
The increased CO2 adsorption performance of chitosan-derived activated carbons with nitrogen-doping†
J.
Fujiki
and
K.
Yogo
*
Chemical Research Group, Research Institute of Innovative Technology for the Earth (RITE), 9-2 Kizugawadai, Kizugawa, Kyoto 619-0292, Japan. E-mail: yogo@rite.or.jp
First published on 21st October 2015
Abstract
Highly porous nitrogen-doped activated carbons (NACs) were prepared by the chemical activation of chitosan using alkali carbonates. The NACs exhibited extremely high CO2 capacities of 1.6 mmol g−1 (15 kPa) and 4.9 mmol g−1 (100 kPa) at 25 °C. Nitrogen atoms doped into carbon frameworks clearly enhanced CO2 adsorption at low partial pressures.
An increase in the atmospheric concentration of CO2 is thought to contribute to global warming. In recent decades, many studies have investigated the reduction of CO2 emissions by separation and recovery of CO2 from large point sources such as coal-fired power plants. Absorption into aqueous amine solutions is the most promising technique for CO2 recovery. However, it has limitations, including high regeneration energy, solvent evaporation/degradation, and equipment corrosion. An alternative technique that overcomes these drawbacks is adsorptive separation using porous materials such as zeolites, carbonaceous materials, siliceous materials, metal–organic frameworks, covalent-organic frameworks and composites.1–4 Recently, amine-functionalized porous materials have received increasing attention for their properties such as high CO2 adsorption capacity, good regenerability, low regeneration energy, and moisture tolerance.1–8 Amine-functionalized materials can be classified into three types of materials, including amine-impregnated (Class 1), amine-grafted (Class 2) and in situ polymerized amine-grafted (Class 3) silica materials.9 Our group has focused on the development of Class 1 and 2 materials.10–16 To date, we have developed highly regenerable amine-based solid sorbents with high CO2 capacity16 and evaluated their performance in a laboratory-scale separation test.
More recently, a new class of materials – nitrogen atom incorporated porous carbon materials (N-doped carbons) – have been investigated as effective CO2 adsorbents.17–22 Generally, carbon materials have advantages over other materials for CO2 capture, including low cost, low regeneration energy, high stability, and hydrophobicity. However, carbon materials, including carbon molecular sieves, exhibit low CO2 adsorption capacity at low CO2 partial pressure. Consequently, many studies have investigated the enhancement of CO2 adsorption on carbon materials. The doping of a heteroatom into the carbon framework is an effective method for enhancing CO2 adsorption. Many studies have reported that N- and O-doping into the carbon framework affects CO2 adsorption.17–24 Nitrogen atoms doped into carbon may increase the number of basic sites in the materials, which could enhance CO2 adsorption. However, it is known that control of the porous structure (i.e., pore size, pore volume and surface area) is more important than N-doping into the carbon framework for enhancing CO2 adsorption.25 Sevilla et al. reported that N-doping did not affect the CO2 adsorption capacity over a wide range of CO2 pressures.26 However, a distinct difference in the CO2 adsorption capacities of carbon materials with and without heteroatom doping may appear at low CO2 partial pressure because the interaction between CO2 and the heteroatom atom doped into the carbon framework is only slightly stronger than CO2-nondoped carbon interactions.20 Accordingly, incorporating heteroatom atoms into porous carbon with well-controlled pore structures must be effective for increasing CO2 adsorption.
Many studies have investigated the development of CO2 adsorbents from waste materials, such as biomass and coal, because of the reduction in the cost of CO2 capture.25,27–29 The present study focused on a simple preparation method for nitrogen-doped activated carbons (NACs) from chitosan for CO2 capture. In addition, the favorable physicochemical properties, such as N content and pore volume, for CO2 adsorption were investigated.
