
Expanding the light absorption of poly(3-hexylthiophene) by end-functionalization with π-extended porphyrins†
Michèle
Chevrier
ab,
Sébastien
Richeter
a,
Olivier
Coulembier
b,
Mathieu
Surin
c,
Ahmad
Mehdi
a,
Roberto
Lazzaroni
c,
Rachel C.
Evans
de,
Philippe
Dubois
b and
Sébastien
Clément
*a
aInstitut Charles Gerhardt, Université de Montpellier, Place Eugène Bataillon, 34095 Montpellier Cedex 05, France. E-mail: sebastien.clement1@umontpellier.fr; Tel: +33 467143971
bService des Matériaux Polymères et Composites (SMPC), Centre d'Innovation et de Recherche en Matériaux et Polymères (CIRMAP), Université de Mons, 20 Place du Parc, 7000 Mons, Belgium
cLaboratory for Chemistry of Novel Materials, CIRMAP, University of Mons UMONS, Place du Parc 20, 7000 Mons, Belgium
dSchool of Chemistry, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
eCentre for Research on Adaptive Nanostructures and Nanodevices (CRANN), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
First published on 22nd October 2015
Abstract
Poly(3-hexylthiophene)s end-functionalized with π-extended porphyrins have been synthesized in a one-pot procedure. The polymers show a broad absorption profile extending to 700 nm and a fibrillar microstructure, which can be tuned through judicious selection of the porphyrin molar ratio.
The development of new conjugated polymers is a major route to improve the power conversion efficiency (PCE) of bulk heterojunction (BHJ)-type polymer solar cells (PSCs), making them one of the most promising candidates as renewable energy sources.1 In this context, considerable attention has been paid to BHJ-PSCs based on poly(3-hexylthiophene) (P3HT) as a standard electron donor material, combined with phenyl-C61-butyric acid methyl ester (PC61BM) as the electron acceptor.2 The synthetic availability of P3HT and its ability to form semicrystalline lamellar microstructures that favour charge transport are major advantages for this application.3 However, its weak absorption in some parts of the visible region and in the near-infrared limits the PCE of P3HT-based devices to about 5%.4 To overcome this challenge, electron donor materials absorbing significant portions of the solar spectrum, in particular at longer wavelengths, are required. An interesting solution consists of combining a dye (polymer/small molecule) with an absorption spectrum complementary to that of P3HT, either by using a ternary blend active layer (P3HT/dye/PCBM)5 or by incorporating the dye directly into the polymer backbone.6,7
As chromophores, porphyrins are ubiquitous in solar energy conversion devices due to their significant optical absorption and photochemical stability, which can be tuned through judicious modifications of their molecular structure.8 An increase in the photocurrent was found in BHJ solar cells using porphyrins directly blended with polythiophenes9 or covalently incorporated in the polymer backbone.6 However, the natural tendency of porphyrins to aggregate can modify the ordered morphology of P3HT, thus negatively impacting the transport of photogenerated charges and decreasing the final PCE.6a,9c To address this limitation, end-group modification of the P3HT chains has emerged as a promising strategy to simultaneously control the active-layer morphology and improve the performance of PSCs.10 Using living chain-growth Kumada Catalyst Transfer Polymerization (KCTP), perfect control over the end-groups on the conjugated polymer can be achieved.11
Here, we report the end-functionalization of P3HT with a controllable number of π-extended porphyrin units using KCTP. Takahashi et al. previously reported that aryl isocyanide bearing a porphyrin pendant group can be converted quantitatively into the corresponding poly(aryl isocyanide).12 More recently, well-defined block copolymers containing P3HT and poly(isocyanide) blocks were synthesized in a one-pot procedure.13 Although the two block polymerizations are mechanistically distinct, the successive polymerization mediated by a common catalyst, proceeded in a controlled manner and led to block copolymers with tuneable molecular weights and compositions.13 Here, by combining these strategies, the modification of the P3HT end-groups by π-extended porphyrins is pursued. To ensure good solubility of the final material, the number of porphyrin attached to the P3HT is limited to few units. The synthesized polymers are found to be soluble in many organic solvents, have significantly broader absorption spectra than pure P3HT and are able to self-assemble into organized microstructures in solution and the solid state. The effect of the porphyrin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thiophene ratio on the optical, thermal and morphological properties of the polymer is also discussed.
