with a broadened Soret peak at 445 nm and a relatively weak, red-shifted band at 760 nm (Fig. 4b). These features are characteristic of a corrole,19 porphyrazine,20 and corrolazine π-radical.9,10,18 Spectral titration showed one equiv. of oxidant was required for complete formation of the π-radical cation complex (Fig. S8, ESI†). EPR spectroscopy revealed a sharp singlet at g = 2.00, similar to MnV(O) and MnV(imido) Cz–π-radical cation species.9,18 Quantitation of the EPR signal showed a 94% yield of the oxidized product. Attempts to isolate this product as a solid were unsuccessful, but taken together the data show that 2 can be oxidized in situ by Ar′3N˙+ to give the monocationic complex ReV(O)(TBP8Cz˙+) (4).
In earlier work, dramatic enhancements in OAT reactivity were seen for MnV(O)(TBP8Cz˙+) compared to its neutral precursor,9 but the ReV analog 4 remained unreactive to OAT. Addition of phosphine derivatives to 4 in CH2Cl2 led only to 1e− reduction, restoring 2 with no evidence of oxo-transfer. However, addition of the H-atom donor 9,10-dihydroanthracene (DHA) to 4 resulted in quantitative conversion to monoprotonated 3. Analysis by GC-FID showed anthracene was produced in 90% yield, confirming that formal HAT occurs via the net reaction in Scheme 2. Although these observations implied that 4 was abstracting an H-atom from DHA, UV-vis time course experiments under pseudo-first-order conditions (excess DHA) showed a surprising linear dependence for the decay of 4 and production of 3 (Fig. S10, ESI†). These data were indicative of a reaction zero-order in 4. The zero-order kinetics can be explained by a mechanism involving back electron-transfer between Ar′3N and 4, establishing the equilibrium shown in eqn (1). The redox potentials for 2 and [Ar′3N˙+][SbCl6−] indicate a relatively small KET = 107. The minor Ar′3N˙+ species can then oxidize DHA to give anthracene and Ar′3N in the rate-determining step (eqn (2)), while the released H+ binds to 2 in preference to Ar′3N (pKa ≤ −4).21a Independent experiments confirm that [Ar′3N˙+][SbCl6−] oxidizes DHA to anthracene (98%) relatively rapidly in a second-order process (k1 = 0.126(9) M−1 s−1) (Fig. S11, ESI†).
| ReV(O)(Cz) + Ar′3N˙+ ⇌ ReV(O)(Cz˙+) + Ar′3N (KET) | (1) |
| Ar′3N˙+ + 0.5 DHA → Ar′3N + 0.5 anthracene + H+ (k1) | (2) |
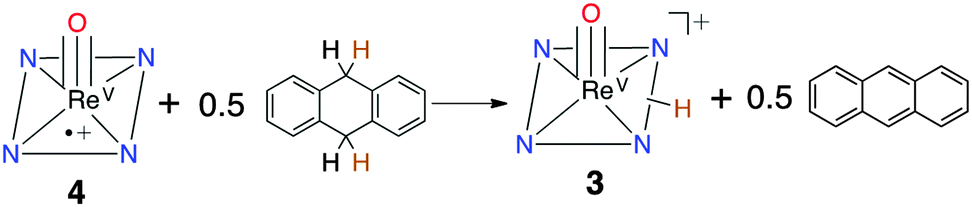 | ||
| Scheme 2 Reaction of 4 with 9,10-DHA to form 3 and anthracene. | ||
Replacement of [Ar′3N˙+][SbCl6−] with the more powerful oxidant CeIV(NH4)2(NO3)6 (CAN, Ered = +1.33 V)17 led to a distinct change in mechanism. Oxidation of 2 with CAN in CH2Cl2/CH3CN (100:1 v/v) gives a UV-vis spectrum similar to 4, and addition of excess DHA results in an exponential decay of 4 with the concomitant formation of 2 (eqn (3)) (Fig. S12, ESI†). These data are consistent with a pseudo-first-order process. The use of the stronger oxidant CAN greatly disfavors back electron-transfer. In this case, the rate-determining step involves the direct reaction of 4 with DHA, and variation of [DHA] leads to a k2 = 6.3(4) × 10−2 M−1 s−1. This reaction likely occurs through an HAT mechanism between DHA and 4 to give 3, which is then rapidly deprotonated by the NO3− counterion21b to give 2. Control experiments show that 3 is rapidly deprotonated by Bu4N+NO3− (Fig. S14, ESI†).
 | (3) |
We have reported the synthesis and XRD characterization of the first third-row metallocorrolazine. This ReV(O) complex is strikingly inert to both H-atom and O-atom transfer reactions, in contrast to its isoelectronic MnV(O) analog. Protonation of 2 gives a cationic ReV(O) complex and no evidence of valence tautomerism, supporting the conclusion that protonation occurs exclusively on the meso-N atom and not on the oxo ligand. Taking advantage of the inertness of the Re–O group, we have provided the first insights into the reactivity of a porphyrinoid π-radical-cation completely decoupled from its high-valent metal–oxo core. This Cz π-radical-cation, which contains a weakly basic meso-N site, appears to be competent to abstract H-atoms from relatively weak C–H substrates. These observations suggest that porphyrin π-radical cations, including those found in heme enzyme metal–oxo intermediates, may have as yet unidentified roles to play in oxidative reactivity.
The authors acknowledge research support by the NIH (Grant GM101153 to D. P. G.). The Kirin cluster at JHU KSAS is thanked for CPU time to J. P. Z. We also thank J. Tang and A. Majumdar (JHU) for NMR assistance and T. Yang and A. Confer for helpful discussions.