NACs were prepared by the chemical activation of chitosan. Although hydroxide, particularly KOH, is a well-known activator for preparing high performance activated carbon, it is highly toxic and corrosive. Alternatively, in this study, alkali metal carbonates were employed as mild activators. Chitosan powder (5 g, low molecular weight, Sigma-Aldrich, St. Louis, MO) was dissolved in 95 g of 5% (mass fraction) lactic acid (Junsei Chemical Co., Ltd, Tokyo, Japan) aqueous solution. The chitosan solution was added dropwise into a 20% (mass fraction) alkali metal carbonate (Na2CO3, K2CO3, Rb2CO3 or Cs2CO3; Junsei Chemical Co., Ltd) aqueous solution to generate porous chitosan beads. After filtration without washing, the beads were dried overnight in an oven at 100 °C to obtain an alkali-metal-carbonate-doped carbon precursor. The dried beads were placed in a tube furnace, and heated to the target temperature (500–700 °C) at a rate of 10 °C min−1 under a N2 flow. This chemical activation was performed for 1 h. The product was washed with pure water until the filtrate was neutral. Then, the product was ground into a powder, and dried overnight in an oven at 120 °C. The obtained carbon materials are denoted by NAC-M(T), where M represents the alkali metal of alkali metal carbonate used to generate the carbon precursor and T is the activation temperature. For example, NAC-K(600) represents the product prepared from a chitosan precursor generated in 20% (mass fraction) K2CO3 with activation at 600 °C.
The prepared NACs were characterized by N2 adsorption–desorption at −196 °C. The properties of the NACs are listed in Table 1. The N2 adsorption–desorption isotherms and pore size distributions determined from the N2 isotherms are shown in Fig. S1 and S2, respectively, in the ESI.† The N2 isotherms of the NACs except NAC-Na were Type I isotherms, indicating the NACs had microporous structures. For NAC-Na, they showed Type III and Type IV isotherms, indicating that NAC-Na(500) was almost non-porous and NAC-Na(600) and NAC-Na(700) had micropores and mesopores. The pore size distributions also suggested the presence of micropores. The physicochemical properties indicated that the pore structure and N content could be controlled by the activation temperature and the alkali activator. Those NACs activated at higher temperatures had higher specific surface areas (SBET) and pore volumes but lower N contents. An increase in the micropore volume with an increase in the activation temperature was also observed in the pore size distributions (Fig. S2, ESI†). NACs with high surface areas (>1000 m2 g−1) could be obtained by activation at a relatively low temperature (approximately 600 °C). At the same activation temperatures and activator loadings, the surface areas, pore volumes and pore sizes of the products increased in the following order: NAC-Na < NAC-K < NAC-Rb < NAC-Cs. This might be caused by the degree of reactivity of each alkali metal carbonate.30–32
| S BET [m2 g−1] | V total [cm3 g−1] | V micro [cm3 g−1] | N content [mmol g−1] | |
|---|---|---|---|---|
| NAC-Na(500) | 10.4 | 0.04 | 0.00 | 5.68 |
| NAC-K(500) | 653.1 | 0.34 | 0.22 | 5.09 |
| NAC-Rb(500) | 873.9 | 0.36 | 0.30 | 4.02 |
| NAC-Cs(500) | 632.7 | 0.28 | 0.22 | 3.74 |
| NAC-Na(600) | 474.5 | 0.32 | 0.16 | 5.16 |
| NAC-K(600) | 1355.0 | 0.60 | 0.47 | 3.23 |
| NAC-Rb(600) | 1496.0 | 0.63 | 0.52 | 3.15 |
| NAC-Cs(600) | 1530.2 | 0.68 | 0.50 | 2.59 |
| NAC-Na(700) | 1328.3 | 0.78 | 0.43 | 2.24 |
| NAC-K(700) | 2044.5 | 0.96 | 0.62 | 1.55 |
| NAC-Rb(700) | 2145.3 | 0.99 | 0.55 | 1.61 |
| NAC-Cs(700) | 1274.4 | 0.60 | 0.26 | 1.39 |
The powder X-ray diffraction patterns of the NACs showed a diffraction peak at around 2θ = 25° for (002), corresponding to a lamellar structure of carbon (Fig. S3 in the ESI†). This peak shifted to higher angle and gradually disappeared as the reactivity of the alkali metal carbonate increased, suggesting that the spacing between the carbon layers, the carbon content, and aromaticity increased. Transmission electron microscopy images of NAC-Rb(600) showed that the NACs had an amorphous layered graphene structure (Fig. 1).
