
Mechanistic insight into proton-coupled mixed valency†
Luke A.
Wilkinson
ab,
Kevin B.
Vincent
a,
Anthony J. H. M.
Meijer
 b and
Nathan J.
Patmore
*a
b and
Nathan J.
Patmore
*a
aDepartment of Chemical Sciences, University of Huddersfield, Huddersfield HD1 3DH, UK. E-mail: n.j.patmore@hud.ac.uk
bDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK
First published on 19th October 2015
Abstract
Stabilisation of the mixed-valence state in [Mo2(TiPB)3(HDOP)]2+ (HTiPB = 2,4,6-triisopropylbenzoic acid, H2DOP = 3,6-dihydroxypyridazine) by electron transfer (ET) is related to the proton coordinate of the bridging ligands. Spectroelectrochemical studies suggest that ET is slower than 109 s−1. The mechanism has been probed using DFT calculations, which show that proton transfer induces a larger dipole in the molecule resulting in ET.
Electron transfer (ET) processes play a critical role in systems found in nature, and across the physical sciences.1 One of the most important, and simplest, models to improve our understanding of these ET processes are mixed-valence (MV) compounds, which typically consist of two identical organic or metal redox active centers bridged by a conjugated organic linker.2 The MV state in these systems is stabilized with respect to the disproportionation products by electron self-exchange. This process can be studied by a variety of theoretical, spectroscopic and electrochemical techniques,3 which provide valuable insight into electron transfer mechanisms. This field is receiving increased attention because of its importance in understanding the numerous electron transfer processes that are critical in biological systems,4 and in developing future technologies, such as molecular electronics.5
More recently, self-complementary hydrogen bonding interactions have been used to link redox active centers, although only a few examples have been reported to date.6 Due the dearth of examples, discussion of mechanisms by which the MV state could be stabilized in these compounds remains limited. However, it is possible to envisage at least three different mechanisms. As seen in covalently linked systems, stabilization could occur via an electronic coupling mechanism, involving ET from the electron donor to electron acceptor through overlap of the donor–bridge–acceptor orbitals, as shown schematically in Fig. 1 (mechanism A). Alternatively, a structural rearrangement such as simple proton transfer (PT, mechanism B) could stabilize the mixed valence state. Finally, proton-coupled mixed valency (PCMV),6d a mechanism by which electron transfer is dependent on the proton coordinate of the bridging ligand (mechanism C), could lead to stabilization of the MV state.
