
A nitrogen-containing carbon film derived from vapor phase polymerized polypyrrole as a fast charging/discharging capability anode for lithium-ion batteries†
Tao
Yuan‡
abc,
Yu-Shi
He‡
 a,
Weimin
Zhang
*ab and
Zi-Feng
Ma
*ab
a,
Weimin
Zhang
*ab and
Zi-Feng
Ma
*ab
aShanghai Electrochemical Energy Devices Research Centre, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: zfma@sjtu.edu.cn; wm_zhang@sjtu.edu.cn
bSinopoly Battery Research Centre, Shanghai, 200241, China
cSchool of Materials Science and Engineering, University of Shanghai for Science and Technology, Shanghai, 200093, China
First published on 15th October 2015
Abstract
A nitrogen-containing carbon (N–C) film was synthesized by pyrolysis of vapor phase polymerized polypyrrole (PPy). This carbon film exhibits excellent rate capability and cyclability as a lithium-ion battery anode. The reversible capacities are 908.4, 825.7, 664.0, 531.6, 415.5 and 325.9 mA h g−1 at 1C, 2C, 5C, 10C, 20C and 40C, respectively.
Rechargeable lithium-ion batteries are used extensively due to their high energy and power densities.1 State-of-the-art lithium secondary batteries typically use graphite as the anode, which has a low theoretical specific capacity (372 mA h g−1) and limited rate capability.2 Thus, new carbon based anode materials such as carbon nanotubes,3 nanofibers,4 nanobeads,5 hollow nanospheres,6 graphene7 and their hybrids8,9 with enhanced Li+ storage capacities and high rate performance have been explored as alternative candidates for anodes of Li-ion batteries.
Another approach to modulate the electrochemical properties of carbon-based anodes is chemical doping of heteroatoms such as N, B, S, P or F.10–12 For example, N-doping can enhance the electrical conductivity, and hence the Li+ storage capacity by electron donors enhancing the redox reactions of N-containing functional groups.13 Devising N-doping methods for carbon-based materials is critical to their future application. One common method is employing conductive polymers (e.g. PPy and polyaniline) as nitrogen doped carbon sources, which can be used to obtain active N-rich carbon materials by pyrolysis at a high temperature.4,14,15
Furthermore, for the development of high energy density Li-ion battery systems and more lightweight and bendable batteries for potential application in flexible electronic devices such as wearable devices, artificial electronic skins, and distributed sensors, soft, free-standing electrode-active materials without binders and conductive agents are significant. Therefore, combining the above needs to construct free-standing and flexible carbon-based anodes with high-electrochemical performance, we propose here an N-rich pure carbon film synthesized by calcination of a vapour phase polymerized PPy film as an anode for Li-ion batteries. The PPy precursor film was synthesized via a simple vapor phase polymerization (VPP) bottom-up assembly method which was first described by Mohammadi et al. using FeCl3 or H2O2 as the oxidant.16 However, the FeCl3 oxidant easily forms crystals when the solvent evaporates, resulting in poor quality conducting polymer films with many grainy particles. Therefore, ferric p-toluenesulfonate (Fe(III) tosylate) was explored as an oxidant which was found to be well-suited for the preparation of the PPy film via a VPP process.17 Using the Fe(III) tosylate oxidant, we can easily obtain a PPy film with a smooth surface and continuously cross-linked structure. After carbonization at a high temperature in an inert atmosphere, we noticed that a continuously cross-linked carbon film doped with nitrogen could be obtained. The as-derived free-standing nitrogen-containing carbon film anode exhibits excellent electrochemical performance, especially, at high charge/discharge rate for lithium-ion batteries. Such a free-standing flexible N–C film anode could be straightforwardly used