Notes and references
- (a) K. P. Gable and E. C. Brown, J. Am. Chem. Soc., 2003, 125, 11018–11026 CrossRef CAS PubMed; (b) M. M. Abu-Omar, Chem. Commun., 2003, 2102–2111 RSC; (c) J. L. Smeltz, C. P. Lilly, P. D. Boyle and E. A. Ison, J. Am. Chem. Soc., 2013, 135, 9433–9441 CrossRef CAS PubMed; (d) S. Das and A. Chakravorty, Eur. J. Inorg. Chem., 2006, 2285–2291 CrossRef CAS; (e) T. Yamamoto, M. Toganoh and H. Furuta, Dalton Trans., 2012, 41, 9154–9157 RSC.
- (a) S. C. Sousa, I. Cabrita and A. C. Fernandes, Chem. Soc. Rev., 2012, 41, 5641–5653 RSC; (b) E. A. Ison, E. R. Trivedi, R. A. Corbin and M. M. Abu-Omar, J. Am. Chem. Soc., 2005, 127, 15374–15375 CrossRef CAS PubMed.
- (a) J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC; (b) C. D. Windle, M. V. Campian, A. K. Duhme-Klair, E. A. Gibson, R. N. Perutz and J. Schneider, Chem. Commun., 2012, 48, 8189–8191 RSC; (c) C. D. Windle, M. W. George, R. N. Perutz, P. A. Summers, X. Z. Sun and A. C. Whitwood, Chem. Sci., 2015 10.1039/C5SC02099A.
- (a) E. John, M. L. Thakur, J. DeFulvio, M. R. McDevitt and I. Damjanov, J. Nucl. Med., 1993, 34, 260–267 CAS; (b) Z.-Y. Jia, H.-F. Deng, M.-F. Pu and S.-Z. Luo, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 734–742 CrossRef CAS PubMed.
- J. W. Buchler and S. B. Kruppa, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 518–530 CAS.
- M. Kin Tse, Z. Zhang and K. S. Chan, Chem. Commun., 1998, 1199–1200 Search PubMed.
- (a) Z. Gross, JBIC, J. Biol. Inorg. Chem., 2001, 6, 733–738 CrossRef CAS PubMed; (b) H. M. Neu, R. A. Baglia and D. P. Goldberg, Acc. Chem. Res., 2015, 48, 2754–2764 CrossRef CAS PubMed; (c) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed; (d) M. T. Green, Curr. Opin. Chem. Biol., 2009, 13, 84–88 CrossRef CAS PubMed.
- (a) R. A. Baglia, K. A. Prokop-Prigge, H. M. Neu, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 10874–10877 CrossRef CAS PubMed; (b) D. E. Lansky and D. P. Goldberg, Inorg. Chem., 2006, 45, 5119–5125 CrossRef CAS PubMed.
- K. A. Prokop, H. M. Neu, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2011, 133, 15874–15877 CrossRef CAS PubMed.
- (a) P. Leeladee, R. A. Baglia, K. A. Prokop, R. Latifi, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 10397–10400 CrossRef CAS PubMed; (b) R. A. Baglia, M. Dürr, I. Ivanović-Burmazović and D. P. Goldberg, Inorg. Chem., 2014, 53, 5893–5895 CrossRef CAS PubMed; (c) J. P. T. Zaragoza, R. A. Baglia, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 6531–6540 CrossRef CAS PubMed.
- (a) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC; (b) P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed.
- (a) T. Yamamoto, M. Toganoh, S. Mori, H. Uno and H. Furuta, Chem. Sci., 2012, 3, 3241–3248 RSC; (b) M. Göldner, L. Galich, U. Cornelisse and H. Homborg, Z. Anorg. Allg. Chem., 2000, 626, 985–995 CrossRef.
- H. M. Neu, J. Jung, R. A. Baglia, M. A. Siegler, K. Ohkubo, S. Fukuzumi and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 4614–4617 CrossRef CAS PubMed.
- R. J. Abraham, B. Evans and K. M. Smith, Tetrahedron, 1978, 34, 1213–1220 CrossRef CAS.
- (a) A. J. McGown, Y. M. Badiei, P. Leeladee, K. A. Prokop, S. DeBeer and D. P. Goldberg, in The Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, New Jersey, 2011, ch. 66, vol. 14, pp. 525–600 Search PubMed; (b) D. E. Lansky, B. Mandimutsira, B. Ramdhanie, M. Clausén, J. Penner-Hahn, S. A. Zvyagin, J. Telser, J. Krzystek, R. Zhan, Z. Ou, K. M. Kadish, L. Zakharov, A. L. Rheingold and D. P. Goldberg, Inorg. Chem., 2005, 44, 4485–4498 CrossRef CAS PubMed.
- T. R. Cundari and P. D. Raby, J. Phys. Chem. A, 1997, 101, 5783–5788 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- D. E. Lansky, J. R. Kosack, A. A. Narducci Sarjeant and D. P. Goldberg, Inorg. Chem., 2006, 45, 8477–8479 CrossRef CAS PubMed.
- P. Schweyen, K. Brandhorst, R. Wicht, B. Wolfram and M. Bröring, Angew. Chem., Int. Ed., 2015, 54, 8213–8216 CrossRef CAS PubMed.
- T. Yoshida, W. Zhou, T. Furuyama, D. B. Leznoff and N. Kobayashi, J. Am. Chem. Soc., 2015, 137, 9258–9261 CrossRef CAS PubMed.
- (a) E. M. Arnett, R. P. Quirk and J. J. Burke, J. Am. Chem. Soc., 1970, 92, 1260–1266 CrossRef CAS; (b) O. W. Kolling, Trans. Kans. Acad. Sci., 1965, 68, 575–581 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic, and computational details. CCDC 1426263 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07956j |
| This journal is © The Royal Society of Chemistry 2016 |