
Formation of colloidal nanocrystal clusters of iron oxide by controlled ligand stripping†
Junxiang
Fu
abc,
Le
He
a,
Wenjing
Xu
a,
Jianle
Zhuang
b,
Xianfeng
Yang
b,
Xiaozeng
Zhang
c,
Mingmei
Wu
*b and
Yadong
Yin
*a
aDepartment of Chemistry, University of California, Riverside, CA 92521, USA. E-mail: yadong.yin@ucr.edu
bMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, State Key Laboratory of Optoelectronic Materials and Technology, School of Chemistry and Chemical Engineering, Sun Yat-Sen (Zhongshan) University, Guangzhou, 510275, P. R. China. E-mail: ceswmm@mail.sysu.edu.cn
cSchool of Metallurgy and Chemical Engineering, Jiangxi University of Science and Technology, 86 Hongqi Road, Ganzhou, 341000, P. R. China
First published on 16th October 2015
Abstract
We report a “ligand stripping” method for the creation of secondary structures of colloidal nanocrystals. Using iron oxide as an example, we demonstrate that the use of diols as “stripping agents” allows the controllable removal of the original capping ligands and induces aggregation of nanocrystals into well-defined clusters.
Colloidal nanocrystals with controlled size and shape are of great interest for researchers from a wide range of disciplines, including materials science, chemistry, physics and engineering.1 Significant progress has been made in the development of robust synthesis protocols which allow precise control over their composition, size, shape, surface properties and uniformity.2 Recently, the focus of synthetic efforts has been devoted to the creation of colloidal nanocrystal clusters (CNCs), the secondary structures of colloidal nanocrystals, which show great potential for the development of advanced materials with novel functions.3 Basically, there are two strategies for the preparation of CNCs:4 (1) one-step processes which integrate the synthesis of nanocrystals and their aggregation into clusters in a single step;5 and (2) multi-step processes which first generate nanocrystals with the desired size, shape and surface functionality, and then assemble them into clusters of designed degree of aggregation in the following steps such as solvent evaporation, electrostatic attraction, or interfacial tension.6 Generally, one step processes are more efficient in producing CNC structures, but realizing fine control over size distribution of the final products remains a challenge.
Peng et al. reported a limited ligand protection (LLP) method to produce complex 3D CNC nanostructures, such as In2O3, CoO, MnO and ZnO crystalline nanoflowers.7 The key is to maintain an appropriate concentration of capping ligands, which is not enough to protect the primary nanocrystals against aggregation but is sufficient to stabilize the resulting 3D nanostructures. Although conceptually elegant, we find it difficult to extend the LLP method to many other systems especially in the cases where the ligands are involved in the production of monomers required for nanoparticle formation. For example, in the classic synthesis of superparamagnetic iron oxide (γ-Fe2O3) nanoparticles, monodisperse particles are produced by thermal decomposition of Fe(CO)5 in the presence of a capping ligand of oleic acid (OA).8 Changing the relative concentration of OA led to variation in the particle size but not in their aggregation state. Lowering the oleic acid amount did not result in maghemite nanoflowers or any other aggregates; instead, only smaller spherical maghemite nanocrystals were produced.9 In this case, OA acts not only as a capping ligand that ensures the colloidal stability of the nanoparticles, but also participates in the reaction with Fe(CO)5 for the production of an iron–oleic acid metal complex which is the needed precursor for nanoparticle growth. In this regard, a more effective and general strategy is preferred to decrease their colloidal stability and induce the formation of secondary structures.
Herein we demonstrate a “ligand stripping” method that enables the precisely controlled aggregation of nanocrystals and produces a series of complex 3D nanostructures with well-defined morphologies. The key strategy involves the destabilization of colloidal nanocrystals by replacing the original capping ligand with a stripping agent (Scheme 1). The stripping agent binds to the surface of nanocrystals weakly, thus inducing their aggregation into nanocrystal clusters. Using iron oxide as an example, we demonstrate that the use of diols as “stripping agents” can controllably remove the original capping ligands (OA) and induce aggregation of nanocrystals into clusters of defined shapes. The degree of aggregation can be controlled using diols with different stripping power. In this communication, we use HO(CH2CH2O)nH including diethylene glycol (DEG) (n = 2), tetraethylene glycol (TEG) (n = 4) and polyethylene glycol 400 (PEG 400) (n ≈ 8) as “ligand stripping” agents. We show that a higher degree of aggregation can be readily achieved by either using diols of lower molecular weight or simply increasing the diol concentration.