 | ||
| Fig. 1 Low-magnification (a) and high-magnification (b) TEM images of NAC-Rb(600). | ||
The CO2 adsorption isotherms for NAC-M(600) measured at three different temperatures (0, 25 and 30 °C) are shown in Fig. 2 and Fig. S4 in the ESI.† All CO2 isotherms showed almost no adsorption–desorption hysteresis, suggesting completely reversible physical adsorption. However, minor hysteresis was observed only in the CO2 isotherms of NAC-Na(600). This might be because of the existence of narrower pores and the partial chemisorption of CO2 onto the surface functional groups of NAC (e.g., remaining amino groups).
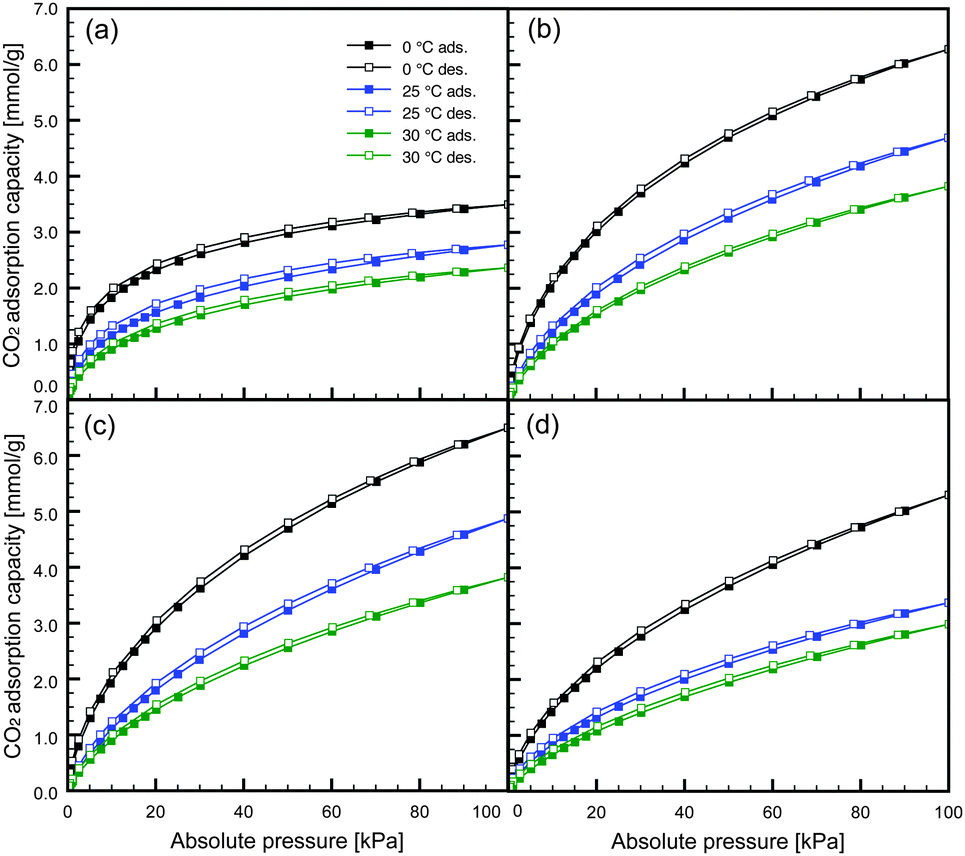 | ||
| Fig. 2 CO2 adsorption isotherms: (a) NAC-Na(600), (b) NAC-K(600), (c) NAC-Rb(600) and (d) NAC-Cs(600). | ||
The CO2 adsorption capacities of NAC-K(600) and NAC-Rb(600) at 25 °C and 100 kPa were 4.69 and 4.88 mmol g−1, respectively. These values are extremely high compared with previously reported chitosan-derived carbons.28,33 Therefore, the simple preparation procedure of activator-doped carbon precursor employed in this study gives a high-performance activated carbon. Moreover, a CO2 capacity of 4.88 mmol g−1 is close to the highest value (5.14 mmol g−1)17 reported to date for all carbonaceous materials.