to fabricate the lithium-ion batteries which are required to deliver high energy and power. The mechanism and typical procedure for the synthesis of the PPy film are shown schematically in Fig. 1. Typically, Fe(III) tosylate solution can be easily coated onto a glass slide to form a uniform oxidant film. Once the Fe(III) tosylate film was dried on a hot plate and transferred into a chamber full of vapor pyrrole under ambient conditions, the pyrrole monomer will polymerize and thus form a conducting PPy film. The reaction includes two steps.17,18 Firstly, single pyrrole monomer molecules were oxidized by Fe3+ to generate cation radicals. Meanwhile, Fe(III) tosylate was produced as a consequence of pyrrole oxidation. During the polymerization process, i.e. two cationic free radicals combine together to form bipyrrole. The p-toluenesulfonate served as a dopant to form the conducting PPy. As the polymerization reaction continued, the chain length kept growing and eventually led to a structure of repeated units and high molecular weight. Typically, the as-obtained PPy film shows good flexibility and a homogeneous morphology of continuously cross-linked sheets with the film fibre diameter in the range of 40–60 nm (Fig. 1 and 2a). After annealing at 700 °C for 2 h under an Ar atmosphere, the as-obtained N–C film was formed with a similar continuously cross-linked web sheet microstructure to the PPy precursor and great flexibility (Fig. 1 and 2b). Such an interconnected surface structure is beneficial for obtaining a high electronic conductivity (5.5 S cm−1) for the anode of lithium-ion batteries. Transmission electron microscopy (TEM) in Fig. 2c reveals that large quantities of mesopores are homogeneously distributed within the carbon film. The uniform mesopores may originate from the release of gas products from the PPy precursor during the pyrolysis process. The specific Brunauer–Emmett–Teller (BET) surface area of the obtained carbon film is 38.8 m2 g−1 and the Barrett–Joyner–Halenda (BJH) pore size distribution in Fig. S1 (ESI†) demonstrates that the carbon film exhibits a mesoporous structure with a mean pore size of 25.9 nm, which is expected to provide very regular pore channels for Li+ charge transfer and storage. This result can be also confirmed by observation of the TEM images.
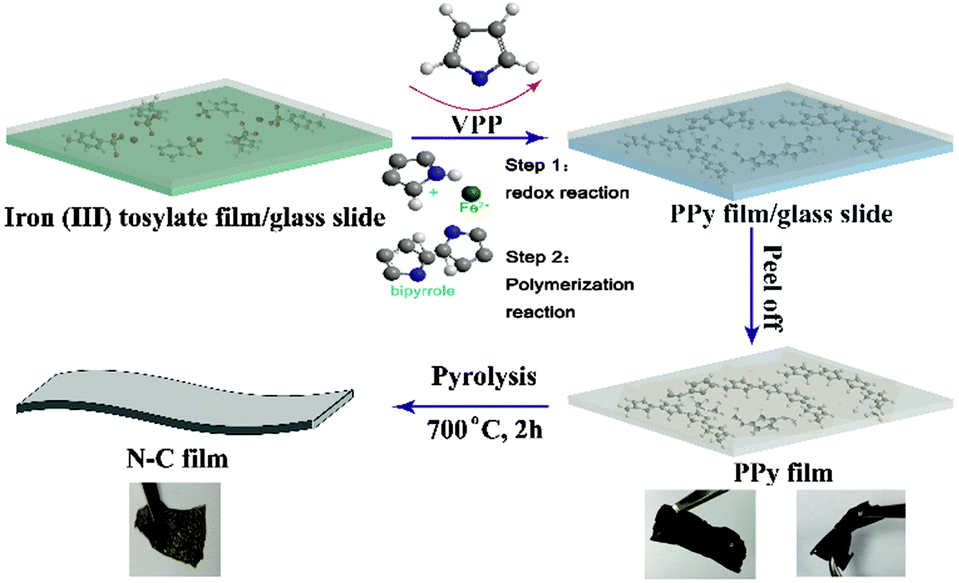 | ||
| Fig. 1 Schematic illustration of the synthesis process of N–C film. | ||
 | ||
| Fig. 2 SEM images and digital photos (inset) of (a) PPy film and (b) the corresponding N–C film, and (c) TEM image of N–C film. | ||
In some earlier reports, polypyrrole has been determined as an amorphous polymer.19 However, a study of the XRD pattern of the present PPy film (Fig. S2a, ESI†) reveals some degree of crystallinity, with the appearance of two broad peaks in the region of about 2θ = 16° and 22.4°. It implies that the synthesized PPy films have a more ordered arrangement, which is also helpful to improve their electrical conductivity.20 After carbonization of the PPy film at a high temperature of 700 °C for 2 h in an Ar atmosphere, the as-prepared carbon film obtained showed two characteristic XRD peaks located at about 25° and 43°. The low intensity peak at around 43° is assigned to the (100) plane of a hexagonal carbon material (JCPDS, Card No. 75-1621). The Bragg reflection at 25° corresponding to the (002) plane shows a broad width, which suggests that the as-obtained carbon film possesses a low degree of graphitization.21
As shown in Fig. S2b (ESI†), both PPy and N–C films feature two Raman shifts. The peak at about 1570 cm−1 of the PPy film is associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds stretching mode of oxidized and neutral species while the other peak located at about 1345 cm−1 corresponds to the ring stretching mode of PPy.22 The Raman spectra of the carbon film also exhibit two peaks at similar Raman shifts of around 1570 cm−1 and 1350 cm−1, which are identified respectively as the D band (defect-induced mode) and G band (E2g mode of graphite) of the carbon film.8 The intensity ratio of ID/IG is calculated to be 0.92, which indicates that the carbon film has a low degree of graphitization, which was also confirmed by XRD studies.