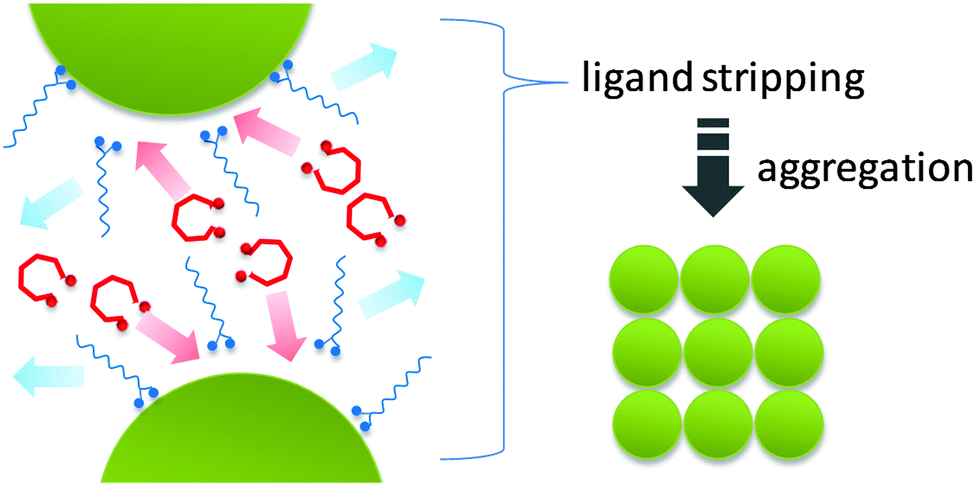 | ||
| Scheme 1 Schematic illustration of the “ligand stripping” method. | ||
The iron oxide CNCs were synthesized by modifying a previously established procedure by Hyeon et al.8 In a typical synthesis of cubic clusters, 0.2 mL of Fe(CO)5 (1.52 mmol) was added to a mixture containing 0.1 mL of DEG (1.11 mmol), 10 mL of ODE and 2 mL of oleic acid (6.41 mmol) at 110 °C. The resulting mixture was heated up to 295 °C. According to Hyeon et al., the initial reaction involves decomposition of Fe(CO)5 and then reaction with oleic acid to form an iron–oleic acid complex. The sudden change of the solution color from clear light yellow to black represents the burst nucleation of FeO nanocrystals. After the solution turned black, the solution was kept at that temperature for 1 h. All the heating processes took place under vigorous stirring in an argon atmosphere. The resulting black solution was cooled to 200 °C, oxidized by bubbling air into the solution for 1 h at 200 °C, and then cooled down to room temperature. Finally, iron oxide cubic nanoclusters were collected by centrifugation and washed with toluene/ethanol. Iron oxide dimers and oligomers were synthesized by using PEG 400 and TEG instead of DEG, respectively (ESI,† Table S1).
Spherical maghemite nanocrystals of 12 nm in diameter were formed without the addition of any diol additives into the Fe(CO)5/OA/ODE solution (Fig. 1a), which is consistent with the previous report by Hyeon et al.8 When 0.1 mL PEG 400 was introduced, most of the products show an elongated nanostructure and some show an apparent dimer structure (Fig. 1b). If 0.1 mL TEG was added instead of PEG 400, most products were composed of several nanoparticles which were termed as “oligomers” in this work (Fig. 1c). And if DEG was applied instead, monodisperse 40 nm sized cubic nanoclusters were formed (Fig. 1d). There were dozens of original particles in each cluster which indicate that the degree of aggregation increases dramatically. If diols of the same volume were used as ligand stripping agents, those molecules with fewer EG repeating units showed stronger ability for causing aggregation. Fig. 1 indicates that both the yield and the morphology uniformity of clusters increased with stripping ligands containing fewer EG repeating units. In particular, with DEG as the ligand stripping agent, uniform cubic clusters with yield near 100% could be obtained (Fig. S2a, ESI†). The presence of a small number of NCs and oligomers (marked by white squares in Fig. S2a, ESI†) suggests that they are probably the building blocks of the larger clusters.2
 | ||
| Fig. 1 TEM images of iron oxide aggregates prepared by using different diols as ligand stripping agents: (a) sphere nanoparticles without diol added; (b) dimers induced by PEG 400; (c) oligomers induced by TEG; (d) cubic clusters induced by DEG. | ||
We have followed the evolution of cubic CNCs by TEM imaging. In the early stage after the solution was heated to 295 °C, the solution was still clear and light yellow. Mainly amorphous complexes together with some nanocrystals and small nanocrystal clusters could be observed (Fig. S3a, ESI†). At this stage, the amorphous complex precursor had not been fully decomposed. Within one minute of the solution turning black, clusters with size ∼40 nm became the principal product (Fig. S3b, ESI†). After that, the overall size of clusters did not change much with prolonging reaction time. In other words, the formation and aggregation processes of nanocrystals happened in a very short period of time. By comparing Fig. S3b (ESI†) with Fig. 1d, we can find that during the later growth stage, the cubic shape of the clusters became clearer and the clusters appeared to be more compact and the size distribution became narrower. In addition, the density of the iron oxide clusters can be further finely tuned by controlling the aging temperature and time (Fig. S4, ESI†). In brief, higher temperature and longer aging time result in more compact structures due to the fusion of nanocrystals within clusters.