:thiophene ratio on the optical, thermal and morphological properties of the polymer is also discussed.
The isocyanide-based porphyrin monomer 1 was synthesized in three steps from the enaminoporphyrin NH-NiTPP, which was previously reported by Callot, Ruppert et al. (Scheme 1).14 Due to the extension of the π-system of the porphyrin over the cyclized phenyl ring and the enamine, NH-NiTPP exhibits a much broader absorption spectrum compared to other non-functionalized meso-tetraarylporphyrins (e.g. see Fig. S22, ESI†). N-Alkylation of NH-NiTPP with 1-(6-iodohexyloxy)-4-nitrobenzene afforded the corresponding NO2-Spacer-NiTPP derivative. The reduction of the nitro group with sodium borohydride (NaBH4) and palladium on carbon (Pd/C 10%), followed by formylation with formic acid, afforded NHCO-NiTPP in 87% yield. Subsequent dehydration with POCl3–NEt3 afforded the desired isocyanide-based porphyrin monomer 1 in 80% yield, which was fully characterized by mass spectrometry and NMR, IR and UV-Vis absorption spectroscopies (see ESI†).
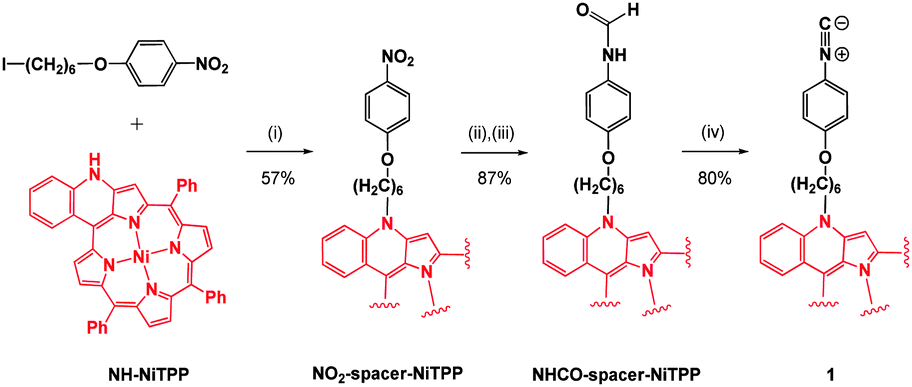 | ||
| Scheme 1 Synthesis of isocyanide-based porphyrin monomer 1. Conditions: (i) NaH, THF, 25 °C 2 h, reflux 16 h; (ii) NaBH4, Pd/C 10%, CH2Cl2/CH3OH, 25 °C 1 h; (iii) formic acid, toluene, reflux 16 h; (iv) POCl3, NEt3, THF, 0 °C 3 h. | ||
With 1 in hand, its sequential copolymerization with 5-chloromagnesio-2-bromo-3-hexylthiophene was investigated (Scheme 2). Using standard KCTP methods, the P3HT macroinitiator with a living nickel end-group (Ni(dppp)Br) was synthesized. The polymerization was followed using Gel Permeation Chromatography (GPC) by removing aliquots from the polymerization mixture. When polymerization of the P3HT block was considered to be achieved, a solution of 1 (0.01 M) in THF was added to the reaction mixture. The orange P3HT solution immediately turned green and subsequently, turned to brown suggesting that copolymerization took place. After 3 hours, the solution was rapidly quenched with HCl (5 M) to prevent interchain coupling reactions from occurring, and thus, to maintain a narrow dispersity.15
 | ||
| Scheme 2 Synthesis of P3HT end-functionalized with π-extended porphyrin in a one-pot procedure via sequential monomer addition. | ||
GPC analysis of the isolated material revealed a higher number-averaged molecular weight (Mn) for 2 compared to the P3HT macroinitiator (Fig. S20, ESI†), indicating successful chain growth polymerization. Both traces present monomodal and symmetric peaks, showing that no chain termination or chain transfer took place during the polymerization. Completion of the polymerization was also inferred from IR spectroscopy, since the isocyanide band of the monomer 1 at ν = 2118 cm−1 disappeared in the IR spectrum of 2 (Fig. S21, ESI†).13,16 As summarized in Table 1, three P3HTs end-functionalized with π-extended porphyrins with different Mn and compositions were synthesized by varying the initial ratio of the aforementioned monomers and catalyst. All synthesized polymers were isolated in good yields (>60% over two steps) with narrow dispersity (<1.4). Their compositions were determined from their 1H NMR spectra (Fig. S14, S16 and S18, ESI†) by integrating the signals observed at δ = 0.90 (terminal methyl group of the hexyl side chain of P3HT) and between δ = 7.30 and 9.00 ppm (β-pyrrolic and meso phenyl protons of the porphyrins). As desired, the GPC data indicate that only a few porphyrin units (3–4) are incorporated as end-groups on the P3HT chains.