C bonds stretching mode of oxidized and neutral species while the other peak located at about 1345 cm−1 corresponds to the ring stretching mode of PPy.22 The Raman spectra of the carbon film also exhibit two peaks at similar Raman shifts of around 1570 cm−1 and 1350 cm−1, which are identified respectively as the D band (defect-induced mode) and G band (E2g mode of graphite) of the carbon film.8 The intensity ratio of ID/IG is calculated to be 0.92, which indicates that the carbon film has a low degree of graphitization, which was also confirmed by XRD studies.
X-Ray photoelectron spectroscopy (XPS) analysis indicated that the PPy film is mainly composed of elements such as carbon, nitrogen and oxygen issued from the pyrrole monomer (C and N) and its chemical oxidative polymerization (O) (Fig. S3 and Table S1, ESI†). The signals at 167 eV (S 2p) and 230 eV (S 2s) show that the PPy polymer chains are S-doped during the polymerization process by the Fe(III) tosylate oxidant. The annealed film yields a relative increase of the C 1s content and a decrease of the N 1s and O 1s content. To further investigate the mechanism involved in the pyrolysis processes, mass spectrometry (MS) combined with a thermogravimetric-differential scanning calorimetry (TG-DSC) was used to monitor the gas products during the process. As shown in the TG-DSC curve of the PPy film in Fig. S4a (ESI†), a significant weight loss was observed before 700 °C which was attributed to the degradation of PPy. As can be seen from Fig. S4b (ESI†), two gas released peaks for SO2 and NO at around 400 °C and 500–700 °C respectively in MS curves are consistent with their weight losses in the TG results and the XPS results, which also demonstrates that the disappeared S signals and part of lost N and O content after annealing are due to the released SO2 and NO gases. Besides, a perceivable Fe signal was detected after annealing of the N–C film, but no Fe 2p signal was detected for the PPy film. That signal was probably covered by the stronger signal of doping species in the PPy film. The N 1s XPS analysis of the PPy film and N–C film can reveal the structure reorganization and distribution of types of nitrogen under annealing treatment for PPy films. As shown in Fig. 3b, the N 1s spectra of the PPy film is dominated by a main peak at 399.5 eV, assigned to pyrrolic-N i.e. uncharged nitrogen atoms in the polymer, which occupied 66.7 at%. In addition, there are another three small peaks at 397.8 eV, 400.5 eV and 401.7 eV, which represent deprotonated N (i.e. uncharged imine nitrogen atoms), protonated nitrogen atoms and high oxidation states of the nitrogen atoms, respectively.23 However, after annealing in an Ar atmosphere at 700 °C for 2 h, the main kinds of nitrogen atoms underwent great changes. As shown in Fig. 3c, the major part of nitrogen is now allocated between pyridinic-N (at 398.0 eV) and quaternary-N (at 400.6 eV). These changes are accompanied by a large decrease of the pyrrolic-N as a result of pyrrole ring decomposition at the high temperature of 700 °C, which demonstrates the incorporation of nitrogen atom within a new carbon structure that provides a better coordination with carbon atoms, stabilizing the nitrogen-bonded carbon structure into an in-plane functional group stable at higher temperature.20 The N content in N–C films is 8.1 at% (Table S1, ESI†), which is much higher than that of N-doped carbon materials using surface chemical post-modification (for example, NH3 treatment of porous carbon at high temperature).