It is worth mentioning that the ligand stripping agent has to be added into the reaction system before the solution turns black. As the burst of nucleation and subsequent growth of nanocrystals completes within minutes, the addition of stripping ligands afterwards did not cause immediate aggregation and the product appeared to still exist as single spherical nanocrystals (Fig. S5a, ESI†). This is because the fully grown nanocrystals have relatively low surface energy and they have a smaller tendency to aggregate. It is also necessary to point out that the size of the primary particles decreased as the degree of aggregation raised. These observations clearly indicate that the stripping agents interact with the nanocrystals in the early stage of their formation, with stronger interaction for smaller diols. This is confirmed by adding ethylene glycol (EG) to the reaction, where larger clusters of about 100 nm were produced. Careful inspection shows that the primary particles are extremely small, only a few nanometers in size. As the boiling point of EG is only 195 °C, as opposed to DEG and TEG which are 245 °C and 330 °C, respectively, the aggregation of the nanocrystals could not be well controlled so that the resultant CNCs were not uniform (Fig. S5b, ESI†).
The detailed crystal structures of iron oxide dimers, oligomers and clusters were studied (Fig. 2). HRTEM images of iron oxide show continuous lattice fringes for all types of the CNCs, indicating single crystallinity which was further confirmed by corresponding FFT patterns of the square area. Maghemite (γ-Fe2O3) has an inverse spinel structure and belongs to the cubic crystallographic system (space group Oh7-Fd3m), with 40 atoms (eight Fe2O3 units) per unit cell with lattice parameters a = b = c = 8.347 Å (JCPDS No. 39-1346).10
 | ||
| Fig. 2 HRTEM images of γ-Fe2O3 aggregates: (a1) dimers induced by PEG 400; (b1) oligomers induced by TEG; (c1) cubic clusters induced by DEG. Images of a2–c2 are corresponding FFT patterns of the square areas. | ||
In addition to the stripping power of the diols, the degree of aggregation could also be controlled by their concentration in the reaction. Here we chose TEG as an example to show such dependence (Fig. 3). When the least amount of 0.01 mL TEG was used, most of the products were single particles or dimers, and the oligomer ratio was rather low. When TEG amount increased, the single particle and dimer ratios kept decreasing and the oligomer ratio kept increasing. Extremely low amounts of TEG resulted in single spherical and dimer like particles while extremely high amounts TEG led to the formation of cube-like clusters.
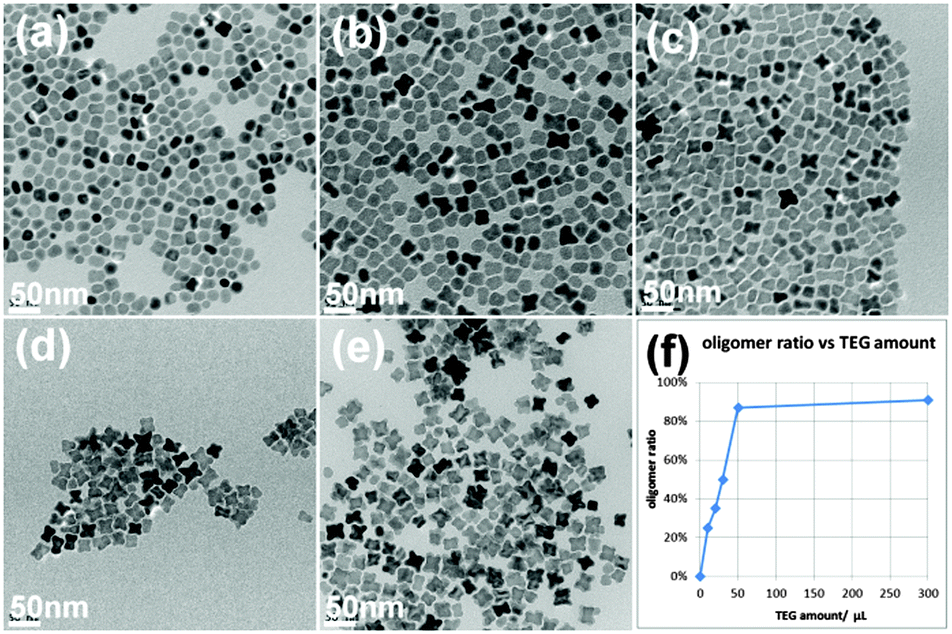 | ||
| Fig. 3 TEM images of iron oxide aggregates made by different amounts of TEG: (a) 0.01 mL; (b) 0.02 mL; (c) 0.03 mL; (d) 0.05 mL; (e) 0.3 mL. And (f) shows the relationship between the oligomer ratio and the TEG amount. | ||
The stripping of OA by diols from the nanocrystal surface occurs in the early stage of nanocrystal growth. At the beginning of the reaction, as the oleate is already part of the precursors and the total amount of OA (6.41 mmol) is significantly more than diols (1.11 mmol), the nucleation and growth of FeO nanocrystals are dominated by the presence of OA so that the reaction proceeds in a similar way to the standard synthesis (without diols), although the diols may act as co-ligands to limit the growth of primary nanocrystals. The decomposition of iron–oleate complex supports the growth of nanocrystals; while at the same time significantly reducing the concentration of OA in the system as the decomposition products evaporate, making it possible for diols to attack the surface of nanocrystals and kick off the attached OA. On the other hand, the adhesion of diols to the surface of oxide nanocrystals is considerably weak at high temperatures, resulting in reduced surface coverage of ligands and therefore destabilization of the colloidal nanocrystals. The coexistence of OA and DEG on the surface of the cubic clusters was confirmed by Fourier transform infrared (FTIR) analysis (Fig. S6, ESI†), and the reduced coverage of capping ligands on the surface of the nanocrystal clusters was clearly revealed by thermogravimetric analysis (TGA, Fig. S7, ESI†). Finally, upon aggregation, the primary nanocrystals bind to each other. Although further verification is needed, we believe that the binding of primary nanocrystals most likely proceeded through the well-known oriented attachment mechanism as each resulting cluster is a single crystal.11 It is necessary to point out that the oxidation of FeO CNCs by bubbling air at 200 °C did not significantly alter their cluster structure and overall morphology (Fig. S8, ESI†).
The bulkiness of the diol molecules is believed to play an important role in determining their diffusion rate in solution and thus their ability for attacking OA molecules on the nanocrystal surface. Even if the concentrations of added diols were kept the same, diols with larger molecular weight still showed lower aggregation ability. For example, although the quantity of –OH groups of 0.3 mL PEG is 1.5 times as that of 0.1 mL DEG, the final products were still oligomers (Fig. 3e), the aggregation degree of which was much lower than clusters produced by DEG (Fig. 1d).
The magnetization hysteresis loops of the iron oxide at 300 and 5 K are shown in Fig. S9 (ESI†). The saturation magnetizations (Ms) of iron oxide single nanoparticles, dimers, oligomers and clusters at 300 K were measured at 67.6, 60.1, 51.4 and 63.7 emu g−1, respectively. All samples possess typical superparamagnetic features at room temperature. However, the magnetization versus field curve exhibits a ferromagnetic behavior at 5 K. The saturation magnetizations (Ms) of iron oxide single nanoparticles, dimers and clusters were 72.0, 54.9 and 58.2 emu g−1, and the coercivity values (HC) were 200, 200 and 430 Oe, respectively. According to the reference, bulk magnetite and maghemite have the highest saturation magnetizations (Ms) among the iron oxides (92–100 emu g−1 and 60–80 emu g−1, respectively).12
In summary, an effective “ligand stripping” approach has been developed for the growth of colloidal nanocrystal clusters. Taking iron oxide as an example, we demonstrate that using diols as “stripping agents” it is possible to controllably remove the original capping ligands (OA) and induce the aggregation of nanocrystals into well-defined clusters, oligomers and dimers. The degree of aggregation can be controlled by using diols with different stripping power or by controlling the concentration of diols. We believe that this “ligand stripping” method represents a general approach for the creation of secondary structures of colloidal nanocrystals. These complex crystalline nanostructures described herein offer unique nanoarchitectures for constructing novel functional nanoscale devices.
