
Asymmetric synthesis of 1H-pyrrol-3(2H)-ones from 2,3-diketoesters by combination of aldol condensation with benzilic acid rearrangement†
Qiang
Sha
ab,
Hadi
Arman
b and
Michael P.
Doyle
*b
aSchool of Chemical Engineering, Nanjing University of Science and Technology, Xiao Ling Wei 200, Nanjing 210094, P. R. China
bDepartment of Chemistry, The University of Texas at San Antonio, San Antonio, Texas 78249, USA. E-mail: michael.doyle@utsa.edu
First published on 16th October 2015
Abstract
An efficient two-step protocol for the asymmetric synthesis of 1H-pyrrol-3(2H)-one derivatives in 99% ee from conveniently accessed 2,3-diketoesters has been developed.
2,2-Disubstituted 1H-pyrrol-3(2H)-ones that possess a chiral center at the 2-position occur widely in natural products,1 and they are also fundamental units which have been used to build molecules with significant biological activities (Fig. 1).2 Due to their importance, a variety of methods for their synthesis have been developed. Cycloaddition strategies are among the most efficient ways used to access the key 1H-pyrrol-3(2H)-ones.3 However, general highly enantioselective addition methods are rare: one begins with chiral starting materials,4 and the other employs catalytic asymmetric cycloaddition.5 Although progress is being made in this area, the narrow substrate scope of reported methods suggests the need for more general enantioselective processes.
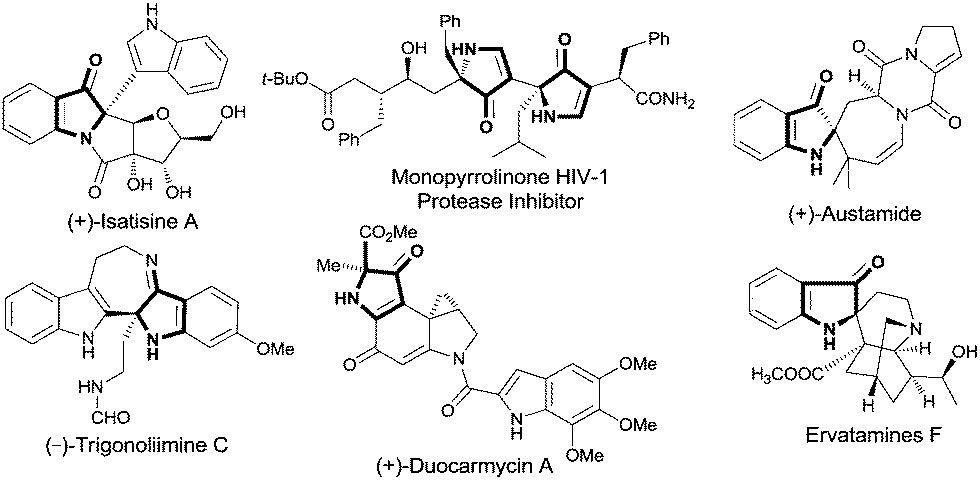 | ||
| Fig. 1 Selected natural products containing the 1H-pyrrol-3(2H)-one unit or its analogue with a tetrasubstituted carbon stereocenter. | ||
Pioneering work by Wasserman and coworkers6 demonstrated wide applications of vicinal tricarbonyl compounds (VTCs) in the synthesis of natural products and synthetic intermediates. Our group has applied VTCs in the synthesis of functionalized furans7 and pyrroles8 and demonstrated their convenient uses as hydrates.8b We also reported the first diastereoselective9 and enantioselective10 nucleophilic addition reactions of VTC compounds. Based on our understanding of the VTC chemistry and reported benzilic acid rearrangement reactions,11 an enantioselective strategy for the synthesis of 1H-pyrrol-3(2H)-ones starting from 2,3-diketoester hydrates was designed as shown in Scheme 1. The mixed asymmetric aldol reaction with VTCs has been unexplored. The enamine formation step is known,12 although not in this system. To form the chiral key intermediate 3, a chiral secondary amine-catalyzed aldol reaction was predicted to have the potential to achieve this goal.
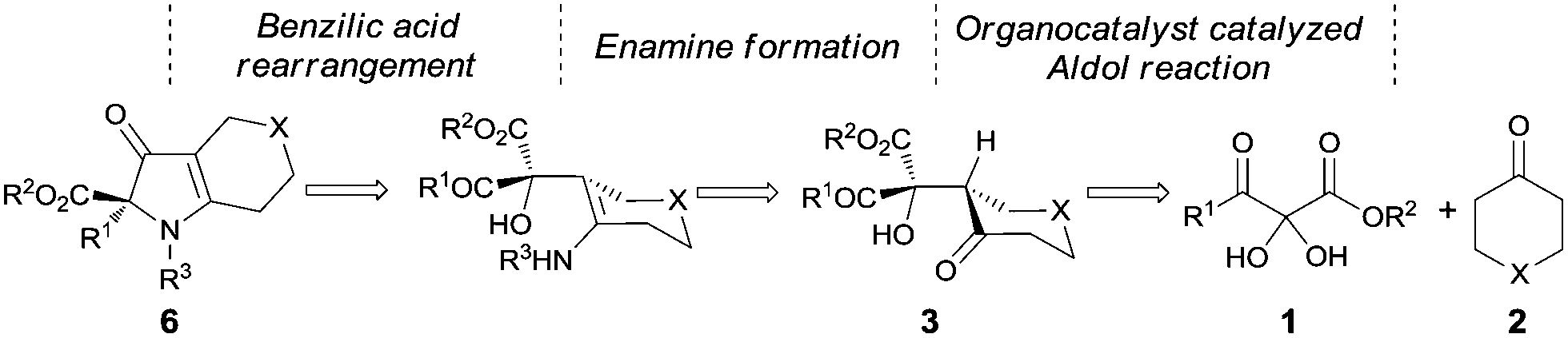 | ||
| Scheme 1 A designed strategy for the asymmetric synthesis of 1H-pyrrol-3(2H)-ones. | ||
The Hajos–Parrish–Eder–Sauer–Wiechert reaction, the first example of asymmetric enamine catalysis, was reported 50 years ago.13 However this powerful reaction was relatively unexplored until List and coworkers14 discovered proline-catalyzed enantioselective intermolecular aldol reaction. Explosive progress ensued during which various organocatalysts were developed to realize the asymmetric aldol reaction.15 Many types of carbonyl substrates have been successfully utilized in aldol reactions, including various aromatic aldehydes, aliphatic aldehydes and activated ketones that include 2-ketoesters. However, 2,3-diketoesters, which are unique and highly activated ketones, have not been investigated. Herein, we present the two-step asymmetric synthesis of 1H-pyrrol-3(2H)-ones that takes unique advantage of the 2,3-diketoester framework to couple an L-proline catalyzed aldol reaction with the benzilic acid rearrangement.
Vicinal tricarbonyl compounds easily absorb water to form their corresponding hydrates that can be dehydrated by heating under vacuum. However, the VTC hydrates were used directly in this study due to their convenience in handling. Based on our initial hypothesis, benzyl 2,2-dihydroxy-3-oxobutanoate hydrate 1a and cyclohexanone 2a were selected for the aldol reaction. Of all the organocatalysts we examined,16L-proline was found to be optimal for diastereoselectivity and enantio control.17 The aldol product was formed in 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :26 d.r., and the major diastereoisomer 3a was conveniently isolated by simple chromatography in 68% yield with 99% ee. All attempts to increase diastereoselectivity with alternative catalysts, lowering reaction temperatures, use of 1a in its anhydrous form, and changing solvents were unsuccessful.16 However, the good yield of the major diastereomer, its ease of isolation, and its excellent enantiomeric excess made this transformation very promising.
:26 d.r., and the major diastereoisomer 3a was conveniently isolated by simple chromatography in 68% yield with 99% ee. All attempts to increase diastereoselectivity with alternative catalysts, lowering reaction temperatures, use of 1a in its anhydrous form, and changing solvents were unsuccessful.16 However, the good yield of the major diastereomer, its ease of isolation, and its excellent enantiomeric excess made this transformation very promising.
With the optimal reaction conditions in hand, we set out to explore the substrate generality of the L-proline catalyzed aldol reactions. Using hydrated 2,3-diketoesters, the effect of different groups on the ester was investigated first (Table 1, entries 1–3). The size of the group did not significantly affect yield or stereoselectivity of products. The major diastereomers were isolated in moderate yields with 99% ee, and the minor diastereomers were also obtained with high ee's. Then 2,3-diketoesters with different groups on the keto side were investigated: replacing the methyl group with ethyl or benzyl gave similar results (Table 1, entries 4 and 5). However, changing the methyl group bound to the keto group to aryl resulted in a significant increase in the d.r. of the aldol products (Table 1, entries 6–12) up to 89:11 (Table 1, entries 7 and 8). The effects of different substituents on the aromatic ring were also investigated showing that electron-withdrawing groups favored this process (Table 1, entry 10). An electron-donating para-substituted methoxy group, however, strongly inhibited the aldol process (Table 1, entry 9). Dihydro-2H-thiopyran-4(3H)-one (2b) worked well in this process as an alternative to cyclohexanone from which the major diastereoisomer 3m was obtained in 56% isolated yield with 99% ee (Table 1, entry 13) (Scheme 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Time (h) | Ratio 3/4b | Yieldc,d (ee) of 3 | Yieldc,d (ee) of 4 |
| a Reaction conditions: 1 (1.0 mmol), 2 (2.0 mmol), L-proline (20 mol%), DCM (5.0 mL), r.t. 24–96 h. b Determined by 1H NMR spectroscopy or HPLC analysis of the reaction mixture. c Yield of the isolated product after column chromatography. d The ee value was determined by HPLC using a chiral stationary phase. | |||||||
| 1 | Me | Bn | CH2 | 24 | 74/26 | 3a/68% (99%) | 4a/24% (97%) |
| 2 | Me | Me | CH2 | 32 | 79/21 | 3b/60% (99%) | 4b/16% (97%) |
| 3 | Me | Cy | CH2 | 48 | 77/23 | 3c/67% (99%) | 4c/20% (98%) |
| 4 | Et | Me | CH2 | 32 | 77/23 | 3d/65% (99%) | 4d/19% (97%) |
| 5 | Bn | Bn | CH2 | 48 | 73/27 | 3e/59% (99%) | 4e/21% (98%) |
| 6 | C6H5 | Et | CH2 | 32 | 81/19 | 3f/51% (99%) | 4f/12% (98%) |
| 7 | p-ClC6H4 | Et | CH2 | 48 | 89/11 | 3g/58% (99%) | 4g/7% (94%) |
| 8 | p-BrC6H4 | Et | CH2 | 48 | 89/11 | 3h/56% (99%) | 4h/6% (95%) |
| 9 | p-OMeC6H4 | Et | CH2 | 96 | 85/15 | 3i/25% (99%) | 4i/4% (98%) |
| 10 | p-CNC6H4 | Et | CH2 | 42 | 84/16 | 3j/70% (99%) | 4j/13% (96%) |
| 11 | 2-Naphthyl | Et | CH2 | 54 | 79/21 | 3k/45% (99%) | 4k/12% (97%) |
| 12 | 2-Thienyl | Et | CH2 | 48 | 88/12 | 3l/67% (99%) | 4l/9% (92%) |
| 13 | Me | Bn | S | 48 | 76/24 | 3m/56% (99%) | 4m/17% (89%) |
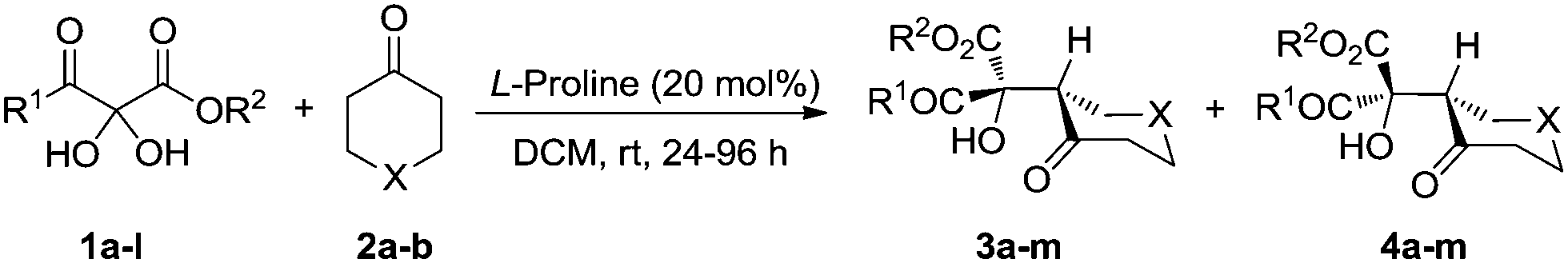
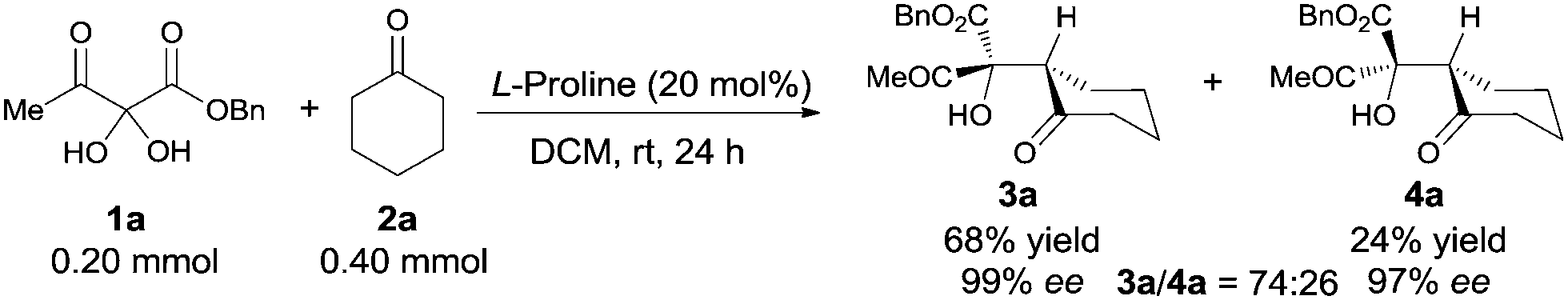 | ||
| Scheme 2 L-Proline catalyzed aldol reaction of benzyl 2,2-dihydroxy-3-oxobutanoate (1a) with cyclohexanone (2a). | ||
The high efficiency and broad generality of the aldol reaction that gave, without exception, the major diastereomers with 99% ee's was evident throughout investigations of substrate scope. A limitation that did appear was that ketone ring sizes other than six, especially cyclopentanone and cycloheptanone, gave intractable mixtures of products.
The high enantio control in these aldol condensation reactions prompted us to investigate the subsequent benzilic acid rearrangement reaction. Initially, we attempted to combine the aldol reaction and the benzilic acid rearrangement reaction in one pot. As shown in Table 2, without adding any additives, 6a was obtained in only 32% yield and 30% ee (Table 2, entry 1). Various additives were examined from which we discovered that by using 20 mol% trifluoroacetic acid the yield of 6a increased significantly. However, 6a was always obtained in only moderate enantiomeric excess no matter what solvent was used (Table 2, entries 2–6) reflecting the uniform conversions of each aldol product diastereomer (3a and 4a) to 1H-pyrrol-3(2H)-one 6a in inverse enantiomeric excesses. This result indicated that a two-step procedure to synthesize 1H-pyrrol-3(2H)-ones in which the major diastereomer from the aldol condensation is used for the benzilic acid rearrangement would be successful.
|
|
||||
|---|---|---|---|---|
| Entry | Additive | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: (1) 1a (0.10 mmol), 2a (0.20 mmol), L-proline (20 mol%), solvent (0.50 mL), r.t. 24 h; (2) 5a (0.11 mmol), CF3COOH (20 mol%), 65 °C, 24 h. b Yield of the isolated product after column chromatography. c The ee value was determined by HPLC using a chiral stationary phase. | ||||
| 1 | — | MeCN | 32 | 30 |
| 2 | CF3COOH | MeCN | 82 | 31 |
| 3 | CF3COOH | DCM | 67 | 47 |
| 4 | CF3COOH | CHCl3 | 64 | 41 |
| 5 | CF3COOH | DCE | 72 | 44 |
| 6 | CF3COOH | Toluene | 55 | 41 |
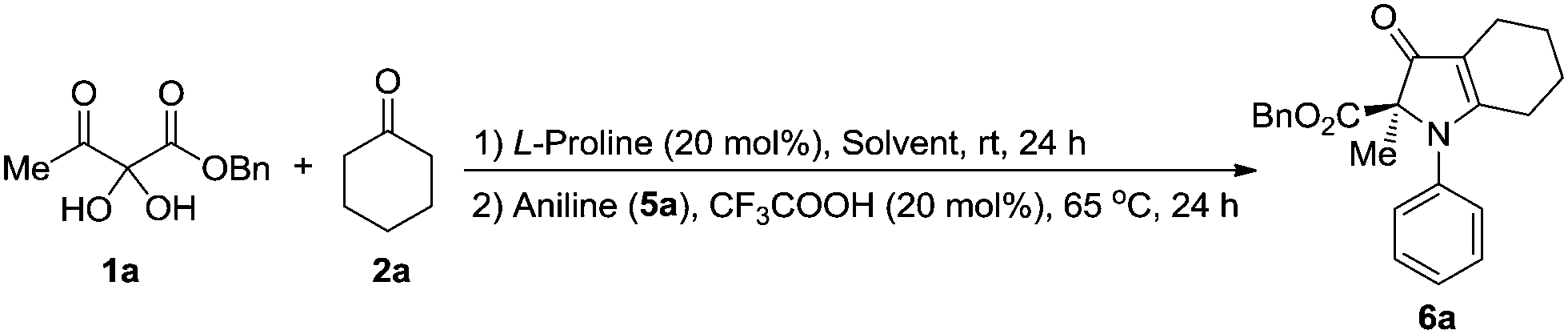
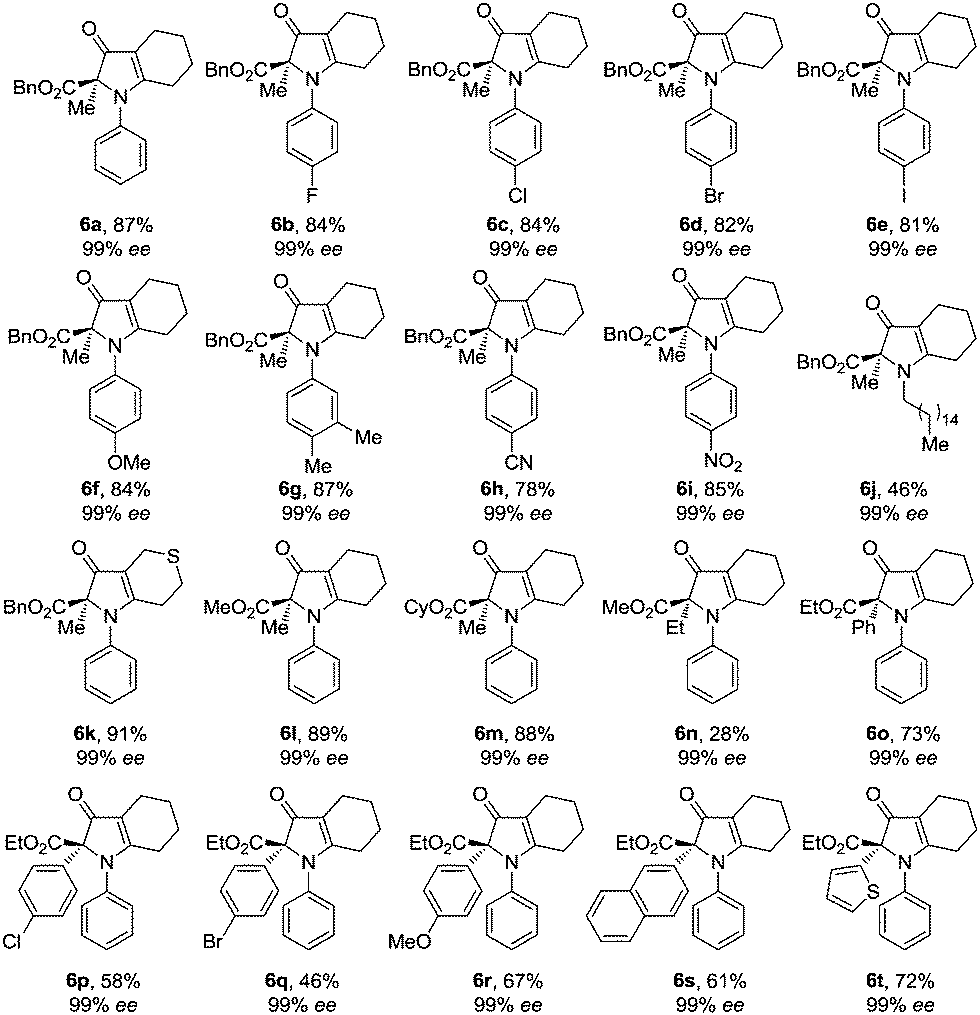
With chromatographic isolation of the major diastereomers from the asymmetric aldol reactions, we examined the benzilic acid rearrangement of these compounds with aniline. By using trifluoroacetic acid as an additive and DCM as the solvent at 65 °C for 24 h, the rearrangement product 6a was isolated in 87% yield with 99% ee (Table 3, 6a). Anilines with halogen atoms (F, Cl, Br, I) all gave the 1H-pyrrol-3(2H)-ones in high yield with 99% ee (Table 3, 6b–6e). Anilines with both electron-donating groups (Me, OMe) and electron-withdrawing groups (CN, NO2) gave the corresponding products in good yields without loss of enantioselectivity (Table 3, 6f–6i). Hexadecylamine, which was chosen as a representative aliphatic amine, gave 6j in 46% yield with 99% ee (Table 3, 6j). The presence of a sulfur atom on the cyclic aliphatic ring (3m), gave the benzilic acid rearrangement product 6k in 91% yield with 99% ee (Table 3, 6k). Different groups on the ester all gave the corresponding 1H-pyrrol-3(2H)-one products in good yields with 99% ee (Table 3, 6l–6m). By changing the substituent from methyl to ethyl on the keto side, a significant decrease of the product yield was observed (Table 3, 6n) and was further limited with other alkyl substituents. However, by replacing the methyl group with phenyl, 6o was obtained in 73% yield with 99% ee (Table 3, 6o). Lastly, the effects from various aryl groups were studied from which the benzylic acid rearrangement products were obtained in moderate to good yields with 99% ee (Table 3, 6p–6t).
|
|
|---|
| a Reaction conditions: 3 (0.10 mmol), 5 (0.11 mmol), DCM (0.50 mL), CF3COOH (20 mol%), 65 °C, 24 h; yields refer to isolated yields after column chromatography; the ee value was determined by HPLC using a chiral stationary phase. |
|
The synthetic utility of the present methodology involving the L-proline catalyzed aldol reaction was examined on a 10 mmol scale.16 Under the optimized reaction conditions, the aldol products were formed in 74:26 d.r. The major diastereoisomer was isolated in 61% yield (1.86 g) with 99% ee. The minor diastereoisomer was obtained in 21% yield (0.63 g) with 98% ee. These separated aldol products were then used to perform the benzylic acid rearrangement. Products 6d and 7d were obtained in 82% yield (2.19 g) and 84% yield (0.77 g) respectively, both with excellent enantiomeric excess.
The relative and absolute configurations of 3g and 6c were assigned (R,S)-3g and R-6c based on their single-crystal X-ray diffraction analysis (Fig. 2).18
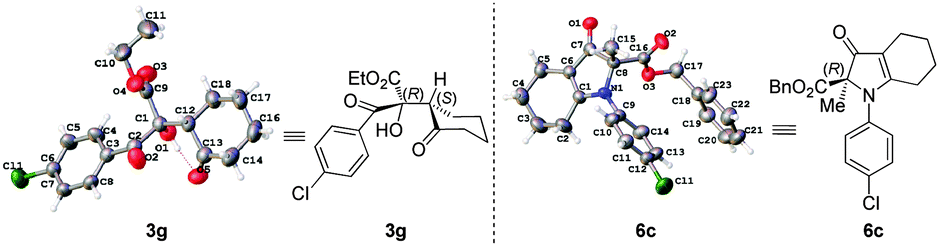 | ||
| Fig. 2 X-ray structures of 3g and 6c. | ||
Reduction of 6d with 4.0 equiv. NaBH4 gave 8d as the sole diastereomer in 81% yield with 99% ee. The configuration of 8d was determined by NOESY experiments. In addition, 6d is conveniently oxidized to 9d in 65% yield with 98% ee, and this process provides convenient access to nearly optically pure indolin-3-ones (Scheme 3).
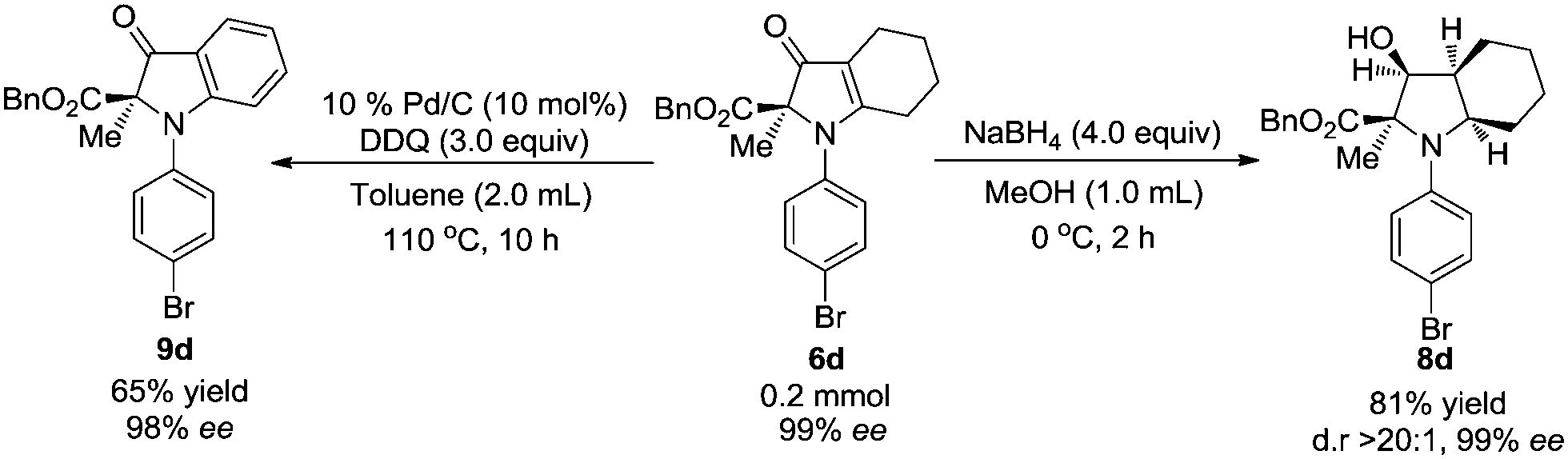 | ||
| Scheme 3 Reduction and oxidation of 1H-pyrrol-3(2H)-one 6d. | ||
In summary, we have developed a two-step method for the highly enantioselective synthesis of 1H-pyrrol-3(2H)-ones. 2,3-Diketoesters have been employed for the first time in asymmetric aldol reactions. By combining the L-proline catalyzed aldol reaction with the benzylic acid rearrangement, 1H-pyrrol-3(2H)-ones are obtained in moderate yields but with notably excellent enantiomeric excess (99% ee for all (R,S)-products). Efforts are underway to examine additional applications of 2,3-diketoesters in highly enantioselective catalytic reactions.
Qiang Sha acknowledges China Scholarship Council (CSC) for his financial support. We acknowledge U.S. National Science Foundation (CHE-1212446), The Welch Foundation, and the University of Texas at San Antonio for supporting this research. The HRMS used in this research was supported by a grant from the National Institutes of Health (G12MD007591).
Notes and references
- For selected examples, see: (a) W. H. Pearson, Y. Mi, I. Y. Lee and P. Stoy, J. Am. Chem. Soc., 2001, 123, 6724 CrossRef CAS PubMed; (b) K. Amada, T. Kurokawa, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2003, 125, 6630 CrossRef PubMed; (c) R. M. Williams, J. H. Cao, H. Tsujishima and R. J. Cox, J. Am. Chem. Soc., 2003, 125, 12172 CrossRef CAS PubMed; (d) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2010, 75, 6830 CrossRef CAS PubMed; (e) D. B. Zhang, D. G. Yu, M. Sun, X. X. Zhu, X. J. Yao, S. Y. Zhou, J. J. Chen and K. Gao, J. Nat. Prod., 2015, 78, 1253 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Brüggemann, A. I. McDonald, L. E. Overman, M. D. Rosen, L. Schwink and J. P. Scott, J. Am. Chem. Soc., 2003, 125, 15284 CrossRef PubMed; (b) M. Mucedda, D. Muroni, A. Saba and C. Manassero, Tetrahedron, 2007, 63, 12232 CrossRef CAS; (c) B. N. Reddy and C. V. Ramana, Chem. Commun., 2013, 49, 9767 RSC; (d) S. Han, K. C. Morrison, P. J. Hergenrother and M. Movassaghi, J. Org. Chem., 2014, 79, 473 CrossRef CAS PubMed; (e) A. B. Smith III, L. D. Cantin, A. Pasternak, L. Guise-Zawacki, W. Q. Yao, A. K. Charnley, J. Barbosa, P. A. Sprengeler, R. Hirschmann, S. Munshi, D. B. Olsen, W. A. Schleif and L. C. Kuo, J. Med. Chem., 2003, 46, 1831 CrossRef PubMed; (f) A. B. Smith III, A. K. Charnley and R. Hirschmann, Acc. Chem. Res., 2011, 44, 180 CrossRef PubMed.
- (a) M. A. Gomaa, J. Chem. Soc., Perkin Trans. 1, 2002, 341 RSC; (b) J. Huang, Y. J. Liang, W. Pan, Y. Yang and D. W. Dong, Org. Lett., 2007, 9, 5345 CrossRef CAS PubMed; (c) I. Kim and K. Kim, Org. Lett., 2010, 12, 2500 CrossRef CAS PubMed; (d) P. X. Zhou, Z. Z. Zhou, Z. S. Chen, Y. Y. Ye, L. B. Zhao, Y. F. Yang, X. F. Xia, J. Y. Luo and Y. M. Liang, Chem. Commun., 2013, 49, 561 RSC; (e) Z. K. Wang, X. H. Bi, P. Q. Liao, X. Liu and D. W. Dong, Chem. Commun., 2013, 49, 1309 RSC; (f) Z. J. Zhang, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Org. Lett., 2013, 15, 4822 CrossRef CAS PubMed; (g) S. R. Mothe, M. L. Novianti, B. J. Ayers and P. W. H. Chan, Org. Lett., 2014, 16, 4110 CrossRef CAS PubMed; (h) X. Sun, P. H. Li, X. L. Zhang and L. Wang, Org. Lett., 2014, 16, 2126 CrossRef CAS PubMed; (i) N. Li, T. Y. Wang, L. Z. Gong and L. M. Zhang, Chem. – Eur. J., 2015, 21, 3585 CrossRef CAS PubMed.
- (a) A. B. Smith III, H. Liu, H. Okumura, D. A. Favor and R. Hirschmann, Org. Lett., 2000, 2, 2041 CrossRef; (b) N. Gouault, M. L. Roch, C. Cornée, M. David and P. Uriac, J. Org. Chem., 2009, 74, 5614 CAS; (c) R. Spina, E. Colacion, B. Gabriele, G. Salerno and J. Martinez, J. Org. Chem., 2013, 78, 2698 CrossRef CAS PubMed.
- (a) J. X. Liu, Q. Q. Zhou, J. G. Deng and Y. C. Chen, Org. Biomol. Chem., 2013, 11, 8175 RSC; (b) Q. Yin and S. L. You, Chem. Sci., 2011, 2, 1344 RSC; (c) M. Rueping, S. Raja and A. Núñez, Adv. Synth. Catal., 2011, 353, 563 CrossRef CAS; (d) A. Parra, R. Alfaro, L. Marzo, A. Moreno-Carrasco, J. L. Garcia Ruano and J. Aleman, Chem. Commun., 2012, 48, 9759 RSC; (e) Y. L. Zhao, Y. Wang, J. Cao, Y. M. Liang and P. F. Xu, Org. Lett., 2014, 16, 2438 CrossRef CAS PubMed; (f) C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232 CrossRef CAS PubMed; (g) R. R. Liu, S. C. Ye, C. J. Lu, G. L. Zhuang, J. R. Gao and Y. X. Jia, Angew. Chem., Int. Ed., 2015, 54, 11205 CrossRef CAS PubMed.
- H. H. Wasserman and H. Parr, Acc. Chem. Res., 2004, 37, 687 CrossRef CAS PubMed.
- P. Truong, X. F. Xu and M. P. Doyle, Tetrahedron Lett., 2011, 52, 2093 CrossRef CAS PubMed.
- (a) P. Truong, M. D. Mandler and M. P. Doyle, Tetrahedron Lett., 2015, 56, 3042 CrossRef CAS; (b) Q. Sha, H. Arman and M. P. Doyle, Org. Lett., 2015, 17, 3876 CrossRef CAS PubMed.
- P. Truong, C. S. Shanahan and M. P. Doyle, Org. Lett., 2012, 14, 3608 CrossRef CAS PubMed.
- P. Truong, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2014, 53, 6468 CrossRef CAS PubMed.
- (a) D. Askin, R. A. Reamer, T. K. Jones, R. P. Volante and I. Shinkai, Tetrahedron Lett., 1989, 30, 671 CrossRef CAS; (b) D. Askin, R. A. Reamer, D. Joe, R. P. Volante and I. Shinakai, Tetrahedron Lett., 1989, 45, 6121 CrossRef; (c) M. J. Fisher, K. Chow, A. Villalobos and S. J. Danishefsky, J. Org. Chem., 1991, 56, 2900 CrossRef CAS; (d) H. H. Wasserman, D. S. Ennis and C. B. Vu, Tetrahedron Lett., 1991, 32, 6039 CrossRef CAS.
- For selected examples, see: (a) W. Schrader, P. P. Handayani, J. Zhou and B. List, Angew. Chem., Int. Ed., 2009, 48, 1463 CrossRef CAS PubMed; (b) L. Ren, T. Lei and L. Z. Gong, Chem. Commun., 2011, 47, 11683 RSC; (c) Y. M. Deng, L. Liu, R. G. Sarkisian, K. Wheeler, H. Wang and Z. H. Xu, Angew. Chem., Int. Ed., 2013, 52, 3663 CrossRef CAS PubMed; (d) Y. M. Deng, S. Kumar, K. Wheeler and H. Wang, Chem. – Eur. J., 2015, 21, 7874 CrossRef CAS PubMed.
- H. Pracejus, Justus Liebigs Ann. Chem., 1960, 634, 23 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- For selected reviews on organocatalyst catalysed aldol reactions, see: (a) M. Movassaghi and E. N. Jacobsen, Science, 2002, 298, 1904 CrossRef CAS PubMed; (b) B. List, Acc. Chem. Res., 2004, 37, 548 CrossRef CAS PubMed; (c) C. Allemann, R. Gordollo, F. R. Clemente, P. H. Cheong and K. N. Houk, Acc. Chem. Res., 2004, 37, 558 CrossRef CAS PubMed; (d) S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570 CrossRef CAS PubMed; (e) M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666 RSC; (f) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (g) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (h) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed; (i) D. W. C. Macmillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (j) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (k) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (l) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed; (m) C. M. Marson, Chem. Soc. Rev., 2012, 41, 7712 RSC.
- See the ESI† for details.
- This finding is consistent with results of Jørgensen and Maruoka for aldol reactions with 2-ketoesters (50 mol% proline with up to 99% ee): (a) A. Bøgevig, N. Kumaragurubaran and K. A. Jørgensen, Chem. Commun., 2002, 620 RSC; (b) O. Tokuda, T. Kano, W.-G. Gao, T. Ikemoto and K. Maruoka, Org. Lett., 2005, 7, 5103 CrossRef CAS PubMed.
- CCDC 1417169 (3g) and 1417170 (6c).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimization of reaction conditions, HPLC and spectral data for all new compounds. CCDC 1417169 (3g) and 1417170 (6c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07780j |
| This journal is © The Royal Society of Chemistry 2016 |