| P3HT | 2 | Yield (%) | Molar 3HT/1 ratioc | |||
|---|---|---|---|---|---|---|
| M n , (kDa) | M w/Mna,b | M n (kDa) | M w/Mnb | |||
| a M n and Mw/Mn of the P3HT moiety were determined by GPC analysis of aliquots removed from the reaction mixture before the addition of 1. b M n and Mw/Mn are reported as their polystyrene equivalents. c The molar ratio between 3-hexylthiophene (3HT) and 1 repeating units was determined by 1H NMR spectroscopy in CDCl3. | ||||||
| 2a | 8.0 | 1.28 | 12.2 | 1.39 | 70 | 90/10 |
| 2b | 5.6 | 1.09 | 9.2 | 1.28 | 62 | 86/14 |
| 2c | 1.7 | 1.24 | 5.0 | 1.18 | 60 | 68/32 |
The optical properties of 2 were subsequently investigated. In chloroform, the UV-Vis absorption spectra of polymers 2a–c are very similar to that of the isocyanide-based porphyrin monomer 1 (Fig. S22, ESI†), with a strong absorption band between 420 and 450 nm and weaker absorption bands between 550 and 636 nm assigned to the Soret and Q bands of 1, respectively. In addition, the absorption band appears broader in the 350–450 nm region due to the concomitant P3HT absorption. Usually, P3HT can be directed to self-assemble into crystalline nanowires by adding a nonsolvent to a polymer dissolved in a good solvent.17 Thus, the optical properties of 2 were studied in chloroform/methanol mixtures at different ratios. Polymer 2a was initially dissolved in chloroform, a good solvent for both P3HT and porphyrin moieties, and aggregation was subsequently induced by adding methanol (MeOH) in increasing ratios. Upon MeOH addition, the colour of the solution turned from green to brown, indicating self-assembly of the polymer chains (Fig. 1). Moreover, further increasing the MeOH content results in a gradual decrease in the absorbance at 426 and 447 nm and the growth of vibronic bands at 518, 556, 601 and 636 nm (Fig. 1). An isobestic point is observed at 480 nm, indicating that two distinct species, i.e. isolated polymer chains and aggregates, contribute to the absorption profile in mixtures from 10/0 to 3/7 (CHCl3/MeOH, v/v).17 These results indicate interchain π–π interactions associated with the formation of semicrystalline aggregates. Dynamic light scattering (DLS) measurements of polymer 2a in CHCl3/MeOH (1:1) revealed that nanostructures with a monomodal size distribution (Fig. S25, ESI†) were formed. The average hydrodynamic diameter of the aggregates was found to be ∼151 ± 26 nm.
