
Nickel as a co-catalyst for photocatalytic hydrogen evolution on graphitic-carbon nitride (sg-CN): what is the nature of the active species?†
Arindam
Indra
a,
Prashanth W.
Menezes
a,
Kamalakannan
Kailasam
b,
Dirk
Hollmann
c,
Marc
Schröder
a,
Arne
Thomas
a,
Angelika
Brückner
c and
Matthias
Driess
*a
aMetalorganic Chemistry and Inorganic Materials, Department of Chemistry, Technische Universität Berlin, Strasse des 17 Juni 135, Sekr. C2, D-10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de
bInstitute of Nano Science and Technology, Mohali, Punjab 160062, India
cLeibniz Institute for Catalysis at the University of Rostock, Albert Einstein-Straße 29A, 18059 Rostock, Germany
First published on 15th October 2015
Abstract
The nature of a nickel-based co-catalyst deposited on a sol–gel prepared porous graphitic-carbon nitride (sg-CN), for photocatalytic H2 production from water, has been investigated. The formation of the active catalytic species, charge separation and recombination of the photogenerated electrons and holes during photochemical H2 evolution has been determined for the first time using in situ EPR spectroscopy.
The use of solar light for photocatalytic water-splitting into dihydrogen (H2) and dioxygen (O2) with semiconductor catalysts has been considered to be a sustainable way of energy production, as an alternative to using the steadily decreasing and environmentally pollutant fossil fuels.1,2 For effective harvesting of visible light, the semiconductor must have a band gap higher than 1.23 eV and the conduction band must be more negative than the reduction potential of H+/H2.3 Moreover, the separation of the photogenerated electrons and holes competing with their recombination is an additional challenge in photocatalytic water-splitting.4
Noble metals, such as Pt for the hydrogen evolution reaction (HER), have been of increasing interest in recent years and widely applied as co-catalysts to hasten the electron transport pathway.5,6 The decorated nanoparticles of noble metals on a catalyst surface act as electron sinks to supply charge separation across the interface or heterojunction. Lately, several CdX (X = S, Se)-based photocatalytic systems have been studied to understand the lifetime of the photogenerated electrons and holes, and their recombination during photocatalytic HER.7 Interestingly, the organic carbon nitride photocatalyst has been effective for hydrogen evolution under visible light irradiation8 whereas, a Pt-containing graphitic carbon nitride (g-CN) system has been investigated to uncover the charge separation and recombination phenomena.9 Several efficient systems with a combination of Pt and g-CN have been examined for enhanced HER,10–14 but a Pt-free approach to give similarly active catalysts remains of particular interest. In the last few years, the Pt- free approach has been demonstrated for HER by combining g-CNs with MoS2,15 CdS,16 CuInS2,17 NiS,18 Ni(OH)2,19 ZnO20 with low to moderate activity. In addition, Ag or Au nanoparticles deposited on g-CN nanocomposites were also studied to achieve plasmonic enhancement in the photocatalytic activity.21,22 Interestingly, the combination of molecular metal complexes with g-CN have been probed for photocatalytic HER as well, however, leading to the attainment of less active photocatalysts than the Pt–g-CN systems.23
The development of H2 evolution catalysts that combine g-CN with cheap and abundant metals, such as Ni as a co-catalyst, is highly desired for effective and stable hydrogen production.24 Although Ni-based catalyst systems have been explored in recent years,24,25 to our knowledge, no attempt has been reported to uncover the nature and structural features of the Ni co-catalyst as well as the possible structural and bonding situation occurring at the co-catalyst active sites during photocatalytic HER. In line with that, the separation and recombination processes of the photogenerated electrons and holes with the Ni–CN system have not been perceived so far. Encouraged by our recent results on the determination of the active sites of oxygen evolution catalysts,26–28 we directed ourselves toward model catalytic systems by depositing non-noble metal co-catalysts to elucidate the role of the co-catalyst during photocatalytic HER.
For this purpose, we have chosen photocatalytically highly active porous sol–gel prepared g-CN6 (abbreviated as sg-CN hereafter) with a photodeposited nickel species as the co-catalyst to achieve long-term HER in the presence of the sacrificial electron donor (SD) triethanolamine (TEOA). After photodeposition, the materials were extensively characterized using various spectroscopic, microscopic and analytical tools. The structural changes of the Ni co-catalyst were thoroughly studied during photocatalytic HER using in situ electron paramagnetic resonance (EPR) and the recovered samples were also subsequently studied by EPR, X-ray photoelectron spectroscopy (XPS), and high resolution transmission electron microscopy (HR-TEM) to uncover the complete structural situation.
First of all, the co-catalyst has been loaded onto sg-CN using a Ni2+ salts under different reaction conditions (Table S1 and the Experimental section in the ESI†). The following abbreviations are used for the prepared samples: Cat-1 (photodeposition, with SD), Cat-2 (deposition in the dark, with SD), Cat-3 (photodeposition, without SD). It is worth mentioning here that the presence of both light and TEOA is necessary for the efficient deposition of Ni0 nanoparticles for enhancing the catalytic hydrogen evolution (see later). TEOA accepts the photogenerated holes and helps in the reduction of Ni2+ to Ni0 whereas in the absence of light, no photoexcitation occurs and only a small amount of the nickel species is deposited on sg-CN.
An extensive characterization of the materials has been performed to understand the structural features of the catalyst and oxidation state of nickel, and its binding interaction with sg-CN. Powder X-ray diffraction (PXRD) of Cat-1 showed two peaks at 2θ values of 13° and 27.4° which correspond to the typical intra-planar arrangement in a sheet and graphitic stacking peak (Fig. S1, ESI†). No new peak was detected after the loading of nickel species onto g-CN under different deposition conditions (Fig. S1, ESI†). The loading of Ni was confirmed by energy dispersive X-ray spectroscopy (EDX, Fig. S2, ESI†) and inductively coupled plasma atomic emission spectroscopy (ICP-AES, Table S1, ESI†). The latter revealed a nickel content of 0.73 wt% in Cat-1 when photodeposition was performed in the presence of SD. However, in the absence of SD or light, a lower loading of nickel (0.1 wt%) was found.
For all catalysts, the ratio of N to C was determined to be ∼1.3 by elemental analyses. The 13C cross-polarization/magic angle spinning solid state (CP/MAS) NMR spectrum of Cat-1 showed a heptazine-based structure of the sg-CN with the CN3 moiety resonating at δ 154 ppm and the C(N)2(NH)x moiety at 162 ppm, respectively. No shift of these peaks was observed after the deposition of the nickel species on this sg-CN material (Fig. S3, ESI†). In addition, the BET (Brunauer–Emmett–Teller) surface area of sg-CN (147 m2 g−1), after photodeposition in Cat-1 (142 m2 g−1), was merely changed. In the UV-Vis spectra, the maximum absorption for Cat-1 was observed at ν = 360 nm but for other catalysts, significant variations in the band positions were found that were reflected in their catalytic activities (Fig. S4a, ESI†). Infrared spectroscopy (IR) confirmed the unchanged sg-CN structure (Fig. S4b, ESI†). TEM and HRTEM measurements displayed the porous structure of sg-CN without any discrete crystallites corresponding to Ni0, NiO or NiOOH in Cat-1 (Fig. S5, ESI†).
What is the nature of the deposited Ni-species as co-catalyst? At first we determined the oxidation state of nickel in Cat-1 after photodeposition. On the surface of the catalyst only a very low amount of nickel could be detected by XPS. The Ni2p3/2 peak at binding energy of 856.8 eV suggests an oxidation state +2. Furthermore, the presence of metallic nickel is indicated by the peak at 853 eV in Cat-1 (Fig. S6a, ESI†).29 It should be mentioned here that in Cat-1, the amount of nickel at the outer surface was relatively low and most of the nickel was present in the bulk of the porous sg-CN (total 0.73%). The N1s XPS peak could be deconvoluted into two peaks corresponding to N-pyridine moieties at 399.8 eV and graphitic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N subunits at 401.7 eV, respectively (Fig. S6b, ESI†).30 The C1s XPS peaks could be attributed to CC, CN and C–N moieties from lower to higher energy (Fig. S6c, ESI†), while the O1s spectrum of Cat-1 represents surface adsorbed oxygen species and C–O bonds (Fig. S6d, ESI†).30
N subunits at 401.7 eV, respectively (Fig. S6b, ESI†).30 The C1s XPS peaks could be attributed to CC, CN and C–N moieties from lower to higher energy (Fig. S6c, ESI†), while the O1s spectrum of Cat-1 represents surface adsorbed oxygen species and C–O bonds (Fig. S6d, ESI†).30
After the initial characterization of the two components of the catalysts (photocatalyst and co-catalysts), we further investigated the catalysts for the photochemical H2 evolution (Fig. 1). Under visible light irradiation (>420 nm), the highest amount of H2 evolution was observed with Cat-1 whereas the other catalysts showed much less activity. Under similar reaction conditions, negligible amount of H2 production could be detected with co-catalyst-free sg-CN. The rate of H2 evolution for Cat-1 was calculated at 0.103 mmolH2 gcat−1 h−1 in the first 4 h for Cat-1. The rate of H2 production for Cat-1 is comparable with related CN-based materials loaded with non-noble metal-containing co-catalysts.31 For example, previously NiS–g-CN has shown a H2 evolution rate of ∼0.4 mmolH2 gcat−1 h−1.18 The photocatalyst Cat-1 was recycled three times to yield a total of 12.2 mL of H2 (Fig. 1) after ∼128 h. A turnover number (TON = mol of H2 produced per mol of Ni) of 79 was achieved in terms of the total amount of loaded nickel. A minor loss of activity, after three cycles was observed in Cat-1 and long-term measurements were performed with the catalyst in the continuous generation of H2 over 8 days to generate ∼11.2 mL of H2 (Fig. S7, ESI†). Further, Cat-2 showed a significant amount of hydrogen evolution after the 1st cycle due to similar types of structural features to that of Cat-1 but with a lower nickel content (Fig. 1).
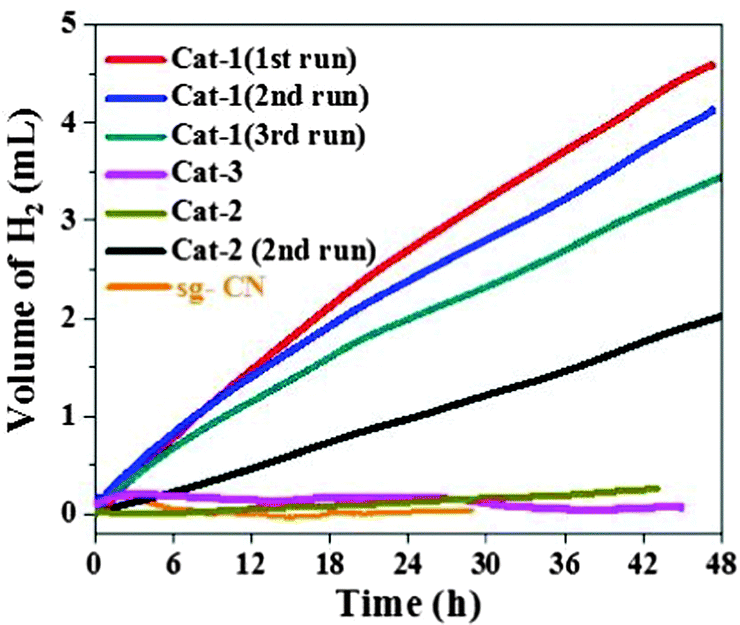 | ||
| Fig. 1 Photocatalytic H2 evolution profiles of various catalysts in the presence of 300 W Xe lamp with a cut off filter of 420 nm using TEOA as the sacrificial agent. | ||
Furthermore, EPR spectroscopy has been applied as a very sensitive and local method for detecting selectively the formation of superparamagnetic Ni0 nanoparticles (NP) which may be too small or too few to be visualized by TEM.32 The EPR spectrum of Cat-2 (Ni deposition in the dark) at 300 K showed no signal, suggesting that all nickel is dispersed as Ni2+ species. After irradiation with visible light (in Cat-1) a broad EPR signal (g = 2.23) could be observed, indicating the formation of ferromagnetic Ni0 particles (Fig. S8, ESI†),33 which illustrate the high sensitivity of EPR spectroscopy for the detection of Ni0 cluster. By investigating the temperature dependence of the EPR signal intensity, an average Ni0 particle diameter could be estimated using the Langevin equation (see ESI†).
Comparison of the experimental data of Cat-1 with the results for different particle diameters, suggests that the mean Ni0 particle size in Cat-1 is around 3 nm (Fig. S9 and eqn (S1), ESI†). When Cat-1 is exposed to air during recovery, the Ni0 signal strongly decreases, broadens and becomes more asymmetric. This might be an indication for the partial oxidation of the Ni NPs to Ni2+ (Fig. S8, ESI†). As mentioned earlier, the main objective of this work is to determine the co-catalyst structure during the photocatalytic hydrogen evolution, in situ EPR spectroscopic studies were performed to prove the formation of the Ni0 NPs under visible light irradiation. Cat-1 has been monitored by in situ EPR in a TEOA/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) solution under irradiation with visible light (>420 nm, Fig. 2). A continuous increase of the Ni0 signal was detected, though, not all Ni2+ species were reduced to Ni0 at the same time (Scheme 1). This could be one of the reasons for the detection of a very small amount of Ni0 in XPS after 48 h of irradiation.
:9) solution under irradiation with visible light (>420 nm, Fig. 2). A continuous increase of the Ni0 signal was detected, though, not all Ni2+ species were reduced to Ni0 at the same time (Scheme 1). This could be one of the reasons for the detection of a very small amount of Ni0 in XPS after 48 h of irradiation.
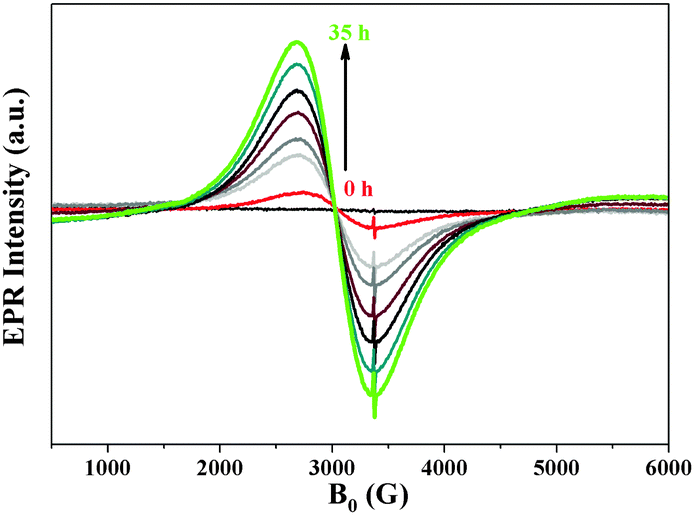 | ||
| Fig. 2 In situ EPR studies of a suspension of Cat-1 in TEOA solution under continuous irradiation with visible light (>420 nm) with variation in time. | ||
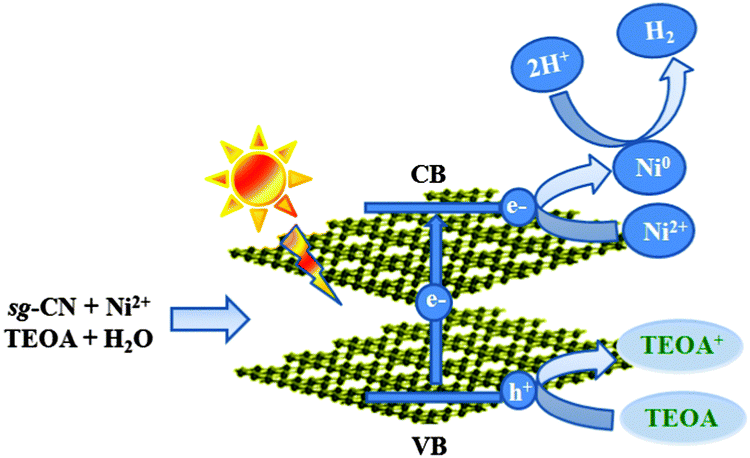 | ||
| Scheme 1 Schematic representation of the charge separation in sg-CN and the formation of Ni0 nanoparticles during photocatalytic hydrogen evolution under visible light irradiation. | ||
Furthermore, EPR spectroscopy has been utilized to study charge separation processes in sg-CN as well as the impact of the co-catalyst on electron transfer. As reported previously, trapped conduction band electrons (CB-e−) in sg-CN can be visualized by EPR during irradiation.34 Charge separation efficiency can also be analyzed from EPR during irradiation (Fig. S10, black, ESI†). The latter corresponds to the photoactive CB-e− trapped at the surface. Furthermore, the recombination efficiency of these CB-e− with holes is indicated by a decrease of the EPR signal after irradiation (Fig. S10, red, ESI†). Highest charge separation as well as lowest recombination efficiency was observed for co-catalyst-free sg-CN. However, with the Ni-co-catalysts, in all samples, even less than half of the CB-e− concentration could be detected compared to ‘free’ sg-CN. This suggests that the CB-e− formed by light-induced charge separation within the support travels quickly to the co-catalyst, from which it is transferred to the protons to evolve dihydrogen.34
Irradiation of sg-CN with visible light generates holes in the valence band and electrons in the conduction band (Scheme 1 and eqn (1)). Migration and separation of the charge was facilitated by the nickel. TEOA is able to quench the photogenerated holes in the valence band and acts as a sacrificial electron donor whereas the conduction band electrons reduce the protons to give dihydrogen. In situ reduction of Ni2+ to Ni0via electron transfer from the conduction band (CB) can also occur which explains the formation of Ni0 NPs (eqn (4)). Under photocatalytic conditions the reduction potential of carbon nitride conduction band (−1.3 V) is large enough to reduce Ni2+ to Ni0 (−0.26 V).35,24b The latter can act as a co-catalyst during proton reduction. Therefore, the actual catalyst system contains both Ni2+ (XPS), and Ni0 (EPR, XPS) species. The present system is significantly different from the Ni2+–fluorescein system reported by Fu and co-workers where irradiation with visible light generated Ni NPs in the reaction medium.24b Transfer of the holes from the valence band of sg-CN to Ni2+ can also occur to generate Ni3+ as reported by Shangguan and co-workers in photodeposited Ni2O3–CdS.36 A similar conclusion was drawn from NiOOH–TiO2 systems, when a Ni2+ salt was photodeposited by UV light on TiO2 in the presence of a surfactant.37 Furthermore, in this study, XPS shows that prolonged irradiation results in the movement of surface Ni0 to the bulk, since only a low amount of nickel could be observed on the surface after a long reaction time. The formation of Ni0 domains could be proved by in situ EPR spectroscopy. Elemental nickel is formed only under photocatalytic reaction conditions and is reoxidized quickly when exposed to air during isolation. Particles with higher contrast have been detected by HRTEM after recycling of Cat-1 after 8 days (Fig. 1b). The lattice spacing and the corresponding fast Fourier transform (FFT) indicates that the particles were oxidized to Ni(OH)2. The nature of the Ni2+ is expected to be similar to Ni(TEOA)2Cl2 as reported by Sun and co-workers.23b
| sg-CN + hν → esg-CN-CB− + hsg-CN-VB+ | (1) |
| TEOA + hsg-CN-VB+ → TEOA+ | (2) |
| 2H+ + 2esg-CN-CB− → H2 | (3) |
| Ni2+ + 2esg-CN-CB− → Ni0 | (4) |
In conclusion, the nature of the active species in a noble metal-free nickel-containing sol–gel prepared mesoporous graphitic carbon nitride (sg-CN) system, for the photocatalytic hydrogen production in the presence of sacrificial agent, has been uncovered for the first time by in situ EPR spectroscopy. In addition, the formed catalytic species were also investigated by ex situ EPR, XPS, TEM, HRTEM. In situ EPR studies clearly proved that Ni2+ has been reduced to Ni0 NPs during the photocatalytic process and acts as a co-catalyst for the reduction of protons to dihydrogen. Quick transportation of the photogenerated electrons to the Ni0 co-catalyst could also be observed by EPR spectroscopy after Ni loading on sg-CN. Photodeposition of Ni2+ onto sg-CN leads to a photocatalytic system for the long-term photochemical proton reduction with activities comparable to other non-noble co-catalyst systems on sg-CN. Finally, the sg-CN system investigated here clearly provides insight into the co-catalyst structure during photochemical H2 evolution and will help in the understanding of the electron–hole separation after the co-catalyst loading.
Financial support by the BMBF (L2H project) and the DFG (Cluster of Excellence UniCat) is gratefully acknowledged. D. H. thanks Dr Radnik for XPS measurements. We would also like to thank Dr Caren Göbel for the TEM measurements.
Notes and references
- H. B. Gray, Nat. Chem., 2009, 1, 1–7 CrossRef PubMed.
- A. Indra, P. W. Menezes, M. Schwarze and M. Driess, New J. Chem., 2014, 38, 1942–1945 RSC.
- J. Xing, W. Q. Fang, H. J. Zhao and H. G. Yang, Chem. – Asian J., 2012, 7, 642–657 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- J. Liu, J. Huang, D. Dontosova and M. Antonietti, RSC Adv., 2013, 3, 22988–22993 RSC.
- K. Kailasam, J. D. Epping, A. Thomas, S. Losse and H. Junge, Energy Environ. Sci., 2011, 4, 4668–4674 CAS.
- (a) T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Dö, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed; (b) J. Zhao, M. A. Holmes and F. E. Osterloh, ACS Nano, 2013, 7, 4316–4325 CrossRef CAS PubMed.
- (a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed; (b) J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015 10.1039/C5EE01895A; (c) Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- D. Hollmann, M. Karnahl, S. Tschierlei, K. Kailasam, M. Schneider, J. Radnik, K. Grabow, U. Bentrup, H. Junge, M. Beller, S. Lochbrunner, A. Thomas and A. Brückner, Chem. Mater., 2014, 26, 1727–1733 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- Z. Lin and X. Wang, ChemSusChem, 2014, 7, 1547–1550 CrossRef CAS PubMed.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- M. Schwarze, D. Stellmach, M. Schroeder, K. Kailasam, R. Reske, A. Thomas and R. Schomäcker, Phys. Chem. Chem. Phys., 2013, 15, 3466–3472 RSC.
- K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed.
- Y. Hou, A. B. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorf, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed.
- L. Ge, F. Zuo, J. Liu, Q. Ma, C. Wang, D. Sun, L. Bartels and P. Feng, J. Phys. Chem. C, 2012, 116, 13708–13714 CAS.
- F. Yang, V. Kuznietsov, M. Lublow, C. Merschjann, A. Steigert, J. Klaer, A. Thomas and T. Schedel-Niedrig, J. Mater. Chem. A, 2013, 1, 6407–6415 CAS.
- J. Hong, Y. Wang, Y. Wang, W. Zhang and R. Xu, ChemSusChem, 2013, 6, 2263–2268 CrossRef CAS PubMed.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Technol., 2013, 3, 1782–1789 CAS.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Energy Environ. Sci., 2011, 4, 2922–2929 CAS.
- X. Bai, R. Zong, C. Li, D. Liu, Y. Liu and Y. Zhu, Appl. Catal., B, 2014, 147, 82–91 CrossRef CAS.
- S. Samanta, S. Martha and K. Parida, ChemCatChem, 2014, 6, 1453–1462 CAS.