Generally, gas physisorption on porous materials is dependent on the porous structure, and materials with large micropore volumes and high surface areas are desirable for superior adsorption performance. As shown in Fig. 3, the CO2 adsorption capacities of the NACs at 100 kPa depended on the specific surface areas and pore volumes, and particularly the micropore volumes. These results indicate that a physisorption process (i.e., pore-filling mechanism) governs the CO2 adsorption on NACs. In contrast, the CO2 capacities at low pressure (1 kPa) were dependent on the N contents of the carbon materials (Fig. 3), and independent of the surface areas and pore volumes. This is because the N contents of the NACs were inversely proportional to the specific surface areas and pore volumes of the NACs (Fig. S5 in the ESI†). In addition, it was obvious that the N content affected the CO2 capacity at pressures less than 15 kPa (Fig. S6 in the ESI†). However, the lower correlation for the higher pressure suggested a decreased influence with the increase in the CO2 partial pressure. Those NACs with high N contents exhibited large CO2 adsorption capacities even with low micropore volumes, suggesting that the doping of N atoms into the carbon framework affects the CO2 adsorption at low partial pressure. This is caused by acid–base34 and/or hydrogen-bonding18 interactions between the N-containing functional groups and the CO2 molecules. When considering CO2 capture from coal-fired power plants, the CO2 partial pressure in the flue gas is approximately 15 kPa. The CO2 adsorption capacities of NAC-K(600) and NAC-Rb(600) at 25 °C and 15 kPa were 1.58 and 1.56 mmol g−1, respectively. Because of the N-doping into their carbon frameworks, these NACs have CO2 adsorption capacities that are high compared with previously reported carbon materials.35 Further incorporation of N-containing functional groups may enhance CO2 adsorption at low partial pressures.
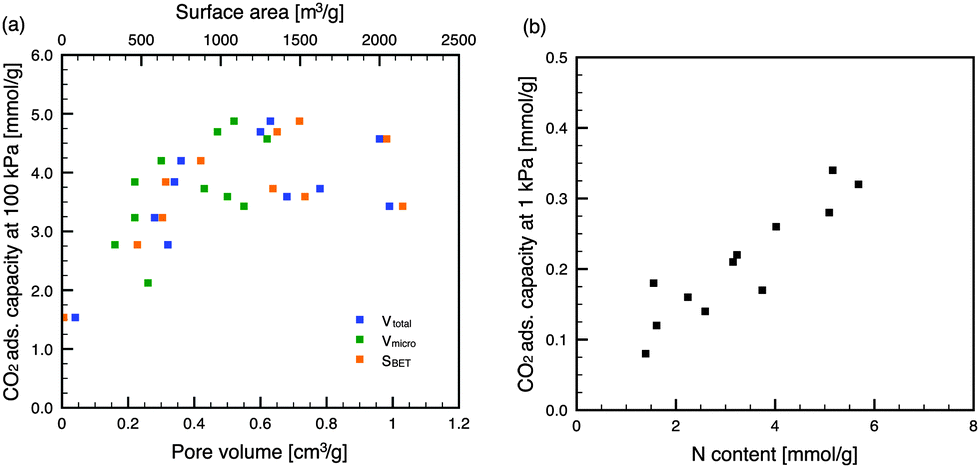 | ||
| Fig. 3 Dependence of CO2 adsorption capacity of NACs (25 °C) at (a) high partial pressure and (b) low partial pressure. | ||
Isosteric heats of adsorption were calculated from the isotherms at three different temperatures using the Clausius–Clapeyron equation. As shown in Fig. 4, NAC-Na(600) exhibited a relatively high heat of adsorption compared with the other NACs. In addition to the hysteresis loop observed in the CO2 isotherm, this result suggests that some CO2 molecules might be chemisorbed on amino groups on the carbon surface. As shown in Fig. 4, a slightly higher heat of adsorption was observed in the first stage of CO2 adsorption (amount of CO2 adsorbed <1 mmol g−1) compared with the later stages. The experimentally obtained initial heats of adsorption for the NACs were almost identical to the computationally estimated value (33.57 kJ mol−1) for N-doped carbon,20 indicating that the primary CO2 adsorption site on the NACs is N-containing functional groups.