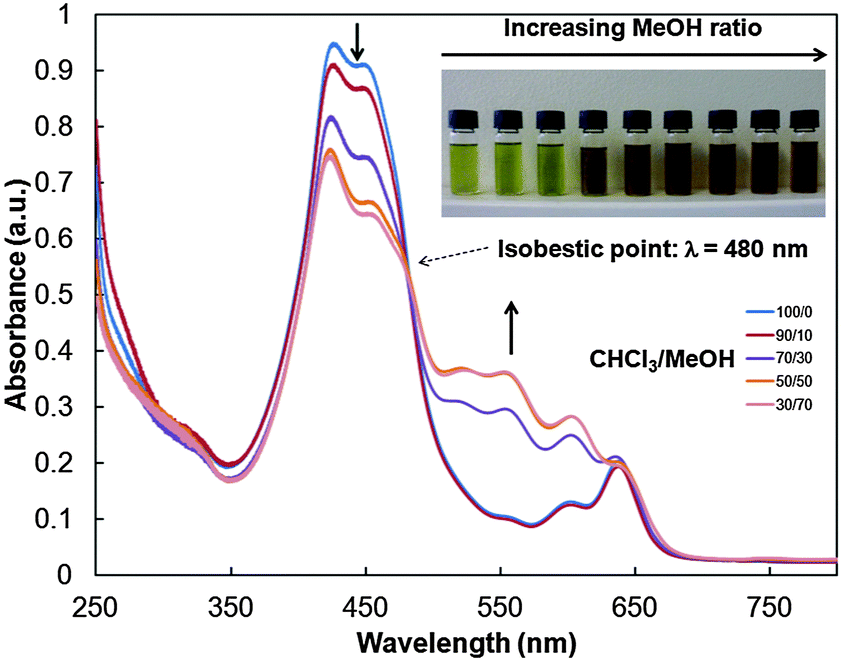 | ||
| Fig. 1 Photographs and UV-Vis absorption spectra of 2a in chloroform/methanol mixtures from 10/0 to 3/7 (CHCl3/MeOH = v/v, C = 0.06 mg mL−1). | ||
To investigate the self-assembly of the polymer chains in the solid state in more details, the UV-Vis absorption spectra of polymer thin films were recorded and compared to pure P3HT and porphyrin 1 thin films (Fig. S23, ESI†). As observed in CHCl3/MeOH (1:1), the polymer 2a thin film exhibits vibronic structure with shoulder peaks around 520, 550 and 600 nm. These peaks correspond fairly well with those observed in pristine P3HT thin films, in which the conjugated backbones are planar and highly organized. Such a similarity indicates that the bulky porphyrin moieties do not disrupt the packing of the P3HT chains.18
The influence of the P3HT:porphyrin molar ratio on the optical and self-assembly properties was then studied. The UV-Vis absorption spectra of 2a and 2b in the solid state exhibit the same profile but with a lower absorbance between 500 and 700 nm due to the higher porphyrin content (Fig. S24, ESI†). In contrast, the absorption profile of 2c appears to be almost identical to that of the porphyrin monomer 1 and exhibits no vibronic band between 500 and 600 nm. This indicates that introducing an excessive number of porphyrin units prevents intermolecular interactions between polymer chains, leading to a reduction in overlap through π–π stacking and disturbed arrangement in the solid films.19 Thus, while the incorporation of π-extended porphyrin units into the P3HT chain-ends enables the absorption profile of the polymer material to be significantly extended, the porphyrin:P3HT ratio must be kept low enough to avoid the loss of the supramolecular organisation of the P3HT segments in the solid state.
To investigate the effect of the porphyrin molar ratio on the thermal properties, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed under an inert atmosphere at a heating rate of 10 °C min−1. Polymers 2a–c display excellent thermal stability, with the first weight losses upon heating occurring only at temperatures exceeding 330 °C (Fig. S26, ESI†). The DSC measurements show different thermal transitions, depending on the porphyrin:P3HT ratio (Fig. S27, ESI†): introduction of the porphyrin leads to a decrease in the melting and, to a lesser extent, crystallization temperatures in comparison with the neat P3HT.20 Upon cooling, a crystallization temperature of 115 °C (compared to 200 °C for P3HT) and, upon heating, a melting temperature of 206 °C (compared to 210 °C for P3HT) are found for 2a. The melting and crystallization temperatures of polymer 2b are lower than for 2a, whereas polymer 2c appears to be fully amorphous. The evolution is a consequence of the increasing porphyrin molar ratio when going from 2a to 2c. These results are consistent with the changes observed in the UV-Vis absorption spectra and confirm that the porphyrin:P3HT ratio needs to be carefully controlled to retain the semicrystalline nature of P3HT.
