
Synthesis of Au@UiO-66(NH2) structures by small molecule-assisted nucleation for plasmon-enhanced photocatalytic activity†
Zhizhi
Gu
,
Liyong
Chen
*,
Binhua
Duan
,
Qiong
Luo
,
Jing
Liu
and
Chunying
Duan
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: lychen@dlut.edu.cn; cyduan@dlut.edu.cn
First published on 26th October 2015
Abstract
We have provided a small molecule-assisted heterogeneous nucleation of MOF route to successfully synthesize Au@UiO-66(NH2) heterostructures. UiO-66(NH2) with a localized electronic state that was characterized by C-AFM exhibited higher photocatalytic activity in the heterostructures via a plasmonic sensitization process.
Heterostructured materials can possess novel properties apart from the individual properties of components.1 In contrast to other solid crystalline materials, metal–organic frameworks (MOFs) composed of metal ions or clusters coordinating with bridged ligands have tuneable porous structures that are allowed to load guest species to achieve intriguing properties for applications in various fields such as catalysis and sensors.2 Until now, to upgrade the performance of MOFs, many nanosized inorganic materials have been implanted into MOF matrices to form core–shell structures; and among these inorganic nanomaterials, metallic nanoparticles (NPs) have become popular guest materials.3 On the other hand, MOFs can serve as a fascinating platform to engineer photocatalysts integrating photo-sensitizers and catalytic sites into a single material.4 The activity of MOF photocatalysts reported, however, is limited by light absorption and carrier stability. In detail, the adsorption edge related to organic linkers transferring the charges to metal or metal clusters has fallen into ultraviolet regions; electrons in a localized state are accountable for easy recombination of photogenerated electrons and holes.5 To enhance the photocatalytic performance of MOFs, we engineered metal NP@MOF heterostructures to improve the ability of collecting solar energy and the stability of carriers based on localized surface plasmon resonance (LSPR) of metal NPs.
Herein, we chose UiO-66(NH2) (Zr6O4(OH)4(ata)6, ATA: 2-aminoterephthalate), exhibiting medium photocatalytic activity for oxidation of alcohols, as host materials loading Au NPs to investigate the plasmon-enhanced photocatalytic performance of MOFs based on little spectral overlap between the absorption edge of UiO-66(NH2) (450 nm) and the LSPR band of Au NPs (>450 nm), which effectively extends the range of adsorption light. Although metal/MOF heterostructures so far have been synthesized by gas-phase loading, solution impregnation, or solid grinding methods, the formation of uniform heterostructures consisted of large Au NPs with a strong LSPR effect and UiO-66(NH2) needed to a seed-mediated heterogeneous nucleation-growth strategy.6 In the preparation of UiO-66(NH2) media, however, Au NPs could be etched due to the presence of Cl− ions and oxygen. On this basis, we developed a small molecule-assisted nucleation-growth route to facilitate the formation of Zr-oxo clusters on Au NPs to achieve Au@UiO-66(NH2) core–shell structures.