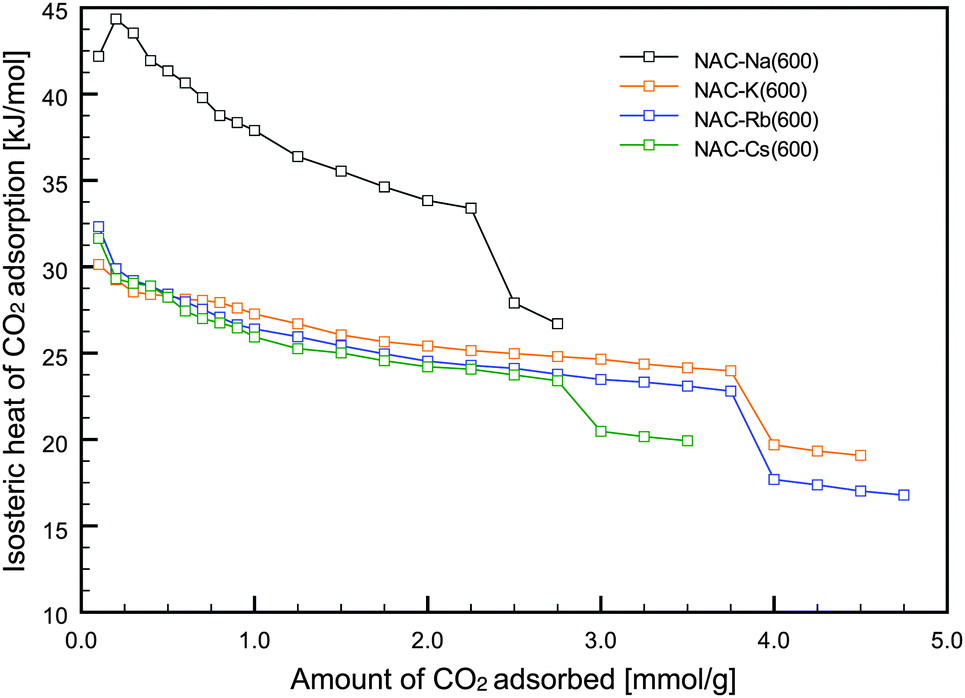 | ||
| Fig. 4 Isosteric heats of adsorption for NAC-M(600). | ||
Therefore, CO2 physisorption proceeds by a pore-filling mechanism, after CO2 molecules are selectively adsorbed on the N-containing functional groups of the NACs in the first stage of adsorption. The interaction energy is low enough to allow easy regeneration because it is in the range of the heat of adsorption for physisorption (25–50 kJ mol−1).2 The porous NACs prepared in this study show potential for efficient CO2 capture because of their CO2 adsorption performance and easy fabrication.
In conclusion, a simple method for the preparation of NAC by the chemical activation of chitosan was developed, and the favorable physicochemical properties of the NACs for CO2 adsorption were investigated. The preparation method provided high-performance CO2 adsorbents. The NACs prepared using K2CO3 and Rb2CO3 exhibited extremely high CO2 adsorption capacities. Furthermore, it was found that the N contents and pore structures (i.e., micropore volumes and surface areas) of the NACs clearly affected the CO2 adsorption at low and high partial pressures, respectively. The prepared NACs are potential adsorbents for efficient CO2 capture because of their high CO2 capture performance and lower heats of adsorption than those of amine-modified materials.5,13
This work was supported by the Ministry of Economy, Trade and Industry (METI), Japan.
Notes and references
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS PubMed.
- M. Nandi and H. Uyama, Chem. Rec., 2014, 14, 1134 CrossRef CAS PubMed.
- K. Shimada, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef PubMed.
- M. Tong, Q. Yang, Y. Xiao and C. Zhong, Phys. Chem. Chem. Phys., 2014, 16, 15189 RSC.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973 CrossRef CAS PubMed.
- Y. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650 CrossRef CAS PubMed.
- W. Liu, S. Choi, J. H. Drase, M. Hornbostel, G. Krishman, P. M. Eisenberger and C. W. Jones, ChemSusChem, 2010, 3, 899 CrossRef PubMed.