To analyze the microscopic morphology of the polymers in the solid state, atomic force microscopy (AFM) in the peak force tapping mode was performed. Polymer 2a is the only sample with a well-defined nanostructured morphology. At a scale of 10 μm (Fig. 2a), we observe a web of few hundred nm-wide tapes. These tapes are composed of aligned fibrils, which can extend over a few micrometers in length (Fig. 2c and d). This fibrillar nanoscale morphology is characteristic of highly regioregular P3HT.21 Powder X-ray diffraction (XRD) measurements were also performed on polymer 2a and confirm the crystalline ordering (Fig. 2b). The diffraction pattern exhibits reflections assigned to pristine P3HT and isocyanide porphyrin monomer 1, indicating the retention of the crystalline structures inherent to both components. The peak at 2θ ∼ 5.0° is typical of a lamellar structure, which is also observed in the P3HT diffraction pattern (2θ ∼ 5.5°).22 In addition, the broad reflection centered at 2θ ∼ 20.3° can be related to diffraction from the isocyanide porphyrin monomer 1 (2θ ∼ 20.8°), which corresponds to the π–π stacking distance (2.8 Å) between two porphyrin units. These similarities, in particular the position of the (100) lattice peak, indicate that the porphyrin moiety, if maintained in relatively low quantity, does not affect the P3HT crystallite structure.
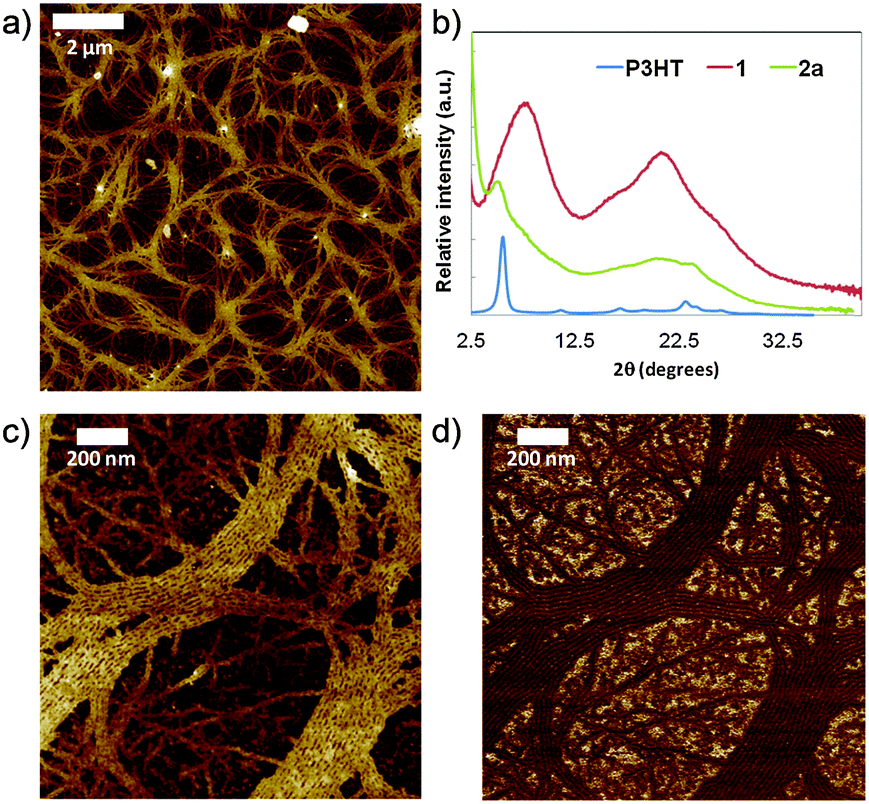 | ||
| Fig. 2 (a) 10 × 10 μm2 peak force AFM height image of 2a drop-cast from xylene onto a mica substrate; (b) X-ray diffraction pattern of isocyanide porphyrin monomer 1, P3HT and 2a; (c and d) 1.5 × 1.5 μm2 peak force AFM height (c) and DMT modulus (d) images of 2a showing the fibrillar morphology. | ||
In summary, P3HTs end-functionalized with π-extended porphyrin have been synthesized using a one-pot procedure based on the distinct polymerization of thiophene and π-extended porphyrin monomers functionalized with a pendant arylisocyanide group. By judiciously adjusting the P3HT:porphyrin molar ratio, self-assembly of the end-functionalized P3HT chains can be achieved, leading to broader absorption profile while maintaining fibrillar nanoscale morphology. The extended visible absorption window, combined with the tunable thin film morphology suggest that these polymers may demonstrate considerable potential as electron donor materials in BHJ polymer solar cells. Further studies will investigate in more detail the aggregation and the self-assembling properties of these polymers and their influence on the photovoltaic performance.