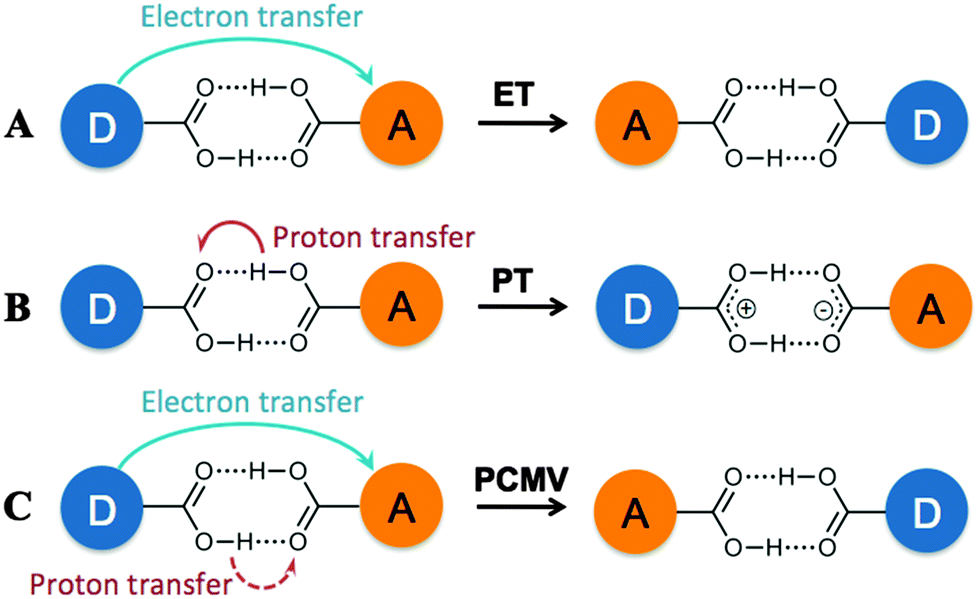 | ||
| Fig. 1 Possible mechanisms for stabilization of a MV state in a hydrogen bonded assembly including electron transfer (A), proton transfer (B) and proton coupled mixed valency (C). | ||
In all instances, stabilization of the MV state would be apparent from the observation of two sequential, one-electron redox processes in the cyclic voltammograms of the compounds. If the hydrogen bonds are sufficiently strong, direct overlap of the bridge π-orbitals through the hydrogen bond could allow electronic coupling between the electron donor and acceptor (mechanism A), which would be distinguished by the appearance of an intervalence charge transfer (IVCT), or charge-resonance, band in the UV-Vis/NIR spectrum. This has been observed in a ferrocene ureido pyrimidinedione complex reported by Kaifer and co-workers that forms a quadruply hydrogen bonded bridged dimer,6b and a triruthenium oxo-centered cluster having a pyridine-4-carboxylic acid ligand, reported by Kubiak, that forms a carboxylic acid bridged dimer.6d,e
An IVCT absorption corresponds to a transition from the ground state to an excited state in which electron transfer has occurred, but there is no change in molecular geometry (or reaction coordinate). As such if the electron transfer is related to the proton coordinate (i.e. changes to the molecular geometry) this transition would be absent, as in the case of proton transfer (mechanism B) or PCMV (mechanism C). This was found to be the case for the hydrogen bonded dimer [ReCl2(PnBu3)2(HBim)2]2 (H2Bim = 2,2′-biimidazole),6c which showed stabilization of the MV state in its cyclic voltammogram, but no IVCT transition in the corresponding UV-Vis/NIR spectrum. DFT calculations on the MV state showed that a proton transfer product was the most stable form of the MV state, suggesting that proton transfer (mechanism B) is responsible for its stabilization.
We have recently reported the synthesis of the MV hydrogen bonded ‘dimer of dimers’ [Mo2(TiPB)3(HDOP)]2+ ([I]2+; TiPB = 2,4,6-triisopropyl-benzoate; H2DOP = 3,6-dihydroxypyridazine) and [Mo2(TiPB)3(HDON)]2+ ([II]2+; H2DON = 2,7-dihydroxy-naphthyridine), shown in Chart 1.7 Cyclic voltammetry indicates stabilization of the MV state in these compounds, but no evidence of an IVCT transition in their UV-Vis/NIR absorption spectra. Unlike the aforementioned systems, there is no evidence for stabilization of a MV state through rearrangement of the system (PT) and as such the ET can be considered as directly related to the proton-coordinate, following a PCMV pathway (mechanism C).