A typical procedure was that ZrCl4, acetic acid, and polyvinylpyrrolidone modified Au (PVP–Au) NPs with uniform size (dAu = 15 nm) (concentrated by centrifugation from 14 mL of Au NPs colloidal solution) were sequentially introduced into a solution of ATA in dimethylformamide (DMF), and the resulting mixture was bubbled by argon (Ar) before being heated at 120 °C for 24 h (synthetic method, ESI†). The main products observed from transmission electron microscopy (TEM) are Au@UiO-66(NH2) nanocrystals with an average diameter of 240 nm accomplished by a few Au NP-free UiO-66(NH2) nanocrystals (Fig. 1a–d). The morphology of most UiO-66(NH2) shells was deviated from octahedral shapes into multifaceted shapes (Fig. S1, ESI†) while large Au NPs were incorporated into the matrices to possibly eliminate the strain energy induced by the lattice mismatch between the Au core and the UiO-66(NH2) shell.7 In spite of Au NPs coated by PVP, from the TEM images, we found that most of the uniform Au NPs were developed into different Au NPs in size after encapsulation. The large Au NPs incorporated UiO-66(NH2) much easily formed irregular multifaceted shapes (Fig. 1d). In previous reports, however, Pt@UiO-66 multi-core heterostructures were achieved via an acetic acid-assisted solvothermal route, and the UiO-66 shell still maintained octahedral shapes, attributed to small Pt NPs.8 While tuning the amount of Au NPs introduced into the reaction systems, uniform Au@UiO-66(NH2) heterostructures with high yield were not able to obtained yet (Fig. S2, ESI†). Powder X-ray diffraction (XRD) patterns (Fig. 1f) revealed that UiO-66(NH2) in the heterostructures had good crystallinity, illustrated by the crystal structures of UiO-66(NH2) observed from its crystallographic 〈110〉 direction (Fig. 1e), and the characteristic diffraction peaks of the {111} crystal plane of Au NPs can be observed (inset of Fig. 1f). In contrast to the typical procedure, if CO2 instead of acetic acid, as well as Ar, was bubbled into the synthetic media, the Au@UiO-66(NH2) core–shell structures with multifaceted shapes were prepared, and size evolution of Au NPs can also be observed (Fig. S3, ESI†).
 | ||
| Fig. 1 As-synthesized Au@UiO-66(NH2) (a) panoramic and (b–d) magnified TEM images with different Au cores in size; (e) schematic illustration of crystal structures of UiO-66(NH2) along the 〈110〉 direction; (f) XRD patterns of (1) Au@UiO-66(NH2) and (2) simulated UiO-66. (Inset is the magnified XRD patterns from 30° to 40°.) | ||
While oxygen was not removed from the synthetic system and acetic acid or CO2 was not introduced into the reaction mixture, however, Au NP-free UiO-66(NH2) nanostructures rather than Au@UiO-66(NH2) heterostructures were formed. Thus, oxygen and acetic acid or CO2 should be the decisive factors in the formation of Au@UiO-66(NH2) heterostructures. To clearly reveal the size evolution of Au NPs, some controlled experiments were performed using conventional heating instead of solvothermal conditions, which is convenient for direct observation of experimental phenomena. When only ZrCl4 and PVP–Au NPs were dispersed in DMF, the pink solution was rapidly changed into colourless solution within several minutes at elevated temperature, and further turned into yellow solution upon heating. In contrast, when the experiment was conducted in an Ar atmosphere, the colour of the solution hardly changed after several hours. Similar experimental phenomena were also observed if ZrCl4 was replaced by HCl. Hence, the process could be attributed to Au NPs oxidized to Au+ and Au3+ by oxygen with the assistance of Cl− ions according to their respective redox potential.9 To retard the effect on oxidative etching, it is needed to remove oxygen from the reaction system. Nevertheless, Au@UiO-66(NH2) heterostructures were not found in the sample yet after removal of oxygen from the solvothermal reaction system if acetic acid or CO2 was not introduced. We speculate that acetic acid or CO2 is favourable for the formation of Zr-oxo clusters and subsequent crystal growth on Au NPs,10 preventing Au from directly contacting with other species. In the heterostructures, hence, the formation of small Au NPs should be ascribed to their dissolution by oxidative etching although oxygen was removed and acetic acid or CO2 was introduced into the preparation system, which can attenuate the etching process but cannot eliminate it completely (Fig. S4, ESI†). In Au@UiO-66(NH2) composites prepared in the presence of acetic acid or CO2, Au NPs are about 2.3% and 2.4%, respectively, by weight with respect to the composites examined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The content of Au was less than the calculative value (3.0%) presumedly due to the loss of Au NPs in the washing process before encapsulation, and also the loss of Au NPs by etching in the encapsulation process.