- N. Hiyoshi, K. Yogo and T. Yashima, Microporous Mesoporous Mater., 2005, 84, 357 CrossRef CAS.
- N. Hiyoshi, K. Yogo and T. Yashima, Chem. Lett., 2008, 37, 1266 CrossRef CAS.
- D. S. Dao, H. Yamada and K. Yogo, Ind. Eng. Chem. Res., 2013, 52, 13810 CrossRef CAS.
- T. Watabe and K. Yogo, Sep. Purif. Technol., 2013, 120, 20 CrossRef CAS.
- J. Fujiki and K. Yogo, Energy Fuels, 2014, 28, 6467 CrossRef CAS.
- J. Fujiki, H. Yamada and K. Yogo, Microporous Mesoporous Mater., 2015, 215, 76 CrossRef CAS.
- H. Yamada, D. S. Dao, F. A. Chowdhury, J. Fujiki, K. Goto and K. Yogo, Energy Procedia, 2014, 63, 2346 CrossRef CAS.
- M. Nandi, K. Okada, A. Dutta, A. Bhaumik, J. Maruyama, D. Derks and H. Uyama, Chem. Commun., 2012, 48, 10283 RSC.
- W. Xing, C. Liu, Z. Zhou, L. Zhang, J. Zhou, S. Zhuo, Z. Yan, H. Gao, G. Wang and S. Z. Qiao, Environ. Sci. Technol., 2012, 5, 7323 CAS.
- L. Wang and R. T. Yang, J. Phys. Chem. C, 2012, 116, 1099 CAS.
- R. Babarao, S. Dai and D. Jiang, J. Phys. Chem. C, 2012, 116, 7106 CAS.
- V. Chandra, S. U. Yu, S. H. Kim, Y. S. Yoon, D. Y. Kim, A. H. Kwon, M. Meyyappan and K. S. Kim, Chem. Commun., 2012, 48, 735 RSC.
- G. Sethia and A. Sayari, Energy Fuels, 2014, 28, 2727 CrossRef CAS.
- Y. Liu and J. Wilcox, Environ. Sci. Technol., 2012, 46, 1940 CrossRef CAS PubMed.
- W. Xing, C. Liu, Z. Zhou, J. Zhou, G. Wang, S. Zhuo, Q. Xue, L. Song and Z. Yan, Nanoscale Res. Lett., 2014, 9, 189 CrossRef PubMed.
- M. Sevilla and A. B. Fuertes, Environ. Sci. Technol., 2011, 4, 1765 CAS.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360 CAS.
- M. Olivares-Marín and M. Maroto-Valer, Greenhouse Gases: Sci. Technol., 2012, 2, 20 CrossRef.
- X. Fan, L. Zhang, G. Zhang, Z. Shu and J. Shi, Carbon, 2013, 61, 423 CrossRef CAS.
- H. Yang, Y. Yuan and S. C. E. Tsang, Chem. Eng. J., 2012, 185–186, 374 CrossRef CAS.
- C. L. Spiro, D. W. Mckee, P. G. Kosky and E. J. Lamby, Fuel, 1983, 62, 180 CrossRef CAS.
- D. W. McKee and D. Chatterji, Carbon, 1975, 13, 381 CrossRef CAS.
- M. S. Masnadi, J. R. Grace, X. T. Bi, C. J. Lim and N. Ellis, Appl. Energy, 2015, 140, 196 CrossRef CAS.
- A. A. Alhwaige, T. Agag, H. Ishida and S. Qutubuddin, RSC Adv., 2012, 3, 16011 RSC.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, N-Doped polypyrrole based porous carbons for CO2 capture, Adv. Funct. Mater., 2011, 21, 2781 CrossRef CAS.
- T. Chen, S. Deng, B. Wang, J. Huang, Y. Wang and G. Yu, RSC Adv., 2015, 3, 48323 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials characterization and adsorption experiments, N2 adsorption–desorption isotherms, pore size distributions and powder X-ray diffraction patterns. See DOI: 10.1039/c5cc06934c |
| This journal is © The Royal Society of Chemistry 2016 |