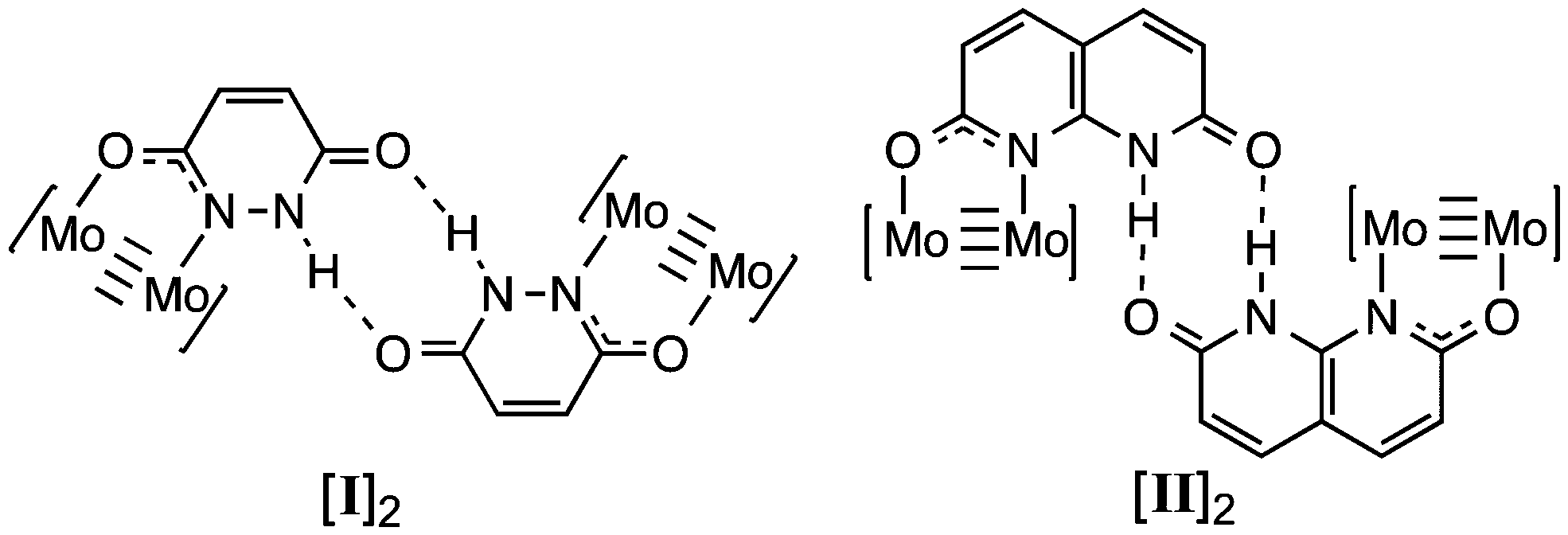 | ||
| Chart 1 Structures of [Mo2(TiPB)3(HDOP)]2 ([I]2) and [Mo2(TiPB)3(HDON)]2 ([II]2). The TiPB− ligands have been omitted for clarity. | ||
Whilst mixed-valency is generally synonymous with electronic coupling, the PT and PCMV examples demonstrate that future studies on hydrogen bonded MV compounds will require new mechanistic frameworks to be developed. An important measure when examining PT and PCMV will be the timescales associated with the proton and/or electron transfer. In this study we probe the timescale associated with PCMV in [I]2+, and propose a possible mechanism for this process, with the aid of DFT calculations.
Coalescence of vibrational bands in the IR spectra of MV compounds can be used to estimate ET rates, if electron transfer is faster than the vibrational timescale, ∼1010 s−1.8 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H stretches of the lactam bridging ligand are convenient handles for IR spectroelectrochemical measurements, which were performed on [I]2 in 0.1 M NBu4PF6/DCM solutions at −30 °C. The N-H stretch for [I]2 appears as a broad band at 3185 cm−1 (Fig. S1 in the ESI†), that upon oxidation to [I]22+ sharpens and shifts to 3247 cm−1. The band for the MV species [I]2+ appears to be a superposition of both the neutral and doubly oxidized compounds, but the bands are broad making comparison difficult. More success can be found from examination of the CO lactam stretches at around 1650 cm−1, shown in Fig. 2. The lactam stretch clearly shifts from 1647 to 1659 cm−1 upon oxidation from [I]2 to [I]22+, accompanied by a slight broadening of this band. The shift to higher wavenumber upon oxidation is due to reduced Mo2–δ → ligand–π* back bonding upon removal of an electron from the δ-orbital.