The large Au NPs produced possibly involves two scenarios: (1) the dissolution and recrystallization of Au NPs and (2) the self-assembly of Au NPs. On the basis of the above discussion, dissolution of Au NPs occurred by oxidative etching. To understand whether recrystallization is mainly responsible for the formation of large Au NPs or not, a parallel experiment of PVP–Au NPs replaced by HAuCl4 was carried out, and HAuCl4 failed to be reduced to form Au NPs under otherwise identical conditions. On the other hand, time-dependent experiments were determined by the TEM technique (Fig. S5, ESI†). Two Au NPs attached to each other were fused together to form a peanut NP with a thin UiO-66(NH2) layer coated on the surface of Au NPs when the reaction mixture was heated for 1 h.11 There was a broader size distribution of Au NPs as compared to that of initial Au NPs. The size of Au NPs engulfed by UiO-66(NH2) shells hardly changed with the reaction proceeding possibly due to UiO-66(NH2) serving the dual roles of a stabilizer and a spacer to impede aggregation and etching of Au NPs. Therefore, we infer that large Au NPs formed are mainly contributed to fusion between Au NPs at the initial encapsulation stage (Fig. S4, ESI†).
The measurement of the current through the UiO-66(NH2) film was done by using conductive atomic force microscopy (C-AFM) to understand the electronic states in UiO-66(NH2) (Fig. S6, ESI†). The ultralow current between the C-AFM tip and UiO-66(NH2) was found while the positive bias voltage 10 V was applied to the tip and the negative bias voltage −10 V was applied to the sample. The result reveals that electrons preferentially remain in the localized state towards UiO-66(NH2), causing the large distance between metal nodes or organic linkers in UiO-66(NH2). Although frontier orbitals of UiO-66(NH2) showed a localized electronic state, a strong photo-absorption band from 290 nm to 460 nm in pure UiO-66(NH2) was also observed in the UV-vis spectrum (Fig. 2a), and attributed to sets of the ligand to Zr-oxo cluster charge transfer (LCCT) process. In comparison with UiO-66, the absorption edge of UiO-66(NH2) was red-shifted, which could be assignable to the amino-substituent providing electrons and donating electron density to the antibonding orbitals of the benzene ring, leading to an increase of the highest occupied molecular orbital (HOMO) level.12 For Au@UiO-66(NH2) heterostructures, besides the absorption bands in the short wavelength region, which were similar to those of UiO-66(NH2), an Au LSPR band peaking at 548 nm was also observed. The absorption edge of UiO-66(NH2) in the heterostructures shows almost no change, so exciton quenching by electron transfer from UiO-66(NH2) to Au NPs is negligible, which was further checked by nanosecond transient absorption (TA) of UiO-66(NH2) and Au@UiO-66(NH2) after 355 nm excitation (Fig. S7, ESI†).13 The similar features of a bleach of UiO-66(NH2) exciton at 420 nm in both TA spectra reveal similar decay pathways of exciton.
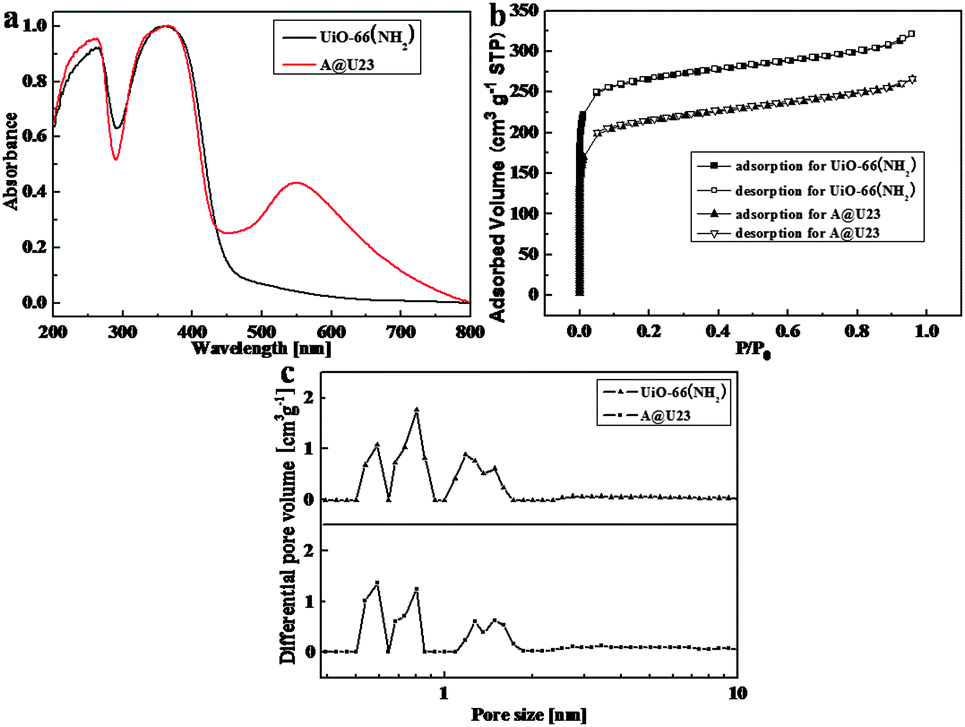 | ||
| Fig. 2 (a) Solid UV-vis absorption spectra of UiO-66(NH2) and A@U23 (Au@UiO-66(NH2) (acetic acid)), (b) nitrogen isotherm at 77 K for UiO-66(NH2) and A@U23, and (c) the corresponding pore size distribution of UiO-66(NH2) and A@U23. | ||