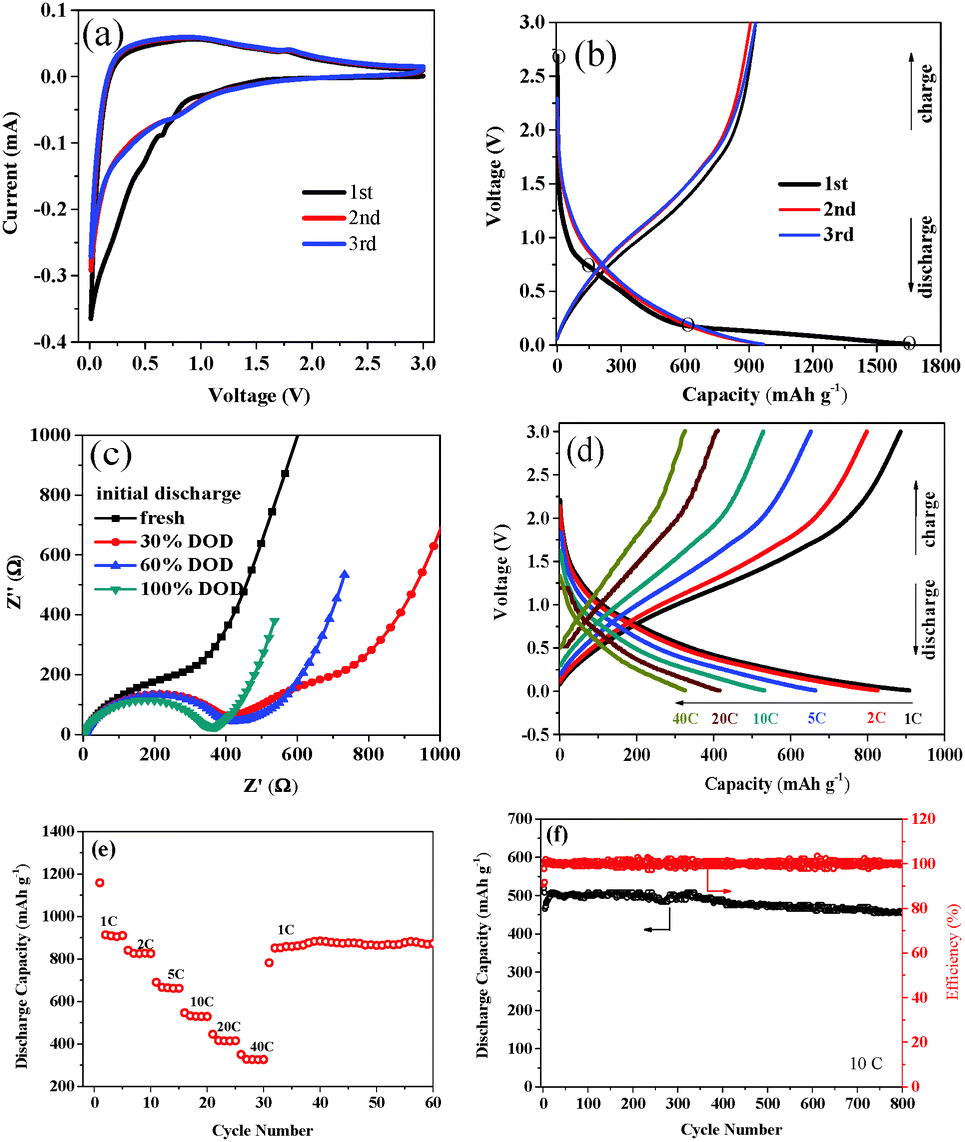 | ||
| Fig. 3 Electrochemical performance of N–C film electrode for lithium-ion batteries. (a) Cyclic voltammetry curves at a scan rate of 0.1 mV s−1; (b) first three discharge/charge profiles at a current density of 500 mA g−1; (c) electrochemical impedance spectra obtained at various charge states; (d) charge/discharge curves at various rates; (e) rate performance at 1C, 2C, 5C, 10C, 20C, and 40C, and back to 1C for additional 30 cycles; (f) cycling and corresponding coulombic efficiency at a rate of 10C. | ||
The N–C film was further investigated as an anode electrode which could be used directly without any conductive and binder agents for lithium ion batteries. The electrochemical performance is presented in Fig. 3. The cyclic voltammogram (CV) plot of the carbon film electrode is shown in Fig. 3a, which exhibits typical curves of the carbonaceous anode materials. From Fig. 3a, the reduction peak area of the first cycle is larger than the subsequent cycles, implying the loss of some irreversible lithium storage sites during the first cycle. Moreover, the CV curves fitted together very well for the 2nd and 3rd cycles. It implies good reversibility and stability of the Li-intercalation and de-intercalation through a carbon film electrode after an initial cycle. Shown in Fig. 3b are the charge/discharge profiles of a nitrogen-containing carbon film electrode for the initial three cycles at a current rate of 0.5C (0.25 A g−1), which are similar to those of the reported nano-sized carbonaceous materials.4,14,24 The initial reversible capacity is as high as 957.8 mA h g−1, more than 2.5 times higher than the theoretical one of graphite (372 mA h g−1). The initial coulombic efficiency for the carbon film anode is 58.0%, which is higher than those of the reported carbon materials (Table S2, ESI†). Also, the capacity becomes stable and reversible after the first cycle.