The authors thank the CNRS and the Université de Montpellier for financial support. Research in Mons is supported by the Science Policy Office of the Belgian Federal Government (BELSPO; PAI 7/05), FNRS-FRFC and Région Wallonne (OPTI2MAT excellence programme). The authors are also grateful to National Fund for Scientific Research (F.R.S.-FNRS) in the frame of the FRFC research program (convention no. 2.4508.12).
Notes and references
- (a) L. Yang, H. Zhou, A. C. Stuart and W. You, in Organic Photovoltaics, ed. C. J. Brabec, U. Scherff and V. Dyakonov, Wiley-VCH, Weinheim, 2nd edn, 2014, p. 61 Search PubMed; (b) J. Youa, L. Doua, Z. Hong, G. Lia and Y. Yang, Prog. Polym. Sci., 2013, 38, 1909 CrossRef; (c) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597 CrossRef CAS PubMed.
- (a) I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202 CrossRef CAS PubMed; (b) Handbook of Oligo- and Polythiophenes, ed. D. Fichou, Wiley VCH, Weinheim, 2007 Search PubMed.
- (a) C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839 CrossRef CAS PubMed; (b) G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS.
- (a) F. Goubard and G. Wantz, Polym. Int., 2014, 63, 1362 CrossRef CAS; (b) Y.-C. Chen, C.-Y. Hsu, R. Y.-Y. Lin, K.-C. Ho and J. T. Lin, ChemSusChem, 2013, 6, 20 CrossRef CAS PubMed.
- (a) L. Angiolini, V. Cocchi, M. Lanzi, E. Salatelli, D. Tonelli and Y. Vlamidis, Mater. Chem. Phys., 2014, 146, 464 CrossRef CAS; (b) L. Angiolini, T. Benelli, V. Cocchi, M. Lanzi and E. Salatelli, React. Funct. Polym., 2013, 73, 1198 CrossRef CAS.
- (a) J. U. Lee, Y. D. Kim, J. W. Jo, J. P. Kim and W. H. Jo, J. Mater. Chem., 2011, 21, 17209 RSC; (b) B. J. Campo, J. Duchateau, C. R. Ganivet, B. Ballesteros, J. Gilot, M. M. Wienk, W. D. Oosterbaan, L. Lutsen, T. J. Cleij, G. de la Torre, R. A. J. Janssen, D. Vanderzande and T. Torres, Dalton Trans., 2011, 40, 3979 RSC.
- (a) J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 Search PubMed; (b) K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, New York, 2000, vol. 6 Search PubMed.
- (a) H. Xu, H. Ohkita, T. Hirata, H. Benten and S. Ito, Polymer, 2014, 55, 2856 CrossRef CAS; (b) D. M. Lyons, J. Kesters, W. Maes, C. W. Bielawski and J. L. Sessler, Synth. Met., 2013, 178, 56 CrossRef CAS; (c) D. M. Lyons, R. J. Ono, C. W. Bielawski and J. L. Sessler, J. Mater. Chem., 2012, 22, 18956 RSC.
- (a) S. L. Fronk, C.-K. Mai, M. Ford, R. P. Noland and G. C. Bazan, Macromolecules, 2015, 48, 6224 CrossRef CAS; (b) Z. Mao, K. Vakhshouri, J. Cherno, D. A. Fischer, R. Fernando, D. M. DeLongchamp, E. D. Gomez and G. Sauve, Macromolecules, 2013, 46, 103 CrossRef CAS; (c) J. S. Kim, Y. Lee, J. H. Lee, J. H. Park, J. K. Kim and K. Cho, Adv. Mater., 2010, 22, 1355 CrossRef CAS PubMed.
- (a) N. V. Handa, A. V. Serrano, M. J. Robb and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 831 CrossRef CAS; (b) R. H. Lohwasser and M. Thelakkat, Macromolecules, 2011, 44, 3388 CrossRef CAS; (c) A. Smeets, K. Van den Bergh, J. De Winter, P. Gerbaux, T. Verbiest and G. Koeckelberghs, Macromolecules, 2009, 42, 7638 CrossRef CAS.