A nitrogen adsorption–desorption isotherm was used to analyze the specific surface area, average pore diameter, and pore volume of composites. Type I nitrogen sorption isotherm behavior for UiO-66(NH2) and Au@UiO-66(NH2) reveals their microporous structures. The specific surface areas were 813 m2 g−1 (Brunauer–Emmett–Teller (BET) model) for UiO-66(NH2) and 659 m2 g−1 (BET model) for Au@UiO-66(NH2), and the total micropore volumes were 0.38 cm3 g−1 for UiO-66(NH2) and 0.35 cm3 g−1 for Au@UiO-66(NH2) (Fig. 2b). Albeit Au NPs implanted in MOF matrices led to the decreased surface area and pore volume of the composite, its pore size distribution almost remained unchanged as compared to pure UiO-66(NH2) (Fig. 2c). The median pore width of 6.93 Å for Au@UiO-66(NH2) was slightly smaller than that of 7.15 Å for UiO-66(NH2) obtained from the Horvath–Kawazoe mode.
The photocatalytic oxidation of alcohol was selected as the model reaction to investigate the photocatalytic activity of UiO-66(NH2) enhanced by LSPR of Au NPs. The aerobic oxidation of benzyl alcohol was performed by photo-irradiation of an acetonitrile solution containing a catalyst (8 mg) and a substrate (50 μmol) using a white fluorescence lamp for 24 h under an O2 atmosphere (photocatalytic test, ESI†). The conversion of benzyl alcohol appeared to be 26.2% and 30.0% while using Au@UiO-66(NH2) (acetic acid/CO2) as a catalyst, and 4.5% when using UiO-66(NH2) as a catalyst. The selectivity of benzyl alcohol for benzaldehyde was as high as >99% in these photocatalytic reactions, and no byproducts were detected by nuclear magnetic resonance (NMR) and gas chromatography (GC). Other UiO-66(NH2) loading different amounts of Au NPs exhibited lower photocatalytic activity compared to aforementioned Au@UiO-66(NH2) (Fig. S8, ESI†). The photocatalytic performance for the same catalyst was strongly dependent on the structure of substrates. While 2,4,6-trimethylbenzyl alcohol used as a substrate, Au@UiO-66(NH2) (acetic acid/CO2) and UiO-66(NH2) showed similarly low photocatalytic activities (< 3%). The possible reason is that small pores less than 6 Å in size (Fig. 2c) obstruct the alcohol molecule to enter the pores to approach Au cores due to its large size (6.1 Å), and was only catalyzed by activated sites located at the external surface of MOFs.14
To elucidate the mechanism of the plasmon-enhanced photocatalytic activity of UiO-66(NH2) by Au NPs, we should understand the active intermediates in the reaction process firstly. Electron spin resonance (ESR) was used to study the formation of Zr3+ species involving active sites for the photocatalytic aerobic oxidation of benzyl alcohol during visible-light irradiation (Fig. S9, ESI†). A week signal at g = 1.989 appears in the ESP spectra of UiO-66(NH2) and Au@UiO-66(NH2) under visible-light irradiation, which could be attributed to Zr3+ species according to previous studies.15 The intensity of the signal from Au@UiO-66(NH2) is slightly higher than that from UiO-66(NH2), so LCCT and LSPR effects should be responsible for the formation of Zr3+ in the heterostructures. To further understand the photocatalytic oxidation process, active oxygen species were determined by quenching experiments. Photocatalytic oxidation of benzyl alcohol did not occur in the presence of 2,6-di-tert-butylmethylphenol, an  scavenger, indicating that the photocatalytic oxidation process was inhibited. Hence,
scavenger, indicating that the photocatalytic oxidation process was inhibited. Hence,  radicals were the dominant active oxygen species for the photocatalytic oxidation of alcohols.