We thank the financial support from the US National Science Foundation (Grant CHE-1308587) and the National Natural Science Foundation of China (21271190), the Government of Guangdong Province for Natural Science Foundation (S2012020011113) and industry (2012B09000026), and the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University (2012-01). Fu also acknowledges the support from the China Scholarship Council.
Notes and references
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545 CrossRef CAS.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664 CrossRef CAS PubMed.
- (a) J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342 CrossRef CAS PubMed; (b) J. P. Ge, Y. X. Hu and Y. D. Yin, Angew. Chem., Int. Ed., 2007, 46, 7428 CrossRef CAS PubMed; (c) R. Fernandes, M. Li, E. Dujardin, S. Mann and A. G. Kanaras, Chem. Commun., 2010, 46, 9270 RSC; (d) D. Baranov, L. Manna and A. G. Kanaras, J. Mater. Chem., 2011, 21, 16694 RSC; (e) A. G. Kanaras, C. Sönnichsen, H. Liu and A. P. Alivisatos, Nano Lett., 2005, 5, 2164 CrossRef CAS PubMed.
- Z. D. Lu and Y. D. Yin, Chem. Soc. Rev., 2012, 41, 6874 RSC.
- (a) Y. Zhu, W. Zhao, H. Chen and J. Shi, J. Phys. Chem. C, 2007, 111, 5281 CrossRef CAS; (b) S. Xuan, F. Wang, Y.-X. J. Wang, J. C. Yu and K. C.-F. Leung, J. Mater. Chem., 2010, 20, 5086 RSC; (c) X.-L. Fang, C. Chen, M.-S. Jin, Q. Kuang, Z.-X. Xie, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, J. Mater. Chem., 2009, 19, 6154 RSC; (d) H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782 CrossRef CAS PubMed; (e) X. F. Yang, J. X. Fu, C. J. Jin, J. A. Chen, C. L. Liang, M. M. Wu and W. Z. Zhou, J. Am. Chem. Soc., 2010, 132, 14279 CrossRef CAS PubMed; (f) J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875 CrossRef CAS PubMed; (g) X. Hu, J. Gong, L. Zhang and J. C. Yu, Adv. Mater., 2008, 20, 4845 CrossRef CAS; (h) J. Geng, Y. Lv, D. J. Lu and J. J. Zhu, Nanotechnology, 2006, 17, 2614 CrossRef CAS PubMed.
- (a) A. Corma, P. Atienzar, H. Garcia and J.-Y. Chane-Ching, Nat. Mater., 2004, 3, 394 CrossRef CAS PubMed; (b) D. E. Gómez, I. Pastoriza-Santos and P. Mulvaney, Small, 2005, 1, 238 CrossRef PubMed; (c) Y. Lin, H. Skaff, T. Emrick, A. Dinsmore and T. Russell, Science, 2003, 299, 226 CrossRef CAS PubMed.
- (a) A. Naravanaswamy, H. Xu, N. Pradhan and X. Peng, Angew. Chem., Int. Ed., 2006, 45, 5361 CrossRef PubMed; (b) A. Narayanaswamy, H. Xu, N. Pradhan, M. Kim and X. Peng, J. Am. Chem. Soc., 2006, 128, 10310 CrossRef CAS PubMed.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. Bin Na, J. Am. Chem. Soc., 2001, 123, 12798 CrossRef CAS PubMed.
- K. Woo, J. Hong, S. Choi, H. W. Lee, J. P. Ahn, C. S. Kim and S. W. Lee, Chem. Mater., 2004, 16, 2814 CrossRef CAS.
- P. Mäkie, G. Westin, P. Persson and L. österlund, J. Phys. Chem. A, 2011, 115, 8948 CrossRef PubMed.
- (a) D. Li, M. H. Nielsen, J. R. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014 CrossRef CAS PubMed; (b) Q. Zhang, S.-J. Liu and S.-H. Yu, J. Mater. Chem., 2009, 19, 191 RSC; (c) M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271 RSC.
- M. Cornell and U. Schwertmann, The Iron Oxides, VCH, New York, 1996, p. 117 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of samples. See DOI: 10.1039/c5cc07348k |
| This journal is © The Royal Society of Chemistry 2016 |