To further investigate the initial discharge process, electrochemical impedance spectroscopy (EIS) was evaluated at four different depths of discharge (DOD) of the carbon film anode. As shown in Fig. 3c, the fresh cell (OCV of ∼3.0 V) shows a single incomplete semicircle in the high-frequency region (>1 kHz), which is attributed mainly to surface-film resistance. When the cell discharged to 30% DOD with the voltage drop at ∼0.8 V, the solid electrolyte interphase (SEI) layer started to form.25 The corresponding EIS spectrum appears as two semicircles in the high-medium frequency range, which suggests that the polarization impedance of the cell has increased. With the increase of DOD, the second semicircle disappears gradually. The spectrum recorded at 0.01 V, the deep discharge limit (100% DOD), shows the smallest diameter semicircle, indicating a better charge transfer. This may be due to the SEI layer playing an important role as a low impedance passivation layer in the N–C film electrode.26
Notably, an excellent high rate-performance was observed for the N–C film anode. Fig. 3d depicts the discharge/charge capability of the N–C film electrode (vs. Li) at 1 to 40C rates. For testing, the cells were first charged and discharged at 0.5C rate to allow the activation of the electrode. The reversible capacities are 908.4, 825.7, 664.0, 531.6, 415.5 and 325.9 mA h g−1 at 1C, 2C, 5C, 10C, 20C and 40C, respectively. At 40C, the reversible capacity is about 35.9% of the capacity of 1C, and is about 87.6% of the theoretical capacity of graphite. The rate capacities of the cycling performance of the N–C film anode are shown Fig. 3e, which display excellent cycling performance with each rate. After a total cycling number of 35 at various rates between 1 and 40C, the specific discharge capacity can be recovered to 839.8 mA h g−1 at 1C rate, with a capacity retention of 92.4%.