radicals were the dominant active oxygen species for the photocatalytic oxidation of alcohols.
Therefore, under visible light irradiation, on the one hand, charge carriers were generated immediately in excited UiO-66(NH2), that is, the photo-generated electrons jumped to the LUMO, consisted of zironium oxo clusters, from the HOMO, composed of C from benzene ring and N from amino group;5,15,16 on the other hand, the excited LSPR of Au NPs decayed into hot electron–hole pairs, followed by the injection of the hot electrons into the LUMO of UiO-66(NH2) with holes near the AuNP Fermi level based on their electronic state structures (plasmonic sensitization process), leading to a charge-separated state (Fig. 3).17 Excited electrons located in the LUMO cannot diffuse into other positions due to the localized electronic state in UiO-66(NH2), but can transfer to O2 molecules that are diffused into pores to adsorb on the Zr3+ sites, accounting for the formation of superoxide radical  and Zr4+ ion recovery. The electrons are transferred from alcohol molecules to electron-deficient UiO-66(NH2) and Au NPs, and the protons from the methylene group and the hydroxyl group of alcohol are removed by
and Zr4+ ion recovery. The electrons are transferred from alcohol molecules to electron-deficient UiO-66(NH2) and Au NPs, and the protons from the methylene group and the hydroxyl group of alcohol are removed by  radicals, which are accountable for oxidation of alcohols to carbonyl compounds.
radicals, which are accountable for oxidation of alcohols to carbonyl compounds. 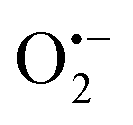 radicals seize electrons from excited UiO-66(NH2) and protons from the dehydrogenated substrate to produce H2O2 molecules. Therefore, the reduction of O2 molecules should be a proton-auxiliary two-step one-electron mechanism in the photocatalytic oxidation of alcohol process (Fig. 3). The photocatalytic activity is related to the concentration of charge carriers that can be enhanced by plasmonic sensitization.
radicals seize electrons from excited UiO-66(NH2) and protons from the dehydrogenated substrate to produce H2O2 molecules. Therefore, the reduction of O2 molecules should be a proton-auxiliary two-step one-electron mechanism in the photocatalytic oxidation of alcohol process (Fig. 3). The photocatalytic activity is related to the concentration of charge carriers that can be enhanced by plasmonic sensitization.
 | ||
| Fig. 3 Schematic illustration of the process of the enhanced photocatalytic performance of Au@UiO-66(NH2) by plasmonic sensitization. | ||
However, there are other possible scenarios of enhancing photocatalysis for plasmonic metal/photocatalyst composites, including the photothermal effect and active sites provided by metal NPs. To carefully check these possibilities, some parallel experiments of photocatalytic oxidation of benzyl alcohols were carried out under different conditions. Without irradiation using a fluorescence lamp, Au@UiO-66(NH2) exhibited low catalytic activity at 80 °C, implying that thermally activated Au NPs hardly work to oxidize benzyl alcohol, inconsistent with the previous report.18 The conversion of benzyl alcohol over Au NPs on UiO-66(NH2) hybrids prepared by the photo-deposition process (Fig. S10, ESI†) was as low as ∼4% with irradiation, excluding Au NPs serving as catalytic sites. Hence, plasmonic sensitization is the dominant pathway to upgrade the photocatalysis of Au@UiO-66(NH2) heterostructures.