O and N–H stretches of the lactam bridging ligand are convenient handles for IR spectroelectrochemical measurements, which were performed on [I]2 in 0.1 M NBu4PF6/DCM solutions at −30 °C. The N-H stretch for [I]2 appears as a broad band at 3185 cm−1 (Fig. S1 in the ESI†), that upon oxidation to [I]22+ sharpens and shifts to 3247 cm−1. The band for the MV species [I]2+ appears to be a superposition of both the neutral and doubly oxidized compounds, but the bands are broad making comparison difficult. More success can be found from examination of the CO lactam stretches at around 1650 cm−1, shown in Fig. 2. The lactam stretch clearly shifts from 1647 to 1659 cm−1 upon oxidation from [I]2 to [I]22+, accompanied by a slight broadening of this band. The shift to higher wavenumber upon oxidation is due to reduced Mo2–δ → ligand–π* back bonding upon removal of an electron from the δ-orbital.
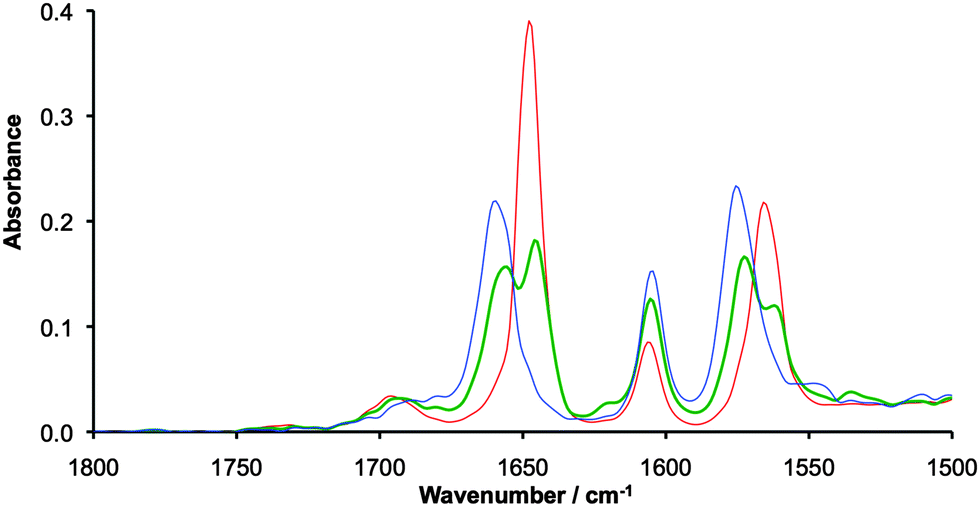 | ||
| Fig. 2 IR spectra of 20 mM solutions [I]2 (red), [I]2+ (green) and [I]22+ (blue) recorded in 0.1 M NBu4PF6/CH2Cl2 at −30 °C using a spectroelectrochemical cell. | ||
The mixed valence species [I]2+ shows two peaks at 1646 and 1654 cm−1, with no evidence of coalescence indicating electron transfer rates slower than 1010 s−1. This is a similar result to that found for the carboxylate bridged triruthenium oxo-centered clusters reported by Kubiak, in which ET was also found to be slower than the vibrational time scale.6e A second resonance associated with the DOP ligand can be seen at around 1575 cm−1 and shows similar behaviour to the NCO stretch at around 1650 cm−1. The resonance at around 1600 cm−1 is consistent with the ring breathing resonance found in the parent Mo2(TiPB)4 compound.