- F. Takei, K. Onitsuka, N. Kobayashi and S. Takahashi, Chem. Lett., 2000, 914 CrossRef CAS.
- (a) M. Su, S.-Y. Shi, Q. Wang, N. Liu, J. Yin, C. Liu, Y. Ding and Z.-Q. Wu, Polym. Chem., 2015, 6, 6519 RSC; (b) R. J. Ono, A. D. Tood, Z. Hu, D. A. Vanden Bout and C. W. Bielawski, Macromol. Rapid Commun., 2013, 35, 204 CrossRef PubMed; (c) Z.-Q. Wu, J. D. Radcliffe, R. J. Ono, Z. Chen, Z. Li and C. W. Bielawski, Polym. Chem., 2012, 3, 874 RSC; (d) N. Liu, C.-G. Qi, Y. Wang, D.-F. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Macromolecules, 2013, 46, 7753 CrossRef CAS; (e) Z.-Q. Wu, R. J. Ono, Z. Chen and C. W. Bielawski, J. Am. Chem. Soc., 2010, 132, 14000 CrossRef CAS PubMed.
- S. Richeter, C. Jeandon, J.-P. Gisselbrecht, R. Graff, R. Ruppert and H. J. Callot, Inorg. Chem., 2004, 43, 251 CrossRef CAS PubMed.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Macromol. Rapid Commun., 2004, 25, 1663 CrossRef CAS.
- A. Thomas, J. E. Houston, N. Vand Den Brande, J. De Winter, M. Chevrier, R. K. Heenan, A. E. Terry, S. Richeter, A. Mehdi, B. Van Mele, P. Dubois, R. Lazzaroni, P. Gerbaux, R. C. Evans and S. Clément, Polym. Chem., 2014, 5, 3352 RSC.
- (a) E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390 CrossRef CAS PubMed; (b) L. G. Liu, G. H. Lu and X. N. Yang, J. Mater. Chem., 2008, 18, 1984 RSC.
- C. Scharsich, R. H. Lohwasser, M. Sommer, U. Asawapirom, U. Scherf, M. Thelakkat, D. Neher and A. Köhler, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 442 CrossRef CAS.
- L. Wang, S. Shi, D. Ma, S. Chen, C. Gao, M. Wang, K. Shi, Y. Li, X. Li and H. Wang, Macromolecules, 2015, 48, 287 CrossRef CAS.
- (a) P. Koh, S. Huettner, H. Komber, V. Senkovskyy, R. Tkachov, A. Kiriy, R. H. Friend, U. Steiner, W. T. S. Huck, J.-U. Sommer and M. Sommer, J. Am. Chem. Soc., 2012, 134, 4790 CrossRef PubMed; (b) D. E. Motaung, G. F. Malgas, C. J. Arendse, S. E. Mavundla, C. J. Oliphant and D. Knoesen, Sol. Energy Mater. Sol. Cells, 2009, 93, 1674 CrossRef CAS.
- (a) H. Yang, T. J. Shin, L. Yang, K. Cho, C. Y. Ryu and Z. Bao, Adv. Funct. Mater., 2005, 15, 671 CrossRef CAS; (b) M. Surin, S. Cho, J. D. Yuen, G. Wang, K. Lee, P. Leclère, R. Lazzaroni, D. Moses and A. J. Heeger, J. Appl. Phys., 2006, 100, 33712 CrossRef; (c) S. Berson, R. De Bettignies, S. Bailly and S. Guillerez, Adv. Funct. Mater., 2007, 17, 1377 CrossRef CAS.
- (a) T. J. Prosa, M. J. Winokur, J. Moulton, P. Smith and A. J. Heeger, Macromolecules, 1992, 25, 4364 CrossRef CAS; (b) K. Tashiro, K. Ono, Y. Minagawa, M. Kobayashi, T. Kawai and K. Yoshino, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 1223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and synthesis, NMR, GPC, IR, UV, TGA, DSC, XRD. See DOI: 10.1039/c5cc06290j |
| This journal is © The Royal Society of Chemistry 2016 |