In summary, we have developed a small molecule-assisted heterogeneous nucleation-growth strategy of MOFs to successfully synthesize Au@UiO-66(NH2) core–shell heterostructures. On the basis of the porous structures and the spectral relation, the heterostructures were used to explore the plasmonic sensitization mechanism for upgrading MOFs' photocatalytic performance in the oxidation of alcohol molecules in the visible region. The restructured surfaces of Au NPs via an etching and fusing process came into direct contact with UiO-66(NH2). Thus, the heterostructures favoured the electrons transfer from Au NPs to UiO-66(NH2) with a localized electronic state characterized by C-AFM, accounting for Au@UiO-66(NH2) higher photocatalytic activity as compared to UiO-66(NH2). Albeit the photocatalytic activity strongly depending on the UiO-66(NH2) nature is not high yet in the case, this provides a different perspective to enhance the photocatalytic performance that could be inaccessible only by changing structures and components of MOFs alone.
This work was supported by the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (21421005), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20130041120025), and the Fundamental Research Funds for the Central Universities (DUT15LK04).
Notes and references
- J. Shi, Chem. Rev., 2013, 113, 2139 CrossRef CAS PubMed.
- (a) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262 RSC; (b) F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761 CrossRef CAS PubMed.
- (a) Y. Li, J. Tang, L. He, Y. Liu, Y. Liu, C. Chen and Z. Tang, Adv. Mater., 2015, 27, 4075 CrossRef CAS PubMed; (b) C. M. Doherty, B. Dario, J. Hill Anita, F. Shuhei, K. Susumu and F. Paolo, Acc. Chem. Res., 2014, 47, 396 CrossRef CAS PubMed.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919 RSC.
- (a) M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701 CrossRef CAS; (b) L. Chen, Y. Peng, H. Wang, Z. Gu and C. Duan, Chem. Commun., 2014, 50, 8651 RSC.
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143 CrossRef CAS.
- K. Na, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2014, 14, 5979 CrossRef CAS PubMed.
- H. Liu and Q. Yang, CrystEngComm, 2011, 13, 5488 RSC.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739 CrossRef CAS PubMed.
- (a) Z. P. Zhang, H. P. Sun, X. Q. Shao, D. F. Li, H. D. Yu and M. Y. Han, Adv. Mater., 2005, 17, 42 CrossRef CAS; (b) H. Colfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef PubMed.
- C. G. Silva, I. Luz, F. X. Llabres i Xamena, A. Corma and H. Garcia, Chem. – Eur. J., 2010, 16, 11133 CrossRef CAS PubMed.
- K. Wu, W. E. Rodriguez-Cordoba, Y. Yang and T. Lian, Nano Lett., 2013, 13, 5255 CrossRef CAS PubMed.
- C. Chizallet, S. Lazare, D. Bazer-Bachi, F. Bonnier, V. Lecocq, E. Soyer, A.-A. Quoineaud and N. Bats, J. Am. Chem. Soc., 2010, 132, 12365 CrossRef CAS PubMed.
- (a) J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wang, Chem. Commun., 2012, 48, 11656 RSC; (b) D. Sun, W. Liu, Y. Fu, Z. Fang, F. Sun, X. Fu, Y. Zhang and Z. Li, Chem. – Eur. J., 2014, 20, 4780 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364 CrossRef CAS PubMed.
- J. B. Priebe, M. Karnahl, H. Junge, M. Beller, D. Hollmann and A. Brueckner, Angew. Chem., Int. Ed., 2013, 52, 11420 CrossRef CAS PubMed.
- K. Leus, P. Concepcion, M. Vandichel, M. Meledina, A. Grirrane, D. Esquivel, S. Turner, D. Poelman, M. Waroquier, V. Van Speybroeck, G. Van Tendeloo, H. García and P. Van Der Voort, RSC Adv., 2015, 5, 22334 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc07042b |
| This journal is © The Royal Society of Chemistry 2016 |