EPR spectroscopy is particularly useful for the determination of electron delocalization between dimolybdenum quadruply bonded compounds as the 95Mo and 97Mo isotopes (25% combined natural abundance) have I = 5/2.2c Hyperfine coupling constants of ca. 28 G indicate delocalization of the odd electron on one dimolybdenum unit, whereas hyperfine coupling constants of ∼14 G are observed if the electron is delocalized over two dimetal fragments.9 The EPR spectra of Mo2(TiPB)4+ and [I]2+ are compared in Fig. 3, and have giso values of 1.937 and 1.939, respectively. The poor resolution of the 95/97Mo hyperfine coupling constants is consistent with a previous study on Mo2(TiPB)4+.10 The 95/97Mo hyperfine coupling for Mo2(TiPB)4+ is 27.3 G, demonstrating delocalization of the odd electron equally over both molybdenum atoms.11 For [I]2+, the Mo–Mo bond is polarized as one Mo atom is coordinated to a nitrogen from the HDOP− ligand, whilst the other is coordinated to an oxygen atom. Therefore, two hyperfine coupling constants are expected. As observed for Mo2(TiPB)4+, the hyperfine coupling is weak, but Aiso values of 25.3 and 32.8 G can be resolved. This shows that the odd electron is localized on one dimetal unit rather than delocalized over both, and indicates that electron transfer is slower than the nanosecond timescale of EPR spectroscopy. This compares to PCET self-exchange rates observed in related metal complexes.12
 | ||
| Fig. 3 EPR spectra of Mo2(TiPB)4+PF6− (top) and [I]2+PF6− (bottom, 20 mM concentration) recorded in CH2Cl2 solutions at −90 °C. Regions of the spectra have been magnified to highlight hyperfine coupling. | ||
Previous studies have shown that electron transfer rates for PCET self-exchange reactions are often located between the EPR and electrochemical timescales, such as [FeII(H2bim)2(Hbim)]2+ + [FeIII(H2bim)3]2+ (H2bim = 2,2′-biimid-azoline) where kPCET = (5.8 ± 0.6) × 103 M−1 s−1 at 298 K,13,14 and [FeIII(H2bip)3]2+ + [FeII(H2bip)2(Hbip)]2+ (H2bip = 2,2′-bi(tetra-hydro)pyrimidine) where kPCET = (1.1 ± 0.2) × 104 M−1 s−1.15
As such, an electrochemical study on a deuterated version of [I]2, in which the bridging lactam hydrogens have been replaced by deuterium [I-D]2 has been performed. The cyclic voltammograms of [1-D]2 showed no variation from [I]2, see Fig. S2 (ESI†), indicating that the PCMV timescale is faster than the electrochemical timescale (∼10−1 s−1). Attempts to study NMR line broadening effects of [I]2+ by oxidation of [I]2 using either FcC(O)MePF6 or AgPF6 in CD2Cl2 proved to be unsuccessful. The generated paramagnetic species gave rise to a spectrum that displayed excessive broadening, from which no characteristic resonances could be identified for the solvate.
DFT calculations have been employed to explore changes associated with proton transfer in the compounds, and gain more insight into possible mechanisms for electron self-exchange in [I]2+. The model compound [(HCO2)3Mo2(HDOP)]2+ ([I′]2+), in which the TiPB− ligands have been replaced by formate ligands, has been used in this study, and geometry optimizations were performed using a PCM solvation model (CH2Cl2). The calculated ground state geometry of the MV compound [I′]2+ shows that the unpaired electron is localized on one dimolybdenum unit (see Fig. S3, ESI†), and the bridging ligands adopt the lactam (NH) tautomeric form. We have examined the potential energy surfaces associated with three possible proton transfer events; concerted double proton transfer (DPT), and, as [I′]2+ is an asymmetric molecule, two single proton transfer processes (SPT1 and SPT2), shown in Scheme 1. These were calculated by constraining the N–H bond distance along the proton transfer pathways, whilst allowing full geometry optimization for the rest of the molecule.