The long-term cycling performance of the N–C film electrode was evaluated at 10C in the voltage range of 0.01–3 V for 800 cycles. As shown in Fig. 3f, the N–C film anode shows excellent cyclability with high capacity. The reversible capacity is ∼500 mA h g−1 at the initial stage while still having a retention of 91.6% after 800 cycles. At the high discharge/charge rate of 10C, the initial coulombic efficiency is 92%. And it increases dramatically upon cycling, reaching nearly 100% during the subsequent cycles. To further understand the excellent cycling properties of the N–C film anode, EIS experiments were carried out after the 10th, 100th, and 800th cycle at 100% DOD. As shown in Fig. S5 (ESI†), with the increase of cycling number of the N–C film anode, the region of semi-circle at the high frequency increased slowly, which indicated that the ohmic resistance change of the N–C film anode is very small even after 800 cycle times. Besides, after the 100th electrochemical cycles, the N–C film still maintained its initial morphological structure as the SEM photo shown in Fig. S6 (ESI†).
For comparison, the rate and cycling capability of the N–C film synthesised in this work and those of carbonized products from PPy and other polymers reported in the literature are summarized in Table S2 (ESI†). At high current density, the rate and cycling capability of the N–C film are higher than those of most of the materials reported. In this work, the ultrahigh capacity and rate capability for the N–C film can be explained with their novel nanostructure, binder-free style, and high-level nitrogen doping. The cross-linked surface structure can serve as channels for electron transportation, whereas large numbers of nanopores in the carbon film may act as reservoirs for storage of Li+. The large surface area and mesopores leads to a sufficiently large electrode/electrolyte interface to absorb Li+ and promote rapid charge-transfer reaction. Moreover, the N doping in the carbon film can enhance the electrochemical reactivity and electronic conductivity, which also results in a better electrochemical performance.
In summary, a free-standing N–C film has been successfully fabricated via carbonization of vapour phase polymerized PPy films. The carbon film is composed of short carbon nano-fibers with a unique continuously cross-linked structure. The carbonization process of the PPy film precursor results in the formation in large numbers of uniform mesopores in the N–C film, which is expected to provide very regular pore channels for Li+ charge transfer and storage. Moreover, the special molecular structure of PPy results in a high N-doping level so as to enhance the electrochemical reactivity and electronic conductivity, which additionally contributes to the exceptional performance. As a free-standing anode for lithium ion batteries, the carbon film electrode shows ultrahigh capacity, excellent rate capability and stable cyclability. The specific capacity is as high as 502 mA h g−1 even at 10C after 50 cycles. Our results show that the N–C film is very promising for the construction of next-generation LIBs which are more flexible, and possess high energy and power density.
We are grateful for financial support for this work from the National Basic Research Program of China (2014CB239700), the Natural Science Foundation of China (21336003, 21403139, 21573147), China Postdoctoral Science Foundation (2013M541510), and the Science and Technology Commission of Shanghai Municipality (14DZ2250800).
Notes and references
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed; A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef PubMed; Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878 CrossRef.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593 CrossRef CAS.
- C. R. Martin, G. Che, B. B. Lakshmi and E. R. Fisher, Nature, 1998, 393, 346 CrossRef.
- L. Qie, W. M. Chen, Z. H. Wang, Q. G. Shao, X. Li, L. X. Yuan, X. L. Hu, W. X. Zhang and Y. H. Huang, Adv. Mater., 2012, 24, 2047 CrossRef PubMed.