- (a) S.-W. Cao, X.-F. Liu, Y.-P. Yuan, Z.-Y. Zhang, J. Fang, S. C. J. Loo, J. Barber, T. C. Sum and C. Xue, Phys. Chem. Chem. Phys., 2013, 15, 18363–18366 RSC; (b) J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS PubMed.
- (a) J.-J. Wang, Z.-J. Li, X.-B. Li, X.-B. Fan, Q.-Y. Meng, S. Yu, C.-B. Li, J.-X. Li, C.-H. Tung and L.-Z. Wu, ChemSusChem, 2014, 7, 1468–1475 CrossRef CAS PubMed; (b) C. Wang, S. Cao and W.-F. Fu, Chem. Commun., 2013, 49, 11251–11253 RSC.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- (a) P. W. Menezes, A. Indra, O. Levy, K. Kailasam, V. Gutkin, J. Pfrommer and M. Driess, Chem. Commun., 2015, 51, 5005–5008 RSC; (b) A. Indra, P. W. Menezes and M. Driess, ChemSusChem, 2015, 8, 776–785 CrossRef CAS PubMed.
- (a) A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed; (b) A. Indra, P. W. Menezes, I. Zaharieva, E. Baktash, J. Pfrommer, M. Schwarze, H. Dau and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 13206–13210 CrossRef CAS PubMed.
- (a) P. W. Menezes, A. Indra, P. Littlewood, M. Schwarze, C. Göbel, R. Schomäcker and M. Driess, ChemSusChem, 2017, 7, 2202–2211 CrossRef PubMed; (b) P. W. Menezes, A. Indra, N. R. Sahraie, A. Bergmann, P. Strasser and M. Driess, ChemSusChem, 2015, 8, 164–171 CrossRef CAS PubMed; (c) P. W. Menezes, A. Indra, D. González-Flores, N. R. Sahraie, I. Zaharieva, M. Schwarze, P. Strasser, H. Dau and M. Driess, ACS Catal., 2015, 5, 2017–2027 CrossRef CAS.
- G. Zhang, G. Li and X. Wang, ChemCatChem, 2015, 7, 2864–2870 CrossRef CAS.
- E. Ech-chamikh, A. Essafti, Y. Ijdiyaou and M. Azizan, Sol. Energy Mater. Sol. Cells, 2006, 90, 1420–1423 CrossRef CAS.
- (a) S.-W. Cao, X.-F. Liu, Y.-P. Yuan, Z.-Y. Zhang, J. Fang, S. C. J. Loo, J. Barber, T. C. Sum and C. Xue, Phys. Chem. Chem. Phys., 2013, 15, 18363–18366 RSC; (b) J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS PubMed.
- (a) A. Brückner, U. Bentrup, H. Zanthoff and D. Maschmeyer, J. Catal., 2009, 266, 120–128 CrossRef; (b) F. Alonso, P. Riente, J. A. Sirvent and M. Yus, Appl. Catal., A, 2010, 378, 42–51 CrossRef CAS.
- T. Isobe, R. A. Weeks and R. A. Zuhr, Solid State Commun., 1998, 105, 469–472 CrossRef CAS.
- J. B. Priebe, M. Karnahl, H. Junge, M. Beller, D. Hollmann and A. Brückner, Angew. Chem., Int. Ed., 2013, 52, 11420–11424 CrossRef CAS PubMed.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, ACS Catal., 2013, 3, 912–919 CrossRef CAS.
- X. Chen, W. Chen, P. Lin, Y. Yang, H. Gao, J. Yuan and W. Shangguan, Catal. Commun., 2013, 36, 104–108 CrossRef CAS.
- L. Zhang, Y. Zhong, Z. He, J. Wang, J. Xu, J. Cai, N. Zhang, H. Zhou, H. Fan, H. Shao, J. Zhang and C. Cao, J. Mater. Chem. A, 2013, 1, 4277–4285 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details and characterization of Ni–CN catalysts before and after photochemical hydrogen evolution. See DOI: 10.1039/c5cc07936e |
| This journal is © The Royal Society of Chemistry 2016 |