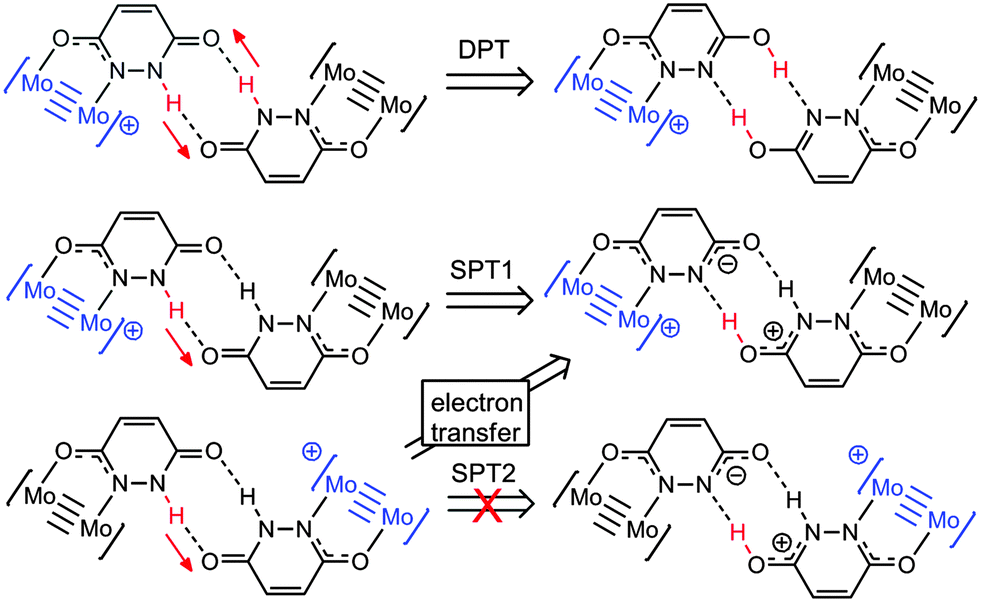 | ||
| Scheme 1 Proton transfer products resulting from concerted double proton transfer (DPT), and the two single proton processes (SPT1 and SPT2). | ||
The potential energy surface for DPT is shown in Fig. S4, with SPT1 and SPT2 compared in Fig. 4. For DPT, the barrier to proton transfer is around 10 kcal mol−1, and the DPT product is 4.0 kcal mol−1 higher in energy than the ground state. For SPT1, the barrier to electron transfer is slightly lower, but the proton transfer product is higher in energy (6.8 kcal mol−1).
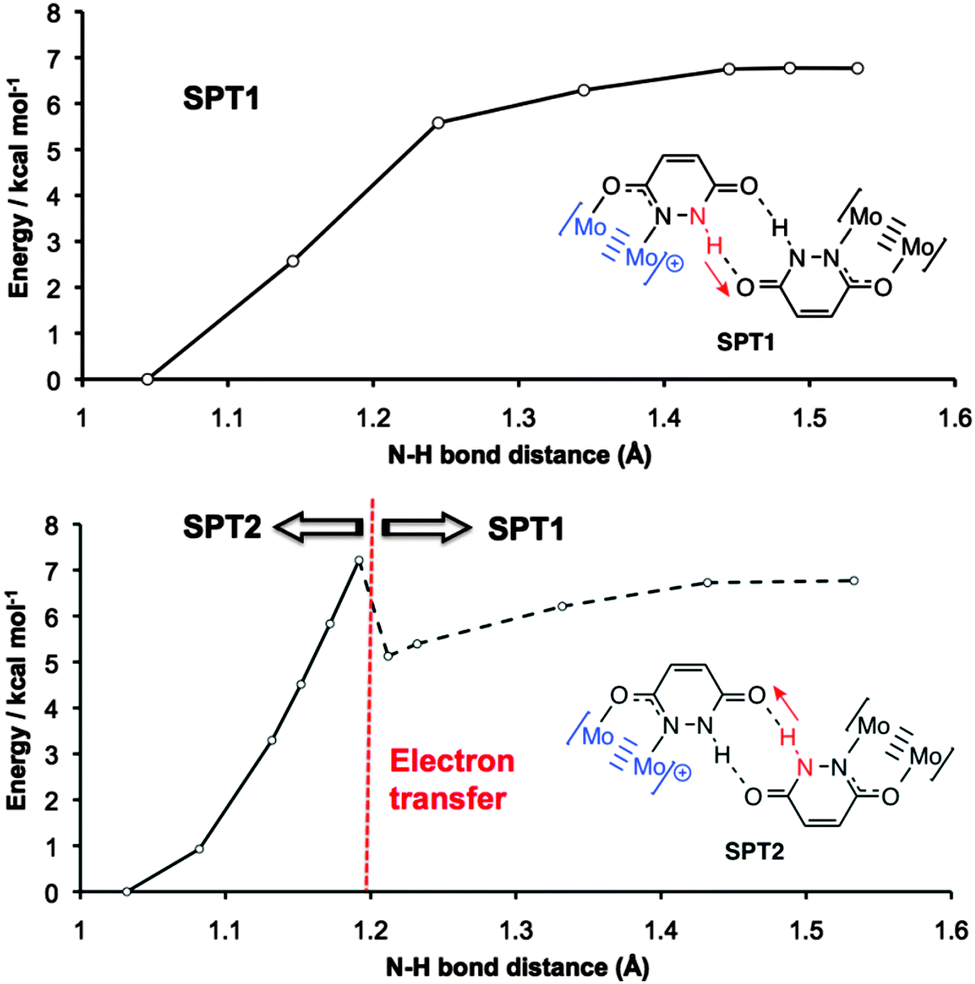 | ||
| Fig. 4 Calculated potential energy surfaces associated with SPT1 (top) and SPT2 (bottom) proton transfer processes in [I]2+. The dotted line for SPT2 indicates that electron transfer has taken place. | ||
The higher energy of the proton transfer products further supports the fact that simple proton transfer (mechanism B, Fig. 1) is unlikely to be responsible for stabilization of the MV state. However, it does not explain why electron transfer should be dependent on the proton coordinate in these systems.
Insight into the PCMV mechanism comes from inspection of the potential energy surface associated with SPT2, shown in Fig. 4. Moving along the proton coordinate, a larger dipole is induced in the molecule by the proton transfer, see Fig. S5 (ESI†). In order to minimize this dipole an electron is transferred from the Mo24+ donor to the Mo25+ acceptor, after moving only a short distance along the potential energy surface. We were unable to calculate any points further along the SPT2 potential energy surface due to the electron transfer that occurs upon geometry optimization. The barrier to this process is around 7.2 kcal mol−1 (Fig. 4), and the dotted line in the graph corresponds to the electron transfer product, which is now identical to the SPT1 species. Although it is unlikely that full proton transfer occurs, these calculations show that electron self-exchange in [I′]2+ is induced by the dipole associated with the change in proton coordinate from SPT2, and it is therefore likely that the movement of proton and electron is concerted as opposed to stepwise.
In summary, the rate constant for electron transfer in [I]2+ is slower than 109 s−1, with a dipole induced electron self-exchange mechanism that is dependent on the proton coordinate of the bridging ligand. This study highlights that the chemistry and physics of hydrogen-bonded mixed valence compounds extends beyond simple electronic coupling mechanisms. Furthermore, these results also suggest that hydrogen bonded materials incorporating redox active units may have interesting charge transport properties.
This work was funded by a Leverhulme Trust Research Project Grant (RPG-2014-303). The Royal Society is also gratefully acknowledged for the award of a University Research Fellowship (N. J. P.). The University of Sheffield thanked for studentship support (L. A. W.). All calculations were performed on the “Sol” cluster of the Theoretical Chemistry Group of the University of Sheffield.
Notes and references
- (a) S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Nature, 1995, 376, 660 CrossRef CAS PubMed; (b) S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673 CrossRef CAS PubMed.
- (a) A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326 CrossRef CAS PubMed; (b) W. Kaim and B. Sarkar, Coord. Chem. Rev., 2007, 251, 584 CrossRef CAS; (c) M. H. Chisholm and N. J. Patmore, Acc. Chem. Res., 2007, 40, 19 CrossRef CAS PubMed; (d) D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424 Search PubMed; (e) J. A. Thomas, Coord. Chem. Rev., 2013, 257, 1555 CrossRef CAS.
- (a) R. F. Winter, Organometallics, 2014, 33, 4517 CrossRef CAS; (b) M. Parthey and M. Kaupp, Chem. Soc. Rev., 2014, 43, 5067 RSC; (c) W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373 RSC.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284 CrossRef CAS PubMed.
- (a) P. J. Low, Coord. Chem. Rev., 2013, 257, 1507 CrossRef CAS; (b) Z. Cao, B. Xi, D. S. Jodoin, L. Zhang, S. P. Cummings, Y. Gao, S. F. Tyler, P. E. Fanwick, R. J. Crutchley and T. Ren, J. Am. Chem. Soc., 2014, 136, 12174 CrossRef CAS PubMed; (c) N. Weisbach, Z. Baranová, S. Gauthier, J. H. Reibenspies and J. A. Gladysz, Chem. Commun., 2012, 48, 7562 RSC.
- (a) M. Pichlmaier, R. F. Winter, M. Zabel and S. Zalis, J. Am. Chem. Soc., 2009, 131, 4892 CrossRef CAS PubMed; (b) H. Sun, J. Steeb and A. E. Kaifer, J. Am. Chem. Soc., 2006, 128, 2820 CrossRef CAS PubMed; (c) M. Tadokoro, T. Inoue, S. Tamaki, K. Fujii, K. Isogai, H. Nakazawa, S. Takeda, K. Isobe, N. Koga, A. Ichimura and K. Nakasuji, Angew. Chem., Int. Ed., 2007, 46, 5938 CrossRef CAS PubMed; (d) J. C. Goeltz and C. P. Kubiak, J. Am. Chem. Soc., 2010, 132, 17390 CrossRef CAS PubMed; (e) G. C. Canzi, J. C. Goeltz, J. S. Henderson, R. E. Park, C. Maruggi and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 1710 CrossRef CAS PubMed; (f) K. Tahara, T. Nakakita, S. Katao and J.-i. Kikuchi, Chem. Commun., 2014, 50, 15071 RSC; (g) Jin-Long, Y. Matsuda, K. Uemura and M. Ebihara, Inorg. Chem., 2015, 54, 2331 CrossRef CAS PubMed.
- (a) L. A. Wilkinson, L. McNeill, A. J. H. M. Meijer and N. J. Patmore, J. Am. Chem. Soc., 2013, 135, 1723 CrossRef CAS PubMed; (b) L. A. Wilkinson, L. McNeill, P. A. Scattergood and N. J. Patmore, Inorg. Chem., 2013, 52, 9683 CrossRef CAS PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Radiochem., 1967, 10, 247 CrossRef CAS.
- T. Ito, T. Hamaguchi, H. Nagino, T. Yamaguchi, J. Washington and C. P. Kubiak, Science, 1997, 277, 660 CrossRef CAS.
- M. H. Chisholm, B. D. Pate, P. J. Wilson and J. M. Zaleski, Chem. Commun., 2002, 1084 RSC.
- F. A. Cotton, L. M. Daniels, E. A. Hillard and C. A. Murillo, Inorg. Chem., 2002, 41, 1639 CrossRef CAS PubMed.
- M. H. Chisholm, J. S. D'Acchioli, B. D. Pate, N. J. Patmore, N. S. Dalal and D. J. Zipse, Inorg. Chem., 2005, 44, 1061 CrossRef CAS PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363 CrossRef CAS PubMed.
- J. P. Roth, S. Lovell and J. M. Mayer, J. Am. Chem. Soc., 2000, 122, 5486 CrossRef CAS.
- J. C. Yoder, J. P. Roth, E. M. Gussenhoven, A. S. Larsen and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 2629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General methods, synthesis of [1-D]2, IR spectra, electrochemical data, computational details. See DOI: 10.1039/c5cc07101a |
| This journal is © The Royal Society of Chemistry 2016 |