- H. Wang, T. Abe, S. Maruyama, Y. Iriyama, Z. Ogumi and K. Yoshikawa, Adv. Mater., 2005, 17, 2857 CrossRef CAS.
- F.-D. Han, Y.-J. Bai, R. Liu, B. Yao, Y.-X. Qi, N. Lun and J.-X. Zhang, Adv. Energy Mater., 2011, 1, 798 CrossRef CAS.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero, V. Pellegrini and B. Scrosati, Nano Lett., 2014, 14, 4901 CrossRef CAS PubMed.
- T. Yuan, W.-T. Li, W. Zhang, Y.-S. He, C. Zhang, X.-Z. Liao and Z.-F. Ma, Ind. Eng. Chem. Res., 2014, 53, 10849 CrossRef CAS.
- X. Zhou, X. Zhu, X. Liu, Y. Xu, Y. Liu, Z. Dai and J. Bao, J. Phys. Chem. C, 2014, 118, 22426 CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 CAS.
- X. Zhou and Y.-G. Guo, ChemElectroChem, 2014, 1, 83 CrossRef CAS.
- P. Wang, B. Qiao, Y. Du, Y. Li, X. Zhou, Z. Dai and J. Bao, J. Phys. Chem. C, 2015, 119, 21336 CAS.
- R. Czerw, M. Terrones, J. C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab, P. M. Ajayan, W. Blau, M. Rühle and D. L. Carroll, Nano Lett., 2001, 1, 457 CrossRef CAS.
- D. Li, L.-X. Ding, H. Chen, S. Wang, Z. Li, M. Zhu and H. Wang, J. Mater. Chem. A, 2014, 2, 16617 CAS.
- J. Yang, J. Xie, X. Zhou, Y. Zou, J. Tang, S. Wang, F. Chen and L. Wang, J. Phys. Chem. C, 2014, 118, 1800 CAS.
- A. Mohammadi, M. A. Hasan, B. Liedberg, I. Lundström and W. R. Salaneck, Synth. Met., 1986, 14, 189 CrossRef CAS.
- B. Winther-Jensen, J. Chen, K. West and G. Wallace, Macromolecules, 2004, 37, 5930 CrossRef CAS.
- J. Kim, D. Sohn, Y. Sung and E.-R. Kim, Synth. Met., 2003, 132, 309 CrossRef CAS.
- H. L. Wang and J. E. Fernandez, Macromolecules, 1993, 26, 3336 CrossRef CAS.
- J. Ouyang and Y. Li, Polymer, 1997, 38, 3997 CrossRef CAS; R. Gangopadhyay and A. De, Eur. Polym. J., 1999, 35, 1985 CrossRef.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717 CAS.
- F. E. Chen, G. Shi, M. Fu, L. Qu and X. Hong, Synth. Met., 2003, 132, 125 CrossRef CAS.
- A. Morozan, P. Jegou, S. Campidelli, S. Palacin and B. Jousselme, Chem. Commun., 2012, 48, 4627 RSC.
- Y. Wang, F. Su, C. D. Wood, J. Y. Lee and X. S. Zhao, Ind. Eng. Chem. Res., 2008, 47, 2294 CrossRef CAS.
- L. G. Bulusheva, A. V. Okotrub, A. G. Kurenya, H. Zhang, H. Zhang, X. Chen and H. Song, Carbon, 2011, 49, 4013 CrossRef CAS.
- D. Aurbach, B. Markovsky, M. D. Levi, E. Levi, A. Schechter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81–82, 95 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, BJH pore size distribution, XRD analysis, XPS spectra, element composition, TG-DSC, and MS measurements, EIS results during cycling processes, and SEM images of N–C film electrode after 100 cycles. See DOI: 10.1039/c5cc06964e |
| ‡ Dr Tao Yuan and Dr Yu